

Abstract
Rice blast disease is a major threat to rice production worldwide, but the mechanisms underlying rice resistance to the causal agent Magnaporthe oryzae remain elusive. Therefore, we carried out a transcriptome study on rice early defense response to M. oryzae. We found that the transcriptional profiles of rice compatible and incompatible interactions with M. oryzae were mostly similar, with genes regulated more prominently in the incompatible interactions. The functional analysis showed that the genes involved in signaling and secondary metabolism were extensively up-regulated. In particular, WRKY transcription factor genes were significantly enriched among the up-regulated genes. Overexpressing one of these WRKY genes, OsWRKY47, in transgenic rice plants conferred enhanced resistance against rice blast fungus. Our results revealed the sophisticated transcriptional reprogramming of signaling and metabolic pathways during rice early response to M. oryzae and demonstrated the critical roles of WRKY transcription factors in rice blast resistance.
Introduction
Plants are frequently challenged by various kinds of pathogens in their living environments. To survive pathogen attack, plants have evolved two layers of immune systems: pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and effector-triggered immunity (ETI)[1]–[3]. PTI is mediated by pattern recognition receptors (PRRs) that recognize PAMPs, whereas ETI is mediated by resistance (R) proteins that recognize pathogen effectors. Generally, PTI and ETI trigger similar defense responses, but ETI is much faster and quantitatively stronger [2]–[4].
As one of the most important staple crops, rice (Oryza sativa) feeds about half of the world’s population. However, 10–30% of annual rice production worldwide is lost to rice blast disease, caused by the fungal pathogen Magnaporthe oryzae [5], [6]. Because rice blast disease poses a serious and recurrent problem, the rice–M. oryzae interaction has been studied for decades. The infection starts with the attachment of fungal spores to plant leaf surfaces. Within 24 h post-inoculation (hpi), fungal spores germinate and form infection-specific appressoria to penetrate leaf cuticles and invade epidermal cells [5], [7]. It has been suggested that 24 hai is a critical point for pathogen invasion [5]. After fungal invasion, the rice–M. oryzae interaction obeys Flor’s “gene-for-gene” hypothesis [8]. If fungal effectors are recognized by cognate rice R proteins, ETI is triggered and culminates in hypersensitive response (HR) that halt fungal growth within 48 hours. If fungal effectors are not recognized, a limited PTI response occurs upon the recognition of fungal PAMPs, such as chitin [9], [10], and fungal hyphae continue to spread through plant tissues, leading to disease symptoms after days [5], [11]–[13]. Thus, the early defense response immediately after M. oryzae invasion is critical to the final outcome of rice blast resistance.
Genetic approaches have identified defense regulators in rice blast resistance. The rice homologue of Arabidopsis Nonexpresser of PR Genes 1 (NPR1), NH1, plays a positive role in rice blast resistance, as knockdown of NH1 impaired rice blast resistance, whereas overexpression enhanced resistance [14]–[16]. Transgenic plants overexpressing OsWRKY13, OsWRKY31, OsWRKY45, and OsWRKY53 showed enhanced resistance to M. oryzae, indicating their important roles in rice blast resistance [17]–[21]. Large-scale approaches have also been used to study rice early response to M. oryzae, including EST sequencing [22], robust-long serial analysis of gene expression [23], proteomics [24], [25], and microarrays [26]. A number of genes potentially involved in rice blast resistance were identified from these studies. In a previous transcriptome study on the incompatible interaction mediated by Pi33, seven genes were up-regulated more than 1.5 fold and 13 were down-regulated more than 2 fold at 24 hpi [26]. Given a broad range of genes responsible for plant defense response [27], [28], the defense transcriptome at early stage of rice–M. oryzae interactions is far from complete and the underlying mechanisms remain largely unclear.
To better understand the defense mechanisms underlying rice early response to M. oryzae, we carried out a whole-genome transcriptional analysis on both compatible and incompatible rice–M. oryzae interactions at 24 hpi. Our results showed that the transcriptional profiles of the compatible and incompatible interactions were mostly similar, with higher transcriptional changes in the incompatible interactions. Functional analysis revealed that the genes involved in signaling, secondary metabolism, and those encoding WRKY transcription factors, were significantly enriched among up-regulated genes. Overexpression of one such WRKY gene, OsWRKY47, greatly enhanced rice resistance to M. oryzae. Gene set enrichment analysis further revealed different regulation patterns among functional categories, suggesting genes with different functions were subjected to distinct regulation programming. This transcriptome study provided a comprehensive overview of transcriptional reprogramming during rice early response to M. oryzae.
Results
Transcriptional Profiles of Rice Compatible and Incompatible Interactions with M. oryzae are Similar Overall
To investigate the defense mechanisms underlying rice early response to M. oryzae, we conducted a microarray-based transcriptome study on compatible and incompatible rice–M. oryzae interactions at 24 hpi. One susceptible rice cultivar, LTH, and two resistant near-isogenic lines (NILs), IRBL18 and IRBL22, were used in this study (Figure 1). In our previous study, IRBL18 (carrying Pi1) and IRBL22 (carrying Pi9) exhibited broad-spectrum resistance to more than 200 M. oryzae strains collected in Yunnan Province of China [29]. Pi9 encodes a typical nucleotide binding and leucine rich repeat (NB-LRR) protein and confers resistance to another 21 blast strains from nine countries [30]. In this study, the incompatible interactions in IRBL18 and IRBL22 were used to investigate the R-mediated ETI response, while the compatible interaction in susceptible LTH represented the PTI response (Figure 1).
Figure 1. The disease symptoms on IRBL18, IRBL22 and LTH leaves at 7 days post-inoculation.
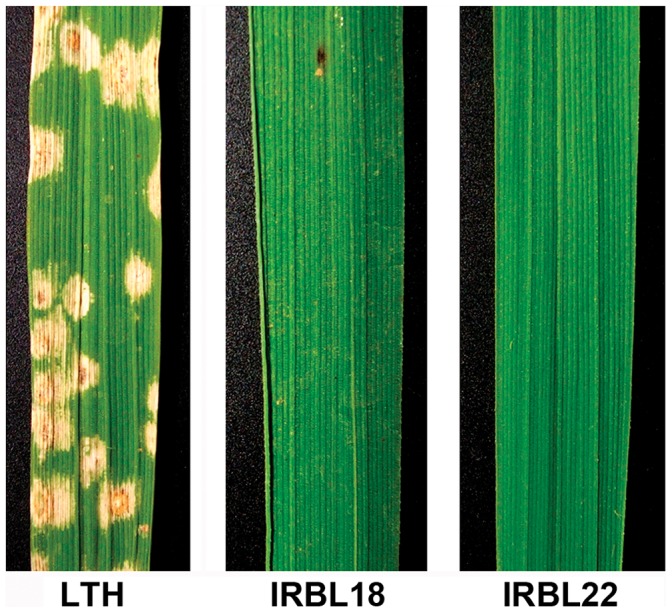
The inoculation experiment was repeated three times (n = 10 seedlings) with similar results.
For microarray analysis, four-week-old rice seedlings were inoculated with fungal spore suspension or mock-inoculated with spore-free water. Microarray analyses were performed using Affymetrix GeneChips with three independent biological replicates. We identified a total of 755 differentially expressed genes (DEGs; fold change ≥2; P<0.05; listed in Table S1) between the fungal- and mock-inoculated IRBL18, IRBL22 or LTH plants. Twenty genes were validated by quantitative RT-PCR (Table S2). Among the 755 DEGs, 551 were differentially regulated in IRBL18, 649 in IRBL22 and only 131 in LTH. The number of DEGs in either IRBL18 or IRBL22 was about four times more than that in LTH (Figure 2A), suggesting that the two incompatible interactions involved more expanded genome-wide transcriptional regulation than did the compatible interaction. We also found that the up-regulated genes greatly outnumbered the down-regulated ones in either rice–M. oryzae interaction, demonstrating that the majority of genes involved in rice early response to M. oryzae were positively regulated (Figure 2B). Interestingly, 451 DEGs were shared by IRBL18 and IRBL22, and 120 of them were also differentially regulated in the susceptible LTH plants (Figure 2A). This reflects some basal similarities between the transcriptomes in rice compatible and incompatible interactions with M. oryzae.
Figure 2. Differentially expressed genes (DEGs) identified from the transcriptome study.

A: The venn diagram of DEGs in IRBL18, IRBL22 and LTH. The numbers of up- and down-regulated genes were shown in red and green, respectively. B: Hierarchical clustering of the log2-transformed fold changes. The scale bar displays log2-transformed fold changes. C: Scatter plot of log2-transformed fold changes.
A hierarchical clustering analysis was conducted on the log2-transformed fold change values (logFCs) of 755 DEGs. The transcriptional profiles in IRBL18 and IRBL22 were similar with comparable amplitudes (Figure 2B), indicating that the Pi1- and Pi9-mediated pathogen recognitions triggered similar ETI responses in the transcriptome term. Interestingly, the transcriptional profile in LTH was also similar to those in IRBL18 and IRBL22, but the amplitudes of gene expression changes were globally lower. Differences in the transcriptional profiles between compatible and incompatible rice–M. oryzae interactions were mostly quantitative, especially in the up-regulated DEGs (Figure 2C). These results suggest that a common set of defense-responsive genes are involved in rice early defense response to M. oryzae, and that they are more prominently regulated in rice incompatible interactions.
Transcriptional Reprogramming in Signaling and Metabolism during Rice Early Response to M. oryzae
As an important step towards understanding the molecular mechanisms underlying rice early response to M. oryzae, we functionally analyzed the 755 DEGs with their Gene Ontology (GO) annotations. The enrichment analysis showed that “kinase activity” was the most significantly enriched GO molecular function term among up-regulated genes in each rice line (P = 5.61×10−21 in IRBL18, 2.38×10−17 in IRBL22, and 4.59×10−4 in LTH), indicating that a significant number of kinase genes were positively regulated during rice early response to M. oryzae (Table 1). Both “RNA binding” and “DNA binding” were depleted among the up-regulated genes in IRBL18 or IRBL22 (P<0.05), suggesting that most genes were not under positive transcriptional regulation. No significantly enriched or depleted GO terms were found in the down-regulated genes, partially because a small number of genes were suppressed.
Table 1. The enrichment analysis of GO in the up-regulated genes.
| GO ID | GO term | Number in reference | IRBL18 | IRBL22 | LTH | |||
| count | P-value | count | P-value | count | P-value | |||
| total | 17,752 | 516 | 573 | 113 | ||||
| Over-represented | ||||||||
| GO:0016301 | kinase activity | 1,086 | 94 | <<0.001 | 93 | <<0.001 | 19 | <0.001 |
| GO:0030246 | carbohydrate binding | 108 | 12 | <0.001 | 14 | <0.001 | 3 | 0.158 |
| GO:0019825 | oxygen binding | 121 | 12 | 0.001 | 11 | 0.007 | 3 | 0.184 |
| GO:0005515 | protein binding | 2,130 | 82 | 0.018 | 95 | 0.003 | 21 | 0.158 |
| GO:0005215 | transporter activity | 911 | 33 | 0.265 | 47 | 0.004 | 9 | 0.492 |
| Under-represented | ||||||||
| GO:0003677 | DNA binding | 879 | 11 | 0.008 | 15 | 0.039 | 3 | 1.000 |
| GO:0003723 | RNA binding | 504 | 1 | <0.001 | 2 | <0.001 | 0 | 1.000 |
P-values were corrected with the Benjamini-Hochberg’s method.
We then performed a more detailed analysis using MapMan with a manually updated mapping file [31]. Among the 755 DEGs, 551 were assigned to 28 known functional categories (Figure 3). The statistical significances of their enrichments in each rice line were assessed with the 17,752 expressed genes as reference. In the resistant IRBL18 and IRBL22, the genes involved in “signaling”, “amino acid metabolism”, “secondary metabolism”, “cell wall synthesis” and “stress response” were significantly enriched in the up-regulated DEGs (P<0.05; Table 2), suggesting that the resistant rice plants went through a transcriptional reprogramming, both in signaling and metabolism, during their early responses to M. oryzae. However, in the susceptible LTH, only three categories, “signaling”, “amino acid metabolism” and “secondary metabolism”, were enriched, partially due to the limited transcriptional reprogramming in compatible interaction.
Figure 3. Overview of the transcriptional changes in IRBL18 at 24 hours post-inoculation.

Genes significantly up- (red) or down-regulated (green) in fungal-inoculated leaf samples relative to mock-inoculated are illustrated. The scale bar displays log2-transformed fold changes.
Table 2. The enrichment analysis of functional categories in the up-regulated genes.
| Functional categories | Number in reference | IRBL18 | IRBL22 | LTH | |||
| count | P-value | count | P-value | count | P-value | ||
| Total | 17,752 | 516 | 573 | 113 | |||
| Over-represented categories | |||||||
| Signaling | 1,275 | 105 | <<0.001 | 107 | <<0.001 | 19 | 0.011 |
| Amino acid metabolism | 229 | 19 | <0.001 | 20 | <0.001 | 6 | 0.045 |
| Secondary metabolism | 255 | 27 | <0.001 | 27 | <0.001 | 8 | 0.008 |
| Cell wall synthesis | 195 | 13 | 0.032 | 14 | 0.029 | 2 | 1.000 |
| Stress response | 481 | 32 | <0.001 | 35 | <0.001 | 5 | 0.830 |
| Transport | 721 | 30 | 0.163 | 42 | 0.001 | 9 | 0.357 |
| Under-represented categories | |||||||
| RNA regulation | 1,903 | 27 | <0.001 | 30 | <0.001 | 8 | 1.000 |
| DNA regulation | 317 | 1 | 0.024 | 1 | 0.009 | 0 | 1.000 |
| Protein metabolism | 2,527 | 38 | <0.001 | 42 | <0.001 | 4 | 0.038 |
P-values were corrected with the Benjamini-Hochberg’s method.
Consistent with GO analysis results, the functional category “RNA regulation” was significantly depleted in the up-regulated DEGs of IRBL18 and IRBL22 (P<0.001; Table 2). However, within this significantly depleted category, the WRKY transcription factor family was the only significantly-enriched one among 14 tested transcription factor families (P = 1.47×10−6 in IRBL18, 3.00×10−5 in IRBL22, and 4.33×10−2 in LTH), suggesting that these WRKYs were likely to play crucial roles in rice early response to M. oryzae (see below).
Genes Encoding Signaling Components were Extensively Up-regulated
Functional analysis indicated that “signaling” was the most significantly enriched functional category among the up-regulated genes in IRBL18 and IRBL22 (P = 5.96×10−21 in IRBL18 and 2.33×10−18 in IRBL22) and the second most significantly enriched in LTH up-regulated genes (P = 1.05×10−2; Table 2). Of the up-regulated DEGs, 20.3% encoded signaling components in IRBL18, 18.7% in IRBL22, and 16.8% in LTH, significantly more than 7.2% in the reference data set. Thus, a broad range of genes involved in signaling pathways were up-regulated during rice early response to M. oryzae.
Further analysis showed that within “signaling”, the sub-category “receptor kinase” was even more significantly enriched in the up-regulated genes (P = 5.00×10−35 in IRBL18, 1.53×10−31 in IRBL22, and 2.33×10−6 in LTH). A total of 103 receptor kinase genes were up-regulated in either rice line, whereas only one was down-regulated (Figure 4B; Table S4). Moreover, this enriched sub-category contained several receptor kinase sub-families, including receptor-like kinases (RLKs) with various extracellular domains, receptor-like cytoplasmic kinases (RLCKs), and wall-associate kinases (WAKs), some of which were also significantly enriched in the up-regulated genes (Table 3). So many receptor kinase genes being up-regulated suggested that signal perception was extensively activated early in M. oryzae infection. In contrast to 103 up-regulated receptor kinases, only nine NB-LRR genes were up-regulated (Figure 4A), although NB-LRRs are usually responsible for specific recognition of pathogen Avr proteins to trigger the ETI response. The different expression patterns between receptor kinase and NB-LRR genes suggested that they are subjected to distinct transcriptional regulation procedures in rice early response to M. oryzae.
Figure 4. The expression patterns of receptor kinase and nucleotide binding and leucine rich repeat (NB-LRR) genes.

A: The expression patterns of NB-LRR genes. B: The expression patterns of receptor kinase genes, with the family names shown on the right side. The scale bar displays log2-transformed fold changes.
Table 3. The enrichment analysis of receptor kinases in the up-regulated genes.
| Sub-family names | Number in reference | IRBL18 | IRBL22 | LTH | |||
| counts | P-value | counts | P-value | counts | P-value | ||
| total | 17,752 | 516 | 573 | 113 | |||
| Leucine rich repeat I | 12 | 3 | 0.031 | 3 | 0.036 | 0 | 1.000 |
| Leucine rich repeat X | 16 | 4 | 0.008 | 2 | 0.343 | 0 | 1.000 |
| Leucine rich repeat VIII | 34 | 5 | 0.022 | 5 | 0.029 | 1 | 0.830 |
| Leucine rich repeat VIII-1 | 31 | 5 | 0.015 | 5 | 0.022 | 1 | 0.830 |
| DUF 26 | 36 | 12 | <0.001 | 10 | <0.001 | 3 | 0.031 |
| Legume-lectin | 35 | 9 | <0.001 | 9 | <0.001 | 3 | 0.031 |
| Wheat LRK10 like | 21 | 7 | <0.001 | 8 | <0.001 | 3 | 0.010 |
| S-locus glycoprotein like | 62 | 10 | <0.001 | 11 | <0.001 | 2 | 0.488 |
| Wall associated kinase | 51 | 14 | <0.001 | 12 | <0.001 | 1 | 0.950 |
| RLCK | 113 | 7 | 0.207 | 10 | 0.027 | 2 | 0.827 |
P-values were corrected with the Benjamini-Hochberg’s method.
Other signaling component genes with known functions in rice resistance were also up-regulated during the early response to M. oryzae. For instance, NH1 (LOC_Os01g09800), a positive regulator in rice blast resistance, was 2.5-, 3.5-, and 2.0-fold up-regulated in IRBL18, IRBL22, and LTH, respectively. Two MAP kinase genes (LOC_Os03g17700 and LOC_Os02g04230) were also up-regulated, in which LOC_Os03g17700 encodes an negative regulator, OsMPK5, in blast resistance [32]. Another five calmodulin-binding protein genes (CBPs) and two calcium/calmodulin-dependent protein kinase genes (CDPKs) were also up-regulated (Table S1), suggesting the calcium signaling was involved as well.
Genes Involved in Both Shikimate and Phenylpropanoid Pathways were Up-regulated
Our data showed that a significant number of genes involved in amino-acid and secondary metabolism were up-regulated during rice early response to M. oryzae (Table 2; Table S5). To better illustrate the transcriptional regulation of rice metabolism during this process, we integrated the up-regulated genes in these pathways into an overall metabolic view (Figure 5). Notably, dozens of genes involved in amino acid and secondary metabolism were up-regulated and formed a metabolic map consisting of the shikimate and phenylalanine biosynthesis pathways as well as the downstream phenylpropanoid biosynthesis pathway. In the shikimate biosynthesis pathway, six out of 14 expressed enzyme genes were up-regulated in either rice line, whose products catalyze five successive reactions. Following the transcriptional activation of shikimate biosynthesis pathway, the genes encoding chorismate mutase and prephenate dehydratase, catalyzing chorismate to form phenylalanine, were also up-regulated. In the downstream, the phenylpropanoid biosynthesis pathway was transcriptionally activated, i.e., 19 genes out of 80 various enzyme genes were up-regulated. Like the shikimate pathway, the enzymes encoded by these 19 genes catalyze a series of eight reaction steps in phenylpropanoid pathway. These results showed that both shikimate and phenylpropanoid pathways were under an expanded transcriptional activation during rice early response to M. oryzae, indicating the transcriptional reprogramming occurred in metabolic pathways.
Figure 5. Transcriptional changes in shikimate and phenylpropanoid biosynthesis pathways.

A: The expression patterns of genes in the shikimate and phenylpropanoid biosynthesis pathway. The scale bar displays log2-transformed fold changes. B: The metabolic view of shikimate and phenylpropanoid biosynthesis pathways. The steps catalyzed by enzymes encoded by up-regulated genes were shown in red. The shikimate and phenylalanine biosynthesis pathways were highlighted with pink background and the phenylpropanoid biosynthesis pathway was highlighted with light blue background. Gene abbreviations were listed in Table S5.
Moreover, eight of 34 enzyme genes involved in jasmonic acid biosynthesis were up-regulated in either rice line (Table S6), suggesting that the jasmonic acid biosynthesis pathway was activated during rice early response to M. oryzae. In addition to the genes involved in established metabolic pathways, other individual enzyme gene families, including cytochrome P450 s and peroxidases, were also significantly enriched in the up-regulated DEGs. In plant defense response, cytochrome P450 s and peroxidases are often involved in modifying a plethora of anti-pathogen proteins. The enrichment of these genes suggested that these enzymes could function as active players in the defense response against rice blast fungus.
Transcriptional Differences Between ETI and PTI Varied Among Functional Categories
In the functional analyses above, we found that differences in the transcriptional profiles between compatible and incompatible rice–M. oryzae interactions were mostly quantitative (Figure 2). This result raised an interesting question, i.e., whether these quantitative differences in transcriptional profiles were of the same scale across functional categories. To address this question, we carried out two complementary analyses. First, we compared the enrichment of functional categories among rice lines. All 17,752 expressed genes were sorted by descending their t-statistic values. The percentage of a given functional category in a 500-gene sliding window was calculated and plotted along the gene list. For the “signaling” category, the lines representing the percentages of signaling components were above the upper threshold (Figure 6A), demonstrating that “signaling” was significantly enriched among the up-regulated genes, consistent with the enrichment analysis above (Table 2). However, among the nearly one thousand most-induced genes, signaling components comprised around 15% in IRBL18 and IRBL22 and only 12% in LTH, reflecting a 3% difference in this functional category between incompatible and compatible interactions (on the left in Figure 6A). This result suggested that the genes encoding signaling components were not only more prominently (Figure 4B) but also more extensively up-regulated in the incompatible interactions than in the compatible interaction.
Figure 6. The sliding window analysis of genes involved in signaling (A) and secondary metabolism (B).

The solid lines represent the percentages of certain functional category in a 500-gene window in IRBL18 (red), IRBL22 (blue) and LTH (green). The blue dotted lines represent the 95% confidence levels.
The pattern was different in “secondary metabolism” (Figure 6B). For each rice line, the percentage of genes involved in secondary mechanism was high within the most up-regulated genes, suggesting that these genes tended to be intensively up-regulated during rice early response to M. oryzae. The difference between IRBL18 or IRBL22 and LTH was about 1% within the most up-regulated genes, also demonstrating that more genes involved in secondary metabolism were up-regulated in the two incompatible rice–M. oryzae interactions. By this method, we found that there were more genes in “amino acid metabolism”, “cell wall synthesis”, “stress response”, and “transporter” up-regulated in the resistant IRBL18 or IRBL22 than in the susceptible LTH (Figure S1). These results demonstrated that the transcriptional regulations in various functional categories differed not only in global patterns but also between compatible and incompatible interactions.
To quantitatively analyze differences within functional categories between interactions, we performed a permutation-based Gene Set Enrichment Analysis (GSEA) [33]. The null hypothesis that genes in a given category had equal transcriptional changes in the compatible and incompatible interactions (see Materials and Methods) was tested. “Secondary metabolism” and “amino acid metabolism” exhibited the most differences between IRBL18 or IRBL22 and LTH (P<0.05; Table 4). The “signaling” and “stress response” differed to a lesser degree between the incompatible and compatible interactions (P<0.1). More importantly, WRKY genes were also more prominently regulated in IRBL18 and IRBL22 than in LTH in a less degree (P = 0.0664 in IRBL18 vs. LTH, 0.0815 in IRBL22 vs. LTH, 0.410 in IRBL18 vs. IRBL22). Given their small number (45), the crucial roles of these WRKYs in rice blast resistance were apparent according to the expression patterns demonstrated here.
Table 4. The result of Gene-set enrichment analysis on functional categories.
| Functional categories | IRBL18 vs LTHP-value | IRBL22 vsLTHP-value | IRBL18 vs IRBL22P-value |
| Secondary metabolism | 0.0079 | 0.0242 | 0.1785 |
| Amino acid metabolism | 0.0255 | 0.0292 | 0.4445 |
| Stress response | 0.0314 | 0.0815 | 0.2068 |
| Lipid metabolism | 0.0429 | 0.1288 | 0.1562 |
| Signaling | 0.0574 | 0.0962 | 0.3255 |
| Protein metabolism | 0.0583 | 0.1571 | 0.1804 |
| Hormone metabolism | 0.07 | 0.3105 | 0.0917 |
| Transporter | 0.0746 | 0.1392 | 0.255 |
| DNA regulation | 0.0972 | 0.2947 | 0.135 |
| Cell wall modification | 0.1084 | 0.1221 | 0.5054 |
| RNA regulation | 0.1142 | 0.3158 | 0.1267 |
| Nucleotide metabolism | 0.1197 | 0.1657 | 0.3717 |
| Redox regulation | 0.1352 | 0.3261 | 0.1332 |
| Development | 0.3478 | 0.4245 | 0.3777 |
| Photosynthesis | 0.4271 | 0.4195 | 0.5273 |
WRKY Transcription Factor Genes play Important Roles in Rice Blast Resistance
Among the 45 WRKY genes expressed during rice early response to M. oryzae, eleven were up-regulated in IRBL18, ten in IRBL22, and only three in LTH (Table 5). WRKY transcription factor genes were significantly enriched in the up-regulated DEGs in each rice line (P<0.001 in IRBL18 and IRBL22, and P = 0.0433 in LTH). Interestingly, these WRKYs showed different expression patterns between compatible and incompatible interactions (Figure 7A). For instance, OsWRKY76 was significantly induced in both resistant rice lines, with 8.8-fold up-regulation in IRBL18 and 4.6-fold in IRBL22, whereas no change occurred in LTH. OsWRKY47 was up-regulated 3.3-fold in IRBL18, 2.4-fold in IRBL22, and only 1.8-fold in LTH (Table S7; also validated in Table S2). Among these WRKYs, OsWRKY45 [19] and OsWRKY55 (designated as OsWRKY31 in [20]) have been demonstrated to play positive roles in rice–M. oryzae interactions, further supporting our microarray results.
Table 5. The summary of differentially regulated transcription factor genes.
| Family names | Number in reference | IRBL18 | IRBL22 | LTH | |||
| up | down | up | down | up | down | ||
| total | 17,752 | 516 | 35 | 573 | 76 | 113 | 18 |
| WRKY | 46 | 11 | 0 | 10 | 0 | 3 | 0 |
| MYB | 51 | 4 | 0 | 3 | 0 | 2 | 0 |
| GARP | 31 | 2 | 0 | 2 | 0 | 0 | 0 |
| HSF | 18 | 2 | 0 | 2 | 0 | 0 | 0 |
| NAC | 55 | 2 | 0 | 2 | 1 | 1 | 0 |
| GRAS | 23 | 1 | 0 | 1 | 0 | 0 | 0 |
| bHLH | 74 | 1 | 1 | 2 | 1 | 1 | 0 |
| DOF | 16 | 1 | 0 | 1 | 0 | 0 | 0 |
| AP2/EREBP | 69 | 0 | 0 | 4 | 1 | 0 | 0 |
| bZIP | 59 | 0 | 1 | 0 | 2 | 0 | 1 |
| Tify | 13 | 0 | 0 | 1 | 0 | 0 | 0 |
| GATA | 14 | 0 | 0 | 1 | 0 | 0 | 0 |
Figure 7. The expression patterns of OsWRKYs (A) and the trans-activation activities of selected OsWRKYs (B).
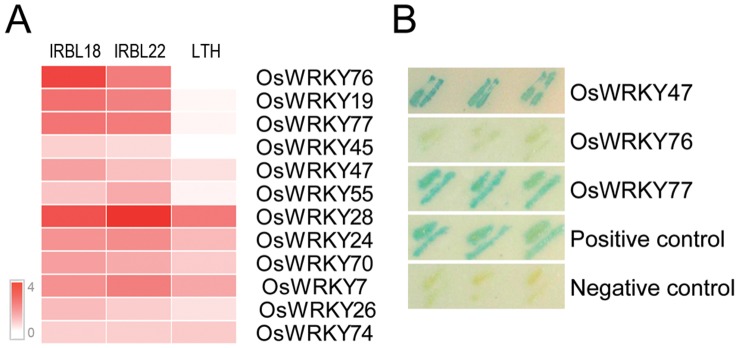
The scale bar in (A) displays log2-transformed fold changes.
To further verify the biological roles of WRKY transcription factors in rice blast resistance, we select three OsWRKYs with different expression patterns, including OsWRKY47, OsWRKY76 and OsWRKY77, for further functional analyses. The yeast assay showed that OsWRKY47 and OsWRKY77 exhibited trans-activation activity, while OsWRKY76 did not (Figure 7B), suggesting these up-regulated OsWKRYs encoded transcription regulators with different functions. We then overexpressed these three OsWRKYs in rice cultivar Taipei 309 (TP309). Interestingly, the transgenic plants overexpressing OsWRKY76 or OsWRKY77 were severely compromised in their viability (data not shown). Twenty-two independent OsWRKY47 overexpression transgenic lines were obtained and two lines (pUbi:OsWRKY47#7 and pUbi:OsWRKY47#16) with constitutively-high expression levels of OsWRKY47 were selected for rice blast resistance assay (Figure 8B). Seven days after inoculation with M. oryzae strain 96-4-1a, typical lesions with grey centers occurred and spread on the leaves of wild-type rice plants, while no visible lesions were observed on the leaves of two pUbi:OsWRKY47 transgenic lines, indicating that the transgenic plants exhibited greatly enhanced blast resistance (Figure 8A; Table S8). Consistent with the enhanced blast resistance, the expression levels of the marker gene Pathogenesis-related 10 (PR10) were significantly higher in the transgenic plants than in the wild-type TP309 (Figure 8C). These results demonstrated that OsWRKY47 played an important role in rice blast resistance.
Figure 8. Overexpression of OsWRKY47 enhanced rice resistance to Magnaporthe oryzae strain 96-4-1a.

A: The disease symptoms on TP309, pUbi:OsWRKY47#16 and #7 transgenic plants at 7 days post-inoculation. The inoculation experiments were repeated three time (n = 15 seedlings) with similar results. B, C: The expression levels of OsWRKY47 and PR10 in TP309 and pUbi:OsWRKY47 transgenic plants determined by quantitative RT-PCR. The expression levels were standardized to OsUBQ and the value in TP309 was set at 1.0. Error bars in (B) and (C) represent the SD from three biological replicates, each containing at least 10 seedlings. The expression levels that differ significantly (P<0.05) from those in TP309 were marked with asterisks.
Discussion
After decades of study of rice–M. oryzae interactions, a number of blast resistance genes have been mapped and identified, and many have been successfully applied in the field. However, because of blast population diversity and genetic instability, many newly released rice cultivars have a short lives of only 2–3 years [6], [12], increasing the urgency of studying the molecular mechanisms underlying rice–M. oryzae interactions. Here, we carried out a transcriptome study on rice compatible and incompatible interaction with M. oryzae at a critical early stage of 24 hpi. A total of 755 differentially-regulated genes were identified from M. oryzae-challenged rice plants, including genes encoding receptors, signaling transducers, transcription factors, and enzymes involved in metabolic pathways. Our study provided an overview of transcriptional reprogramming during rice early defense response to blast fungus.
Rice resistance against blast fungus obeys the “gene-for-gene” hypothesis [8]. In this study, we used two resistant NILs, IRBL18 and IRBL22, to investigate rice early response in incompatible interactions with M. oryzae, and used their susceptible parent LTH to investigate a compatible interaction. By comparing these three transcriptomes, we found that the Pi1- and Pi9-mediated ETI responses in IRBL18 and IRBL22 shared similar transcriptional profiles to the PTI response in LTH, with stronger transcriptional changes during the ETI responses (Figure 2). These patterns resembled those in the Arabidopsis thaliana–Pseudomonas syringae interaction, in which the transcriptional profiles of compatible and incompatible A. thaliana–P. syringae interactions at the same time points were similar and the differences in profiles were mostly quantitative [4]. In another well-studied barley–powdery mildew interaction, similar transcriptional regulation patterns were also found between compatible and incompatible interactions at an early stage [28], [34]. In the innate immune system, plants detect the potential pathogens via PRRs and R proteins, activating PTI and ETI, respectively. Although triggered by different mechanisms, the transcriptional signatures in PTI and ETI are largely similar, suggesting that their responses are the same overall but differ in magnitude [2]–[4]. Our results indicate that the rice–M. oryzae interaction shows a quantitative nature similar to other plant–pathogen interactions.
During the functional analysis, we noticed that some functional categories, such as “signaling”, comprised a greater percentage of DEGs in IRBL18 and IRBL22 than in LTH. This raised the interesting question of whether the quantitative differences were of the same scale across the whole defense transcriptome. As functional enrichment analyses could be only conducted with a certain gene list with a given cutoff, we carried out two complementary analyses with all expressed genes. Assuming that all transcriptional changes were proportionally higher in the incompatible interactions, the genes in any given category should have a similar distribution when ranking according to their significance of transcriptional changes. Interestingly, this was not the case here. The percentages of enriched categories identified in functional analyses, including “signaling”, “stress response”, “secondary metabolism” and “amino acid metabolism”, in the most-induced genes were higher in the resistant IRBL18 and IRBL22 plants than in the susceptible LTH plants, and the differences in their transcriptional changes were significant to different extents (Table 4). These results suggested that the genes in these categories were under broader (Figure 6; Figure S1) as well as stronger (Figure 2) regulation in ETI than in PTI. Moreover, different functional categories seemed to exhibit different regulation patterns. For instance, the genes involved in signaling were broadly up-regulated among the first one thousand genes, while those involved in secondary mechanism were mainly enriched in only a few hundred (Figure 6B). Given that “secondary mechanism” was the most significantly different functional category between ETI and PTI, and enriched in the most up-regulated genes, it indicated that the secondary metabolism genes were highly induced during rice early response to M. oryzae (Table 4). The extensive regulation in signaling and intensive regulation in metabolism suggested that different functional categories within rice immune system were under distinct transcriptional regulation mechanisms, which might reflect the amplification of defense signals via signaling cascades.
Plant immunity is governed by sophisticated systems involving perception receptors, signaling mediators, transcriptional regulators, and a variety of anti-pathogen proteins [2], [3], [13], [27]. Among the functional categories enriched among up-regulated genes, we found that genes involved in signaling were the most significantly enriched in IRBL18 and IRBL22, in which a significant number (103) of receptor kinase genes were up-regulated (Figure 4; Table S4). Receptor kinases, which comprise one of the largest plant gene families, are often responsible for perceiving internal and external signals [35]–[37]. In Arabidopsis, FLS2 functions as a PRR receptor to recognize bacterial flagellin, leading to a PTI response [38]–[40]. OsFLS2, the rice homolog of FLS2, has been also identified as a flagellin perception receptor [41], and its encoding gene was up-regulated 1.9- and 1.7-fold in IRBL18 and IRBL22, respectively. OsWAK25 (LOC_Os03g12470) encodes a receptor with a positive role in resistance to the bacterial pathogen Xanthomonas oryzae pv. oryzae [42] and was also up-regulated during rice early response to M. oryzae. The extensive up-regulation of receptor kinases genes, including those responding to other pathogens, suggested that rice plants challenged by M. oryzae might activate a broad range of perception receptors to prepare for further potential infections by pathogens, such as X. oryzae.
The WRKY transcription factor family was one of the most notably enriched gene families identified in this study, because it was the only enriched transcription factor family within the significantly depleted “RNA regulation” category (Table 2). Our results indicated that WRKY transcription factors play important roles in rice blast resistance, as was previously demonstrated by genetic approaches. For example, overexpressing OsWRKY45, OsWRKY55, and OsWRKY47 enhanced rice blast resistance, indicating their positive roles in rice defense response to M. oryzae [19], [20]; this study). Knockdown of OsWRKY28 led to an increase in blast resistance, indicating its negative role [43]. Moreover, Xie et al. demonstrated that OsWRKY72 and OsWRKY77 played synergistic roles with abscisic acid (ABA) in aleurone cells, and OsWRKY24 and OsWRKY45 repressed ABA induction of downstream genes, reflecting OsWRKYs involvement in plant development [44]. In Arabidopsis, WRKY transcription factors with both positive and negative regulatory roles form a complex transcriptional network to modulate plant defense responses [45]. Therefore, the up-regulation of these OsWRKY genes, whose products possess distinct trans-activation activity and biological roles, suggested that a complex network of rice WRKY transcription factors could also be activated to fine-tune the early responses to M. oryzae.
In the downstream of defense signaling pathways, many defense-related genes are transcriptionally regulated to actively respond to invasive pathogens [2], [3], [27]. The phenylpropanoid biosynthesis pathway is of great importance in this process, because phenylpropanoids are important antimicrobial compounds and lignification plays an important role in fungal resistance [46], [47]. Down-regulation of phenylpropanoid biosynthesis genes compromises plant resistance to fungal pathogens [46], [48]–[50]. These biosynthesis genes are under coordinated transcriptional regulation, particularly by MYB transcription factors [51]–[53]. In this transcriptome study, we found that many enzyme genes involved in the phenylpropanoid biosynthesis pathway were up-regulated, as were those in the upstream shikimate and phenylalanine biosynthesis pathways, reflecting a transcriptional reprogramming in metabolism during rice early response to M. oryzae. This transcriptional reprogramming was correlated with massive metabolic changes during rice–M. oryzae interactions, in which dozens of metabolites accumulated in rice leaves within 48 h of challenge by M. oryzae [54]. Moreover, nearly 40% of the “explanatory” m/z signals were predicted to derive from phenylpropanoid metabolites, highlighting our finding of transcriptional reprogramming in plant metabolism prior to an active metabolic response.
In summary, we demonstrated that a transcriptional reprogramming in signaling and metabolism happened during rice early response to M. oryzae. By comparing the transcriptomes in compatible and incompatible rice–M. oryzae interactions, we revealed that the defense response to rice blast fungus was quantitative in nature, with distinct transcriptional regulation mechanisms in various functional categories. This study enhanced our understanding of the complex network of transcriptional regulation during rice early response to the blast fungus.
Materials and Methods
Plant Material and Growth Conditions
Oryza sativa L. ssp japonica cultivar LTH and two near isogenic lines with LTH background, IRBL18 and IRBL22 bred by International Rice Research Institute (IRRI), were used in this study. All rice seedlings were grown in a greenhouse at Yunnan Agricultural University from April to May in the growing season. Seeds were surface-sterilized, washed repeatedly and soaked in water for germination. Five days later, 30 well-germinated seeds of each rice line were planted in 35×25×8.5-cm trays filled with sterile soil. The trays were put in water tanks and watered every three days. Twenty grams of nitrogen equivalent were fertilized in each tray two weeks before inoculation. The same growth conditions were applied to both fungal- and mock-inoculated groups of rice seedlings.
Fungal Inoculation and RNA Preparation
The rice blast fungus (Magnaporthe oryzae) strain CH63 was cultured as previously [29], and conidia were harvested by rinsing the cultural plates with sterile distilled water and filtering through two layers of gauze. For the inoculation group, four-week-old rice seedlings were inoculated by spraying a conidial suspension of 1×105 conidia mL−1 with 0.02% Tween-20. For the control group, sterile water with 0.02% Tween-20 was sprayed instead. The fungal- and mock-inoculated rice seedlings were kept in dark inoculation chambers with 100% humidity at 26°C.
At 24 hours after the inoculation, the fully-expanded third and fourth leaves of 20 rice seedlings from each rice line were harvested, pooled together and immediately frozen in liquid nitrogen. The remaining ten seedlings were left for disease assay at 7 days post-inoculation (dpi). Total RNA was extracted from leaf samples with TRIzol agent (Invitrogen) and purified with RNeasy kit (Qiagen). The quality of RNA was assessed by determining the A260/A280 ratio of RNA and by gel electrophoresis. The whole experiment was repeated three times with two-day intervals, resulting in three independent biological replicates of RNA samples.
Microarray Analysis
Affymetrix GeneChip Rice Genome Arrays (Affymetrix, Santa Clara, USA) were used in this transcriptome study. RNA quality assessment, RNA labeling and microarray hybridization were performed at Capitalbio Ltd. (Beijing, China) according to the manufacturer’s instructions. After hybridization, the Affymetrix GeneChip Scanner 3000 was used for microarray scan and the Affymetrix GeneChip Operating Software Version 1.0 was used for image analysis and data extraction.
For microarray analysis, we used a series of R/Bioconductor packages (http://www.bioconductor.org) [55]. Briefly, the CEL files were imported into R environment and the robust multi-array average (rma) methodology, as implemented in the affy package, was used for microarray normalization [56], [57]. The Pearson’s correlation coefficiencies of log2-transformed expression values between replicates ranged from 0.985 to 0.995, indicating high consistency between biological replicates (TableS9). Following normalization, a non-specific filtering step was carried out. The probe sets called “Present”, which were determined by the mas5 algorithm, in at least two among three replicates in at least one rice sample were regarded as expressed and included in further analyses. Bacterial control probe sets and ambiguous probe sets that match no gene or multiple genes annotated in MSU Rice Genome Annotation Project database release 7.0 (http://rice.plantbiology.msu.edu/; [58]) were also removed, resulting in a final 21,593-data set representing 17,752 expressed rice genes (see below).
Subsequently, the limma package was used to identify differentially expressed probe sets between the fungal-inoculated samples with mock-inoculated ones [59]. The resulting P-values were adjusted by Benjamini and Hochberg’s method to control the false discovery rates [60]. A cutoff of P-value of 0.05 and fold change of two was used as the criterion for significantly differentially expressed probe sets. Hierarchical clustering was performed using the Cluster program (version 3.0; http://www.falw.vu/~huik/cluster.htm) with the Pearson’s correlation similarity metric and the average linkage clustering method, and illustrated by the TreeView program [61]. The raw data and processed results were submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database under accession number GSE41798.
Gene Annotation and Functional Analysis
Due to the great changes in rice genome annotation after the release of Affymetrix GeneChip Rice Genome Array, we updated the probe sets annotations in the first place. Probe sequences were aligned against the latest version 7.0 of MSU-annotated gene models using BLASTN program [62]. The probe sets with more than half of its entire set of probes perfectly matched with one transcript were considered, as previously described [63]. The probe sets matching no gene or multiple genes were discarded as ambiguous probe sets. By this means, we re-annotated a total of 40,539 probe sets, representing 33,814 rice genes. For functional analysis, GOSlim assigned to these genes were retrieved from the MSU database [58] and used for an enrichment analysis based on the hypergeometric distribution, with P-values adjusted by the Benjamini-Hochberg’s method [60]. As rice GO classification was too general, we therefore carried out a more detailed analysis using the MapMan program (version 3.5.0) [31]. The mapping file, which assigned genes into functional categories, was also updated to the latest MSU rice genome annotation version 7.0, and integrated with annotations from additional databases: PlantTFDB [64] and plnTFDB [65] for rice transcription factors; rice kinase database [37] for rice kinases, and rice gene families information from MSU database. Among the 17,752 expressed genes, 10,617 genes were assigned to known functional categories. The statistical significances of the enrichment or depletion in functional categories were tested using a hypergeometric test, with the P-values adjusted by the Benjamini-Hochberg’s method [60].
Gene Set Enrichment Analysis
To compare the transcriptional changes between the compatible and incompatible interactions, two complementary analyses were performed using all 17,752 expressed genes. Firstly, we carried out a sliding-window analysis. For each rice line, all expressed genes were sorted according to their t-statistic values, provided by the Bayesian models fitted in the limma package, in a decreasing order. The top of the sorted gene list included the most up-regulated DEGs and the bottom included the most down-regulated. The percentage of the genes in a given MapMan functional category within a 500-gene sliding-window was calculated and plotted along their ranks.
Secondly, we performed a gene-set enrichment analysis to quantitatively assess the differences in transcriptional changes between compatible and incompatible interactions [33]. The null hypothesis is that the difference in transcriptional changes between compatible and incompatible interactions was equal to zero, i.e., (IT−IM)−(CT−CM) = 0, where IT, IM represent the log2-transformed gene expression values in fungal- and mock-inoculated incompatible rice line; CT, CM represent those in fungal- and mock-inoculated compatible rice line. As comparing the transcriptional changes between two groups, we used the Bayesian moderate t-statistic tn, implemented by the limma package, and the statistic zN for a given functional category containing N genes, in which 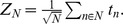 For each functional category, the observed zN statistic was calculated and the reference distribution of zN was generated with a ten thousand times of permutation of sample labels. The P-values were therefore calculated by comparing the observed zN statistic with its reference distribution.
For each functional category, the observed zN statistic was calculated and the reference distribution of zN was generated with a ten thousand times of permutation of sample labels. The P-values were therefore calculated by comparing the observed zN statistic with its reference distribution.
Quantitative RT-PCR
RNA samples used for the quantitative RT-PCR assay were the same as those for microarray hybridization. RNA treatment and cDNA synthesis was performed as previously described [66]. The quantitative RT-PCR was performed using the SYBR Green real-time PCR Master Mix (TOYOBO) on an Option 2 Continuous Fluorescence Detector (Bio-Rad), as previously described [67]. For each pair of primers, the amplification efficiency was assessed using LinRegPCR software [68] and the product specificity was examined by the melting curve and gel electrophoresis. Three independent biological replicates were performed for each sample. Cycling conditions were 1 min at 95°C, followed by 40 cycles of 30 sec at 95°C, 20 sec at 55°C, 20 sec at 72°C and plate read at an optimal temperature. OsUBQ was used as the internal control, and the 2−ΔΔCT method was used to calculate relative expression levels [69]. Primer sequences for the quantitative RT-PCR were listed in Table S3.
Generation of Transgenic Rice Plants
The coding sequence (CDS) of OsWRKY47 was amplified from rice cDNA by RT-PCR using primers (5'-ATG GCG TCT CCT GAT GGT GG-3', 5'-TTA AGG ATC GAA GCC AAA CA-3'). The PCR products were then cloned into pBluescript vector and confirmed by sequencing, resulting in the pBS-WRKY47 construct. To drive the constitutive expression of OsWRKY47, a maize Ubiquitin promoter was released by digesting pAHC25 [70] with Hind III/BamH I and ligated into the same restriction site of pBluescript vector, resulting the pBS-pUbq construct. Subsequently, pBS-WRKY47 was digested by Spe I/Kpn I, and the fragments including OsWRKY47 CDS were ligated with Hind III/Kpn I digested pWM101 vector and the maize Ubiquitin promoter released from Hind III/Spe I digested pBS-pUbq, resulting in the pWM-WRKY47 overexpression construct. To generate transgenic plants for blast disease assay, we transformed the pWM-WRKY47 construct into the susceptible LTH cultivar using an Agrobacterium-mediated method [71]. However, the transformation efficiency for LTH was too low to obtain transformants. Therefore, we transformed the construct into another broadly-used blast-susceptible rice cultivar TP309. The pWM-WRKY47 construct was introduced into A. tumefaciens strain EHA105 and then transformed into TP309 calli, generating 22 independent transgenic lines with hygromycin resistance. T1-generation transgenic seedlings were screened for a possible single T-DNA insertion according to a 3∶1 (hygromycin-resistant/hygromycin-sensitive) segregation ratio. Homozygous T2-generation transgenic plants were used for blast disease assay. As described above, four-week-old wild-type TP309 and pUbi:OsWRKY47 transgenic rice plants were inoculated with M. oryzae strain 96-4-1a and the disease symptom were assessed at 7 dpi,. The expression levels of OsWRKY47 and PR10 were examined by quantitative RT-PCR (primers listed in Table S3).
Trans-activation Activity Assay
The CDS of OsWRKYs were cloned into pYF503 vector using primers listed below: OsWRKY47-F 5'-ATG GCG TCT CCT GAT GGT G-3' and OsWRKY47-R 5'-TTA AGG ATC GAA GCC AAA CA-3'; OsWRKY76-F 5'-ATG GAC GCG GCG TGG CGC-3' and OsWRKY76-R 5'-GAA TTC GGG CAG CTT CTG GAG G-3'; OsWRKY77-F 5'-ATG TCG TCG CTG TAC CCG TC-3' and OsWRKY77-R 5'-GTC AAG GAA GCA GCA GCG AG-3'. Trans-activation activity assay was conducted as previously described [72].
Supporting Information
The 500-sliding window analysis of the genes involved in amino acid metabolism (a), cell wall metabolism (b), stress response (c), transporter (d), lipid metabolism (e) and RNA metabolism (f). The solid lines represent the percentages of certain functional category in a 500-sliding window in IRBL18 (red), IRBL22 (blue) and LTH (green). The blue dotted lines represent the 95% significance levels of greater or smaller than the reference.
(TIF)
List of 755 differentially regulated genes during rice early responses to Magnaporthe oryzae .
(XLS)
List of quantitative RT-PCR results of selected genes.
(XLS)
List of primers for quantitative RT-PCR.
(XLS)
List of differentially regulated receptor kinase genes and nucleotide binding and leucine rich repeat ( NB-LRR ) genes.
(XLS)
List of differentially regulated genes involved in jasmonic acid biosynthesis pathway.
(XLS)
List of differentially regulated transcription factor genes.
(XLS)
The result of rice blast disease assay on pUbi:OsWRKY47 transgenic plants.
(XLS)
Pearson’s correlation efficiencies between biological replicates.
(XLS)
Acknowledgments
We thank Prof. Ge Gao (Center for Bioinformatics, Peking University) for his valuable advices and discussion in microarray data analysis. We thank Jing Yang, Lin Liu and Yuan Su (Yunnan Agricultural University) for their assistance in experiments. And we also thank Baolong Zhang (Jiangsu Academy of Agricultural Sciences) for kindly providing pAHC25 construct and Agrobacterium EHA105 strain.
Funding Statement
This work was supported by the National Transgenic Research Project (Grant No. 2011ZX08009-003-001-003). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chisholm ST, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: Shaping the evolution of the plant immune response. Cell 124: 803–814. [DOI] [PubMed] [Google Scholar]
- 2. Jones JDG, Dangl JL (2006) The plant immune system. Nature 444: 323–329. [DOI] [PubMed] [Google Scholar]
- 3. Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant-pathogen interactions. Nature Reviews Genetics 11: 539–548. [DOI] [PubMed] [Google Scholar]
- 4. Tao Y, Xie ZY, Chen WQ, Glazebrook J, Chang HS, et al. (2003) Quantitative nature of Arabidopsis responses during compatible and incompatible interactions with the bacterial pathogen Pseudomonas syringae . Plant Cell 15: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Talbot NJ (2003) On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea . Annu Rev Microbiol 57: 177–202. [DOI] [PubMed] [Google Scholar]
- 6. Skamnioti P, Gurr SJ (2009) Against the grain: safeguarding rice from rice blast disease. Trends Biotechnol 27: 141–150. [DOI] [PubMed] [Google Scholar]
- 7. Ebbole DJ (2007) Magnaporthe as a model for understanding host-pathogen interactions. Annu Rev Phytopathol 45: 437–456. [DOI] [PubMed] [Google Scholar]
- 8. Flor HH (1971) Current status of gene-for-gene concept. Annu Rev Phytopathol 9: 275–296. [Google Scholar]
- 9. Kaku H, Nishizawa Y, Ishii-Minami N, Akimoto-Tomiyama C, Dohmae N, et al. (2006) Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc Natl Acad Sci USA 103: 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shimizu T, Nakano T, Takamizawa D, Desaki Y, Ishii-Minami N, et al. (2010) Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J 64: 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kankanala P, Czymmek K, Valent B (2007) Roles for rice membrane dynamics and plasmodesmata during biotrophic invasion by the blast fungus. Plant Cell 19: 706–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu J, Wang X, Mitchell T, Hu Y, Liu X, et al. (2010) Recent progress and understanding of the molecular mechanisms of the rice-Magnaporthe oryzae interaction. Mol Plant Pathol 11: 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen X, Ronald PC (2011) Innate immunity in rice. Trends Plant Sci 16: 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan Y, Zhong S, Li Q, Zhu Z, Lou Y, et al. (2007) Functional analysis of rice NPR1-like genes reveals that OsNPR1/NH1 is the rice orthologue conferring disease resistance with enhanced herbivore susceptibility. Plant Biotechnol J 5: 313–324. [DOI] [PubMed] [Google Scholar]
- 15. Sugano S, Jiang CJ, Miyazawa SI, Masumoto C, Yazawa K, et al. (2010) Role of OsNPR1 in rice defense program as revealed by genomewide expression analysis. Plant Mol Biol 74: 549–562. [DOI] [PubMed] [Google Scholar]
- 16. Feng J-X, Cao L, Li J, Duan C-J, Luo X-M, et al. (2011) Involvement of OsNPR1/NH1 in rice basal resistance to blast fungus Magnaporthe oryzae . Eur J Plant Pathol 131: 221–235. [Google Scholar]
- 17. Chujo T, Takai R, Akimoto-Tomiyama C, Ando S, Minami E, et al. (2007) Involvement of the elicitor-induced gene OsWRKY53 in the expression of defense-related genes in rice. BBA-Gene Struct Expr 1769: 497–505. [DOI] [PubMed] [Google Scholar]
- 18. Qiu DY, Xiao J, Ding XH, Xiong M, Cai M, et al. (2007) OsWRKY13 mediates rice disease resistance by regulating defense-related genes in salicylate- and jasmonate-dependent signaling. Mol Plant-Microbe Interact 20: 492–499. [DOI] [PubMed] [Google Scholar]
- 19. Shimono M, Sugano S, Nakayama A, Jiang CJ, Ono K, et al. (2007) Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 19: 2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J, Peng YL, Guo ZJ (2008) Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res 18: 508–521. [DOI] [PubMed] [Google Scholar]
- 21. Tao Z, Liu HB, Qiu DY, Zhou Y, Li XH, et al. (2009) A pair of allelic WRKY genes play opposite roles in rice-bacteria Interactions. Plant Physiol 151: 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jantasuriyarat C, Gowda M, Haller K, Hatfield J, Lu GD, et al. (2005) Large-scale identification of expressed sequence tags involved in rice and rice blast fungus interaction. Plant Physiol 138: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gowda M, Venu RC, Li H, Jantasuriyarat C, Chen S, et al. (2007) Magnaporthe grisea infection triggers RNA variation and antisense transcript expression in rice. Plant Physiol 144: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim ST, Kim SG, Hwang DH, Kang SY, Kim HJ, et al. (2004) Proteomic analysis of pathogen-responsive proteins from rice leaves induced by rice blast fungus, Magnaporthe grisea . Proteomics 4: 3569–3578. [DOI] [PubMed] [Google Scholar]
- 25. Ryu HS, Song MY, Kim CY, Han MH, Lee SK, et al. (2009) Proteomic analysis of rice mutants susceptible to Magnaporthe oryzae . Plant Biotechnol Rep 3: 167–174. [Google Scholar]
- 26. Vergne E, Ballini E, Marques S, Mammar BS, Droc G, et al. (2007) Early and specific gene expression triggered by rice resistance gene Pi33 in response to infection by ACE1 avirulent blast fungus. New Phytol 174: 159–171. [DOI] [PubMed] [Google Scholar]
- 27. Nimchuk Z, Eulgem T, Holt BE, Dangl JL (2003) Recognition and response in the plant immune system. Annu Rev Genet 37: 579–609. [DOI] [PubMed] [Google Scholar]
- 28. Wise RP, Moscou MJ, Bogdanove AJ, Whitham SA (2007) Transcript profiling in host-pathogen interactions. Annu Rev Phytopathol 45: 329–369. [DOI] [PubMed] [Google Scholar]
- 29. Li JB, Li CY, Chen Y, Lei CL, Ling ZZ (2005) Evaluation of twenty-two blast resistance genes in Yunnan using monogenetic rice lines. Acta Phytophylacica Sinica 32: 113–119. [Google Scholar]
- 30. Qu SH, Liu GF, Zhou B, Bellizzi M, Zeng LR, et al. (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172: 1901–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, et al. (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939. [DOI] [PubMed] [Google Scholar]
- 32. Xiong LZ, Yang YN (2003) Disease resistance and abiotic stress tolerance in rice are inversely modulated by an abscisic acid-inducible mitogen-activated protein kinase. Plant Cell 15: 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gentleman R, M M, Huber W (2008) Gene Set Enrichment Analysis. In: Hahne F, Huber W, Gentleman R, Falcon S, editors. Bioconductor Case Studies. New York: Springer. 193–195.
- 34. Caldo RA, Nettleton D, Wise RP (2004) Interaction-dependent gene expression in Mla-specified response to barley powdery mildew. Plant Cell 16: 2514–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shiu SH, Karlowski WM, Pan RS, Tzeng YH, Mayer KFX, et al. (2004) Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 16: 1220–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morillo SA, Tax FE (2006) Functional analysis of receptor-like kinases in monocots and dicots. Curr Opin Plant Biol 9: 460–469. [DOI] [PubMed] [Google Scholar]
- 37. Dardick C, Chen J, Richter T, Ouyang S, Ronald P (2007) The rice kinase database. A phylogenomic database for the rice kinome. Plant Physiol 143: 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gomez-Gomez L, Boller T (2000) FLS2: An LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis . Mol Cell 5: 1003–1011. [DOI] [PubMed] [Google Scholar]
- 39. Chinchilla D, Bauer Z, Regenass M, Boller T, Felix G (2006) The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity of flagellin perception. Plant Cell 18: 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chinchilla D, Zipfel C, Robatzek S, Kemmerling B, Nurnberger T, et al. (2007) A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448: 497–U412. [DOI] [PubMed] [Google Scholar]
- 41. Takai R, Isogai A, Takayama S, Che FS (2008) Analysis of flagellin perception mediated by flg22 receptor OsFLS2 in rice. Mol Plant-Microbe Interact 21: 1635–1642. [DOI] [PubMed] [Google Scholar]
- 42. Seo YS, Chern M, Bartley LE, Han MH, Jung KH, et al. (2011) Towards establishment of a rice stress response interactome. PLoS Genet 7: e1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delteil A, Blein M, Faivre-Rampant O, Guellim A, Estevan J, et al. (2012) Building a mutant resource for the study of disease resistance in rice reveals the pivotal role of several genes involved in defence. Mol Plant Pathol 13: 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xie Z, Zhang ZL, Zou XL, Huang J, Ruas P, et al. (2005) Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol 137: 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10: 366–371. [DOI] [PubMed] [Google Scholar]
- 46. Naoumkina MA, Zhao QA, Gallego-Giraldo L, Dai XB, Zhao PX, et al. (2010) Genome-wide analysis of phenylpropanoid defence pathways. Mol Plant Pathol 11: 829–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dixon RA, Achnine L, Kota P, Liu CJ, Reddy MSS, et al. (2002) The phenylpropanoid pathway and plant defence - a genomics perspective. Mol Plant Pathol 3: 371–390. [DOI] [PubMed] [Google Scholar]
- 48. Maher EA, Bate NJ, Ni WT, Elkind Y, Dixon RA, et al. (1994) Increased disease susceptibility of transgenic tobacco plants with suppressed levels of preformed phenylpropanoid products. Proc Natl Acad Sci USA 91: 7802–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kruger WM, Carver TLW, Zeyen RJ (2002) Effects of inhibiting phenolic biosynthesis on penetration resistance of barley isolines containing seven powdery mildew resistance genes or alleles. Physiol Mol Plant Pathol 61: 41–51. [Google Scholar]
- 50. Bhuiyan NH, Selvaraj G, Wei YD, King J (2009) Gene expression profiling and silencing reveal that monolignol biosynthesis plays a critical role in penetration defence in wheat against powdery mildew invasion. J Exp Bot 60: 509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen YH, Zhang XB, Wu W, Chen ZL, Gu HY, et al. (2006) Overexpression of the wounding-responsive gene AtMYB15 activates the shikimate pathway in Arabidopsis . Journal of Integrative Plant Biology 48: 1084–1095. [Google Scholar]
- 52. Zhou JL, Lee CH, Zhong RQ, Ye ZH (2009) MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis . Plant Cell 21: 248–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Q, Dixon RA (2010) Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci 16: 227–233. [DOI] [PubMed] [Google Scholar]
- 54. Parker D, Beckmann M, Zubair H, Enot DP, Caracuel-Rios Z, et al. (2009) Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea . Plant J 59: 723–737. [DOI] [PubMed] [Google Scholar]
- 55. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biology 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264. [DOI] [PubMed] [Google Scholar]
- 57. Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy - analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- 58. Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, et al. (2007) The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res 35: D883–D887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smyth GK (2005) Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer. 397–420.
- 60. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Ser B-Methodol 57: 289–300. [Google Scholar]
- 61. Saldanha AJ (2004) Java Treeview-extensible visualization of microarray data. Bioinformatics 20: 3246–3248. [DOI] [PubMed] [Google Scholar]
- 62. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jung KH, Dardick C, Bartley LE, Cao PJ, Phetsom J, et al. (2008) Refinement of light-responsive transcript lists using rice oligonucleotide arrays: evaluation of gene-redundancy. PLoS One 3: e3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang H, Jin JP, Tang LA, Zhao Y, Gu XC, et al. (2011) PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res 39: D1114–D1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Perez-Rodriguez P, Riano-Pachon DM, Correa LGG, Rensing SA, Kersten B, et al. (2010) PInTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38: D822–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu J, Zhang Y, Qin G, Tsuge T, Sakaguchi N, et al. (2008) Targeted degradation of the cyclin-dependent kinase inhibitor ICK4/KRP6 by RING-type E3 ligases is essential for mitotic cell cycle progression during Arabidopsis gametogenesis. Plant Cell 20: 1538–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao Y, Wei T, Yin KQ, Chen ZL, Gu HY, et al. (2012) Arabidopsis RAP2.2 plays an important role in plant resistance to Botrytis cinerea and ethylene responses. New Phytol 195: 450–460. [DOI] [PubMed] [Google Scholar]
- 68. Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339: 62–66. [DOI] [PubMed] [Google Scholar]
- 69. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 70. Christensen AH, Quail PH (1996) Ubiquitin promoter-based vectors for high-level expression of selectable and/or screenable marker genes in monocotyledonous plants. Transgenic Res 5: 213–218. [DOI] [PubMed] [Google Scholar]
- 71. Toki S, Hara N, Ono K, Onodera H, Tagiri A, et al. (2006) Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J 47: 969–976. [DOI] [PubMed] [Google Scholar]
- 72. Yang XY, Li JG, Pei M, Gu H, Chen ZL, et al. (2007) Over-expression of a flower-specific transcription factor gene AtMYB24 causes aberrant anther development. Plant Cell Rep 26: 219–228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 500-sliding window analysis of the genes involved in amino acid metabolism (a), cell wall metabolism (b), stress response (c), transporter (d), lipid metabolism (e) and RNA metabolism (f). The solid lines represent the percentages of certain functional category in a 500-sliding window in IRBL18 (red), IRBL22 (blue) and LTH (green). The blue dotted lines represent the 95% significance levels of greater or smaller than the reference.
(TIF)
List of 755 differentially regulated genes during rice early responses to Magnaporthe oryzae .
(XLS)
List of quantitative RT-PCR results of selected genes.
(XLS)
List of primers for quantitative RT-PCR.
(XLS)
List of differentially regulated receptor kinase genes and nucleotide binding and leucine rich repeat ( NB-LRR ) genes.
(XLS)
List of differentially regulated genes involved in jasmonic acid biosynthesis pathway.
(XLS)
List of differentially regulated transcription factor genes.
(XLS)
The result of rice blast disease assay on pUbi:OsWRKY47 transgenic plants.
(XLS)
Pearson’s correlation efficiencies between biological replicates.
(XLS)