

Summary
Abnormal basal activity and stress-evoked reactivity of the hypothalamic-pituitary-adrenal (HPA) axis are often seen in depression, implicating HPA axis dysfunction as a potentially causative or exacerbating factor. Chronic stress is also a factor in depression, but it is not known what may underlie the shift from adaptive to maladaptive HPA activity over the course of chronic stress. Interleukin 6 (IL-6), a stress-inducible cytokine that signals through gp130 and IL-6R! receptors to activate the JAK/STAT3 signaling cascade, is elevated in some subtypes of depression, and may have a modulatory effect on HPA activation, raising the possibility that IL-6 contributes to depression through effects on the HPA axis. In this study, we examined the effects of three different stress modalities, acute footshock, chronic intermittent cold (CIC) stress and chronic unpredictable stress (CUS) on IL-6 signaling in the hypothalamus. We also investigated whether IL-6 modulates the HPA response to chronic stress, by blocking IL-6 signaling in the brain during CIC stress using either a neutralizing antibody or an inhibitor of STAT3 phosphorylation. We show that IL-6 and STAT3 in the hypothalamus are activated in response to footshock and CUS. We also found that basal IL-6 signaling through the JAK/STAT3 pathway is required for the sustained CORT response to chronic, but not acute, cold stress and therefore is a potential determinant of plasticity in the HPA axis specifically during chronic stress exposure.
Keywords: HPA axis, chronic stress, interleukin 6, JAK/STAT signaling, hypothalamus
1. Introduction
The hypothalamic-pituitary-adrenal (HPA) axis coordinates the primary physiological response to stress through the secretion of glucocorticoids. While HPA axis responses to acute stress are generally considered adaptive for re-establishing homeostasis, there is now substantial evidence that repeated exposure to stress induces persistent changes in HPA axis functionality that may produce maladaptive outcomes (McEwen, 2007). Indeed, stress is a prevalent risk factor in many psychopathologies, and several of these disturbances are linked to altered glucocorticoid secretion. For example, depression is often associated with increased glucocorticoid secretion (Gillespie and Nemeroff, 2005), whereas, post-traumatic stress disorder patients often present with lower cortisol levels than controls (Yehuda, 2006). Dysregulation of the HPA axis is also found in panic disorder, chronic fatigue syndrome, generalized anxiety disorder, obsessive-compulsive disorder and bipolar disorder (Daban et al., 2005; Abelson et al., 2007; Gustafsson et al., 2008; Van Houdenhove et al., 2009; Lenze et al., 2011). This wealth of evidence points to HPA axis dysfunction as a possible etiological link between stress and development of psychiatric disease. However, it is not known what underlies the switch between adaptive and maladaptive HPA axis responses to different modalities and degrees of stress.
There is also a growing recognition of the potential role of neuroinflammation in the development of many psychiatric disorders (Koo et al., 2010; Haroon et al., 2011; Monje et al., 2011). Interleukin 6 (IL-6) is a pleiotropic cytokine secreted by cells of the innate immune system during the first phases of an immune challenge. Elevated IL-6 levels appear to be consistently associated with some subtypes of depression (Dantzer et al., 2008; Schmidt et al., 2011), and depressed patients subjected to acute stress have higher levels of IL-6 in their plasma than non-depressed individuals (Pace et al., 2006). Peripheral IL-6 levels are elevated in humans and rodents exposed to acute stress (Zhou et al., 1993; Steptoe et al., 2007). Stress also alters IL-6 expression in the rat brain (Girotti et al., 2011), but the functional implications of these effects have not been elucidated.
IL-6 is produced in the brain by activated glia, in response to peripheral immune challenge, and is also expressed in neuronal populations (Vallieres and Rivest, 1997; Ghorbel et al., 2003; Frank et al., 2010; Jankord et al., 2010). IL-6 signals through a specific trans-membrane receptor (IL-6Rα) and the ubiquitous transducer molecule gp130. The IL-6Rα is also present as a soluble protein that acts agonistically and confers IL-6 responsivity to cells that do not express the trans-membrane form of the receptor, but do express gp130 (Schobitz et al., 1995). This may be an especially important mechanism in the CNS, where soluble IL-6Rα has been detected (Marz et al., 1999). The IL-6/IL-6Rα/gp130 ternary complex activates Janus kinase 1 and 2 (JAK1, JAK2) that phosphorylate the transcription factor STAT3 on Tyr705 residues, inducing STAT3 dimerization and transcriptional activation. One target gene of activated STAT3 is Suppressor of Cytokines 3 (SOCS3), an E3 ubiquitin ligase that binds activated JAK and STAT and targets them to the proteosome, acting therefore as a negative regulator of JAK/STAT signaling. In addition, IL-6-activated JAKs can stimulate the ERK1/2 pathway as well as the PI3 kinase/Akt pathways through phosphorylation of Src-homology phosphatase 2 (SHP2) (Ernst and Jenkins, 2004).
There is evidence that both acute (Jankord et al., 2007; Jankord et al., 2010) and chronic stress (Girotti et al., 2011a) can increase levels of IL-6 mRNA in the rat hypothalamus. In addition, chronic intermittent cold (CIC) stress sensitizes brain responses to a peripheral immune challenge, with consistent and robust effects on IL-6 expression (Girotti et al., 2011a). These data indicate that IL-6 is a stress-responsive cytokine, and that some of the detrimental effects of stress in the brain could be due to increased expression of this inflammatory molecule. However, not all models of stress alter hypothalamic IL-6 expression (Hueston et al., 2010).
IL-6 and its receptors (IL-6R! and gp130) have been localized in the PVN (Vallieres and Rivest, 1997; Jankord et al., 2010). In addition, early investigations using ex vivo, in vitro or overexpression models suggested that IL-6 may have a modulatory effect on HPA axis outputs, both at the level of the pituitary gland (Lyson and McCann, 1992; Sarlis et al., 1993) and the adrenal gland (Gonzalez-Hernandez et al., 1994), raising the possibility that IL-6 contributes to stress-related psychopathology through its effects on HPA axis activation. However, very little functional data is available to suggest a mechanism of action for IL-6 as a possible modulator of HPA axis plasticity. If IL-6 signaling is shown to regulate HPA axis activity following chronic stress, it may be possible to target IL-6 signaling as a novel therapeutic approach, to effectively reduce HPA axis hyperactivity in depression and other psychopathologies.
Thus, in this study, following up on our previous observations that acute footshock stress robustly induces IL-6 expression in the hypothalamus (Girotti et al., 2011a), we determined whether this effect was accompanied by functional activation of the IL-6 signaling pathway. In addition, to assess the generality of stress effects on IL-6 signaling to chronic as well as acute stress, we compared the induction of IL-6 expression following acute footshock to that obtained with two other qualitatively different stress stimuli. The first was a homotypic metabolic stressor, chronic intermittent cold (CIC) stress. The second was chronic unpredictable stress (CUS), which has been shown to produce depressive-like behaviors in rats (Bondi et al., 2008; Hill et al., 2012). We then investigated a potential role for IL-6 in the plastic changes the HPA axis undergoes during chronic stress, by blocking IL-6 signaling in the brain during CIC stress using either a neutralizing antibody or a pharmacological inhibitor of STAT3 phosphorylation. Portions of this work have been presented in abstract form (Girotti, 2011b).
2. Methods
2.1. Animals
Adult male Sprague-Dawley rats (Harlan), weighing approximately 220-240 g upon arrival, were housed 3 per cage and maintained on a 12/12-h light cycle (lights on at 07:00h) with access to food and water ad libitum. Animals were allowed to acclimatize to the facility for a minimum of four days before any experimental procedures began. A total of 182 animals were used in these experiments. All procedures were conducted according to NIH guidelines, and were reviewed and approved by the UTHSCSA Institutional Animal Care and Use Committee. All efforts were made to minimize unnecessary pain and distress, and the number of animals used.
2.2. Surgery
For intracerebroventricular infusions, rats were anesthetized (ketamine 43 mg/ml, acepromazine 1.4 mg/ml, xylazine 8.6 mg/ml, in a volume of 1.0 ml/kg IM, with 25% supplement as needed) and a local analgesic (1mg/kg bupivacaine) was applied SC at the site of incision. Animals were placed in a stereotaxic apparatus and a bilateral injection cannula (28GA, 4.5mm, Plastics One) was implanted aimed at the lateral ventricles (coordinates from bregma: AP −0.9 mm, ML ± 1.4 mm, DV –4.5 mm) and affixed to the skull with dental acrylic. Sterile vinyl tubing connected the cannula to two osmotic minipumps (Alzet model 1004, flow rate 0.11! l/h, for up to 28 days) implanted sub-cutaneously at the base of the neck. After surgery, the rats were treated prophylactically with penicillin G (300,000 IU/ml, 1.0 ml/kg, SC), and housed singly for at least one week before any experimental procedures began.
2.3. Experiment 1
Acute footshock was applied as described previously (Girotti et al., 2011a). Briefly, rats were placed in a footshock chamber (Habitest Model H10-24, Coulbourn) and subjected to a series of footshocks consisting of 3 × 5 sec scrambled footshocks (1.5 mA) with 5 sec between shocks, followed by 150 sec with no shock, repeated 5 times in 15 min. The 15 min shock period was followed by 15 min of no shock, and the entire 30 min sequence, 15 min footshock + 15 min no shock, was repeated three times for a total of 90 min. The last 15 min were no footshock. Rats were then either decapitated immediately (i.e., with no home cage recovery time, 0hr) or returned to their home cage for recovery time of 0.5hr, 3.5hr or 24hr before sacrifice (See Figure 1). No Footshock (No FS) control rats were left undisturbed in their home cages and sacrificed along with stressed rats at each time point. Since there was no effect of time in the No FS animals, the data for this group were pooled. In addition, to investigate any potential effect of exposure to the footshock chamber alone on the IL-6 or P-STAT3 levels, an experiment was performed in which animals were placed in the footshock chamber for 90 min without delivering any shock, and were sacrificed 30 min after returning to their home cage along with a small number of animals that remained in the home cage (n=4). After sacrifice, hypothalami were dissected for qPCR and western blot analysis.
Figure 1. Schematic representation of the experimental outlines.

CIC= chronic intermittent cold stress, CUS= chronic unpredictable stress, Sac= sacrifice.
2.4. Experiment 2
Chronic intermittent cold (CIC) stress was administered as described previously (Girotti et al., 2011a). Animals were transported in their home cage with food, water and bedding to a 4°C cold room for 6 h each day for 14 consecutive days. Control animals remained undisturbed in their home cages in the housing room for the same period of time. Different groups of rats were sacrificed at the end of 6h cold exposure on day 1, day 7 or day 14 of CIC (see Figure 1), together with subsets of non-stressed (NS) control rats. Since there was no effect of time alone in the NS rats, the data were pooled into a single control group. Trunk blood was collected, and hypothalami were dissected for qPCR, ELISA and western blot analysis.
2.5. Experiment 3
Chronic unpredictable stress consists of a battery of different mild stressors, one per day for 14 days, according to the list in Figure 1, essentially as described previously (Bondi et al., 2008) except that we introduced acute cold as one of the stress stimuli, to allow comparison with the CIC protocol in Experiment 2. Thus, as in the CIC stress procedure, rats were sacrificed at the end of a 6h acute cold stress, either on day 7 or day 14 of CUS (Figure 1). An additional control group of rats were exposed only to the two acute cold stress treatments seven days apart, with no other stressors applied (Cold x2). This group was compared to the 14 days CUS group to differentiate effects due simply to two repeated cold exposures from effects due to the chronic unpredictable stress treatment (Figure 1). Groups of non-stress control rats were left undisturbed in their home cages and sacrificed at the same time as stressed animals. Since no effect of time was observed, the NS data was pooled into a single control group. After sacrifice, hypothalami were dissected for qPCR and western blot analysis.
2.6. Experiments 4 and 5
For experiment 4, animals were implanted with osmotic minipumps delivering goat anti-rat IL-6 antibody (150 ng/2.64! l/side/day, ICV) or an equivalent amount of normal goat IgG control (R&D Systems). Following 7-8 days of recovery, one group of animals from each drug treatment was subjected to CIC stress for 14 days (CIC 14d, see Figure 1). At the end of the 6h cold exposure on day 14, animals were sacrificed together with a group of rats that had received the drug treatments and were exposed to just 6h of cold stress on that day (CIC 1d) and a group that had received treatment but was never stressed.
Experiment 5 was identical in design to Experiment 4, except that the animals received 100ng/2.64! l/side/day of the JAK/STAT inhibitor, JSI-124 (Cucurbitacin I, Indofine Chemical Company). This concentration of JSI-124 is 75! M, which corresponds to 10x the IC50 for inhibition of STAT3 phosphorylation in vitro (Sun et al., 2005). The compound was made as a 10 mg/ml stock in 100% ethanol, then diluted to the desired concentration with sterile saline, resulting in a final ethanol concentration of 0.33%. Vehicle animals were infused ICV with 0.33% EtOH/saline. In both experiments, after sacrifice trunk blood was collected for HPA axis hormone measures and hypothalami dissected for western blot analysis of P-STAT3. Hypothalamic STAT3 phosphorylation was used as an index of treatment effectiveness of the neutralizing antibody and of JSI-124. There were no visible signs of gross physical or behavioral changes in any of the animals receiving either IL-6 antibody or JSI-124 compared to controls and to non-operated animals.
2.7. Tissue and plasma collection
The brain was rapidly removed and dissected on ice with the aid of a brain matrix. For the hypothalamus, a 5 mm coronal slab was first cut, approximately between bregma −0.3 mm and −5.3 mm. The hypothalamus was then dissected from this slab, by cutting 3 mm laterally from the midline at each side and 3 mm dorsally from the base of the brain (just below the thalamus). The hypothalamus was then dissected into two halves along the midline; one side was used for mRNA extraction and the other for ELISA or western blot procedures. The brain dissections were immediately frozen in a bath of 2-methylbutane on dry-ice, then stored at −80°C. Trunk blood was collected into a tube containing 100 μl 0.5M EDTA on ice. Plasma was separated by centrifugation (4000 x g, 15 min, 4°C), and stored in aliquots at −80°C.
2.8. Real-time qPCR
Quantitative real-time PCR was performed essentially as described (Girotti et al., 2011a). Briefly, total RNA was extracted and purified from hemi-hypothalami using Trizol reagent (Invitrogen) and the PureLink RNA Mini Kit (Invitrogen Carlsbad, CA), with the addition of an on-column DNAse purification step, according to manufacturer’s instructions. Equal amounts of total RNA (2.5! g per sample) were used in a cDNA synthesis reaction using the High Capacity cDNA RT Kit (Applied Biosystems, Foster City, CA). No reverse transcriptase controls were also run and used in qPCR to verify the complete removal of genomic DNA from the RNA samples. Real time PCR reactions were assembled using diluted cDNA and 400 nM of each specific forward and reverse primer (Integrated DNA Technology) with Sso Fast EVAgreen reaction mix (SYBR green, Biorad). Samples were run in triplicate on a BioRad CFX384 Real Time System with the following conditions: one cycle at 95°C for 2 min followed by 40 cycles of denaturation (95°C, 5 sec), annealing and elongation (60°C, 10 sec). Relative gene expression was calculated using the 2−ΔΔCT method. Primer sets for IL-6 and GAPDH were as previously described (Girotti et al., 2011a). Primer sequences for SOCS3 (Genebank acc# NM_053565.1) were: Forward: 5′-ACCTTCCTTTGAGGTTCAGGAGCA-3′, Reverse: 5′-TGACCGTTGACAGTCTTCCGACAA-3′; they were validated for efficiency and absence of primer dimers as described previously (Girotti et al., 2011a).
2.9. Western blotting
Frozen tissue was sonicated (12 sec, 50% power) in lysis buffer (50mM Tris pH 7.4, 150mM NaCl, 1% Nonident-P40, 0.1% SDS, 0.5% deoxycholate) containing protease and phosphatase inhibitors (Sigma), incubated on ice for 10 min with occasional mixing and centrifuged for 10 min at 18000x g at 4°C. Forty micrograms of total protein were subjected to SDS-PAGE and transferred to PVDF membrane (immobilon P, Millipore). After blocking (Prime ECL blocking agent, GE Healthcare), blots were incubated overnight at 4°C with a rabbit anti-phospho-STAT3 (Tyr 705) monoclonal antibody (1:1000, Cell Signaling) followed by HRP-secondary antibody and ECL detection with Prime ECL reagent (GE Healthcare). The blots were stripped and re-probed with anti-STAT3 antibody (1:1500, sc-8019, Santa Cruz) to normalize the P-STAT3 signal, and with anti-GAPDH (1:20000, Cell Signaling) to normalize for loading. No effect of treatment was found on total STAT3 expression. For IL-6 detection, blots were blocked with 5% BSA and incubated overnight at 4°C with goat anti-IL-6 antibody (1:400, M19, sc-1265, Santa Cruz) followed by secondary goat-HRP incubation and ECL detection (Ghorbel et al., 2003).
2.10. Enzyme-linked immunosorbent assay
For experiment 2, hypothalamic lysates were prepared with Tissue Extraction Reagent I (Invitrogen, Cat # FNN0071) and half of the sample was used in western blots (as described above) and the other half was used in a high sensitivity ELISA assay for the determination of rat IL-6 (Invitrogen) according to manufacturer’s instructions. Absorbance at 450 nm (with correction at 570 nm) was measured on an ELx808 instrument (Biotek Instruments). The assay sensitivity was 0.5 pg/ml and the coefficient of variability was 8.8%. Hypothalamic IL-6 was expressed as a function of total protein, as determined by the Bradford Assay (Sigma).
2.11. Radioimmunoassays
Plasma corticosterone and ACTH levels were determined using the ImmuChem Double Antibody 125I RIA Kit (MP Biomedicals) according to manufacturer’s instructions. The inter-assay coefficient of variation was 10% for Corticosterone and 6% for ACTH. The sensitivity of the assay is approximately 8 ng/ml for CORT and 6 pg/ml for ACTH.
2.12. Data analysis
All data were analyzed by one-or two-way ANOVA. Where significant main effects or interactions were detected, the Dunnett’s or Newman-Keuls tests were used for post hoc comparisons. Significance in all analyses was determined at p < 0.05.
3. Results
3.1. Experiment 1
We have previously shown that acute footshock (90 min) elevates the levels of IL-6 transcript in the hypothalamus (Girotti et al., 2011a). In Figure 2A, we show that IL-6 protein levels were also significantly increased in the hypothalamus 3.5h following the end of stress compared to No FS controls (F4,22=3.71, p<0.05, n=4-6). IL-6 mRNA levels were also significantly increased in these rats at 0.5h of recovery (F4,22= 3.10, p<0.05; 0.5h recovery vs No FS, p<0.05, Dunnet’s post-hoc comparison, n= 4-6, data not shown). In parallel, phosphorylation of STAT3, a primary transcriptional effector of IL-6/gp130 receptor activation, was significantly increased 0.5h after the end of stress compared to No FS (Figure 2B; F4,21=3.47, p<0.05, n=4-6). Expression of SOCS3, a target of P-STAT3 activation, was also significantly elevated at 0.5 and 3.5h of recovery compared to No FS (Figure 2C; F4,50= 3.49, p<0.05, n= 10-14). Western blot analysis revealed no significant differences in levels of IL-6 or P-STAT3 in the hypothalamus of rats that were simply exposed to the footshock chamber for 90 min with no shock, then sacrificed 30 min after returning to the home cage compared to rats that remained in the home cage (Optical density ratio of IL-6/tubulin: chamber 0.43 ± 0.05, cage 0.41± 0.08; Optical density ratio of P-STAT3/STAT3: chamber 0.82 ± 0.03, cage 0.87± 0.28; n=4; data not shown). These data suggest that acute footshock induced the expression of IL-6 and activated the downstream signaling effectors of the IL-6/gp130 pathway in the hypothalamus.
Figure 2. Effect of acute footshock on IL-6 protein, STAT3 phosphorylation and SOCS3 mRNA levels in the hypothalamus.
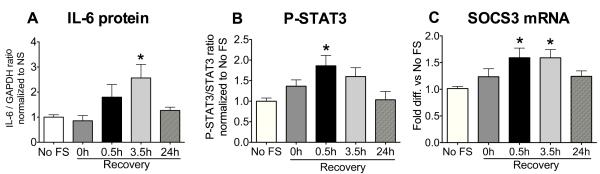
Rats were sacrificed immediately after 90 min of footshock (0h) or at 0.5h, 3.5h and 24h of recovery. The hypothalami were rapidly dissected and one half used for determination of IL-6 protein levels (A, n=4-6) and STAT3 phosphorylation (P-STAT3, n=4-6) (B) with western blot; the other half of the hypothalamus was used to determine SOCS3 mRNA levels by qPCR (C, n= 10-14). No FS= no footshock. *significantly different from No FS control by Newman-Keuls post-hoc test at p<0.05.
3.2. Experiment 2
We then investigated whether chronic intermittent cold stress (CIC), a homotypic chronic metabolic stressor that sensitizes the HPA axis to novel acute stressors (Ma and Morilak, 2005), could also produce long term changes in IL-6 expression and signaling in the hypothalamus. There was a significant main effect of CIC stress on IL-6 mRNA expression (F3,44=7.61, p<0.001, n=12). As shown in Figure 3A, expression of IL-6 mRNA was elevated at the end of 6h cold stress exposure on the 1st day of stress as well as on the 7th day of repeated exposure compared to No Stress (NS) controls, but not on day 14. Throughout this time course, however, we did not observe changes in IL-6 protein, STAT3 phosphorylation or SOCS3 expression (Figure 3B-D). IL-6 protein levels were verified by two independent methods, western blot (data not shown) and high-sensitivity ELISA (shown in Figure 3B), obtaining similar results in both.
Figure 3. Effect of chronic intermittent cold (CIC) stress on IL-6 mRNA, IL-6 protein, P-STAT3 and SOCS3 mRNA levels in the hypothalamus.

Rats were sacrificed at the end of 6h cold stress on day 1, 7, or 14 of CIC, or at the same time of day for non-stressed (NS) rats. One half of the hypothalamus was used to determine levels of IL-6 mRNA (A, n=12) and SOCS3 mRNA (D, n=12) and the other half was used to measure IL-6 protein (B, n=9-11) and P-STAT3 (C, n=10-12) content. IL-6 protein was determined using an ELISA assay, but similar results were seen by western blot. *significantly different from NS control by Newman-Keuls post-hoc test at p<0.05.
3.3. Experiment 3
Next, we examined the effects of chronic unpredictable stress (CUS), a stress paradigm more intense than CIC stress and known to elevate basal corticosterone levels (Herman et al., 1995), on hypothalamic IL-6 signaling. In this experiment, for comparison with the CIC stress time course, we used 6h cold stress as one of the variable stressors of the CUS and sacrificed the animals at the end of this stress (see Methods and Figure 1). Controls were represented by a No Stress group (NS) and a group of rats that were exposed twice to 6h cold stress, 7 days apart (Cold x2). This group was compared to the 14 days CUS group to differentiate effects due simply to two cold exposures from effects due to the variable stress treatment. As shown in Figure 4A, there was a significant increase in IL-6 mRNA levels in animals receiving CUS compared to NS controls (F3,18=6.71, p<0.01, n=4-6). IL-6 mRNA expression was elevated after the cold stress applied on day 7 of CUS, and was still significantly elevated by acute cold stress after 14 days of CUS. However, this was not significantly different from the increase in IL-6 mRNA observed after two exposures to cold stress alone (Cold x2). Nonetheless, unlike the effect of CIC, IL-6 protein levels were also increased at day 14 by CUS (Figure 4B; F3,13=3.33 p=0.052, n=4-5). Further, P-STAT3 levels were also elevated and were significantly different from both the NS and Cold x2 controls (Figure 4C; F3,17= 4.36, p<0.05, n=4-6). This suggests that a history of chronic unpredictable stress can prime an enhanced IL-6 signaling response to cold stress more robustly than repeated homotypic cold stress. These data further suggest that activation of IL-6 signaling in the hypothalamus is not confined to acute stress, and is not simply an idiosyncratic response to the specific metabolic challenges imposed by cold stress exposure. A notable difference in the responses to acute footshock and CUS was the failure of CUS to induce SOCS3 (Figure 4D).
Figure 4. Effect of chronic unpredictable stress (CUS) on IL-6 mRNA, IL-6 protein, P-STAT3 and SOCS3 mRNA levels in the hypothalamus.

Rats were sacrificed at the end of 6h cold stress on day 7 and day 14 of a CUS paradigm detailed in Figure 1. NS= no stress control. Cold x2= rats were exposed to 6h cold stress on two days separated by six days of no stress (see Figure 1). One half of the hypothalamus was used to determine levels of IL-6 mRNA (A, n= 4-6) and SOCS3 mRNA (D, n= 4-6) and the other half was used to measure IL-6 protein (B, n= 4-5) and P-STAT3 (C, n=4-6) content. *significantly different from NS control and # significantly different from Cold 2x, by Newman-Keuls post-hoc test at p<0.05.
3.4. Experiment 4
Repeated exposure to homotypic stress often results in habituation of the HPA axis (Girotti et al., 2006). Despite the repetitive nature of CIC stress, and in agreement with previous observations (Bhatnagar and Meaney, 1995), the corticosterone response during CIC stress did not habituate and was still significantly elevated at the end of 6h cold stress on day 14 of CIC stress (Figure 5; F3,39= 11.52, p<0.0001, n=10-11). In addition, basal CORT levels on the morning after day 14 of CIC stress were indistinguishable from controls (data not shown), ruling out the possibility that the elevated CORT levels at the end of day 14 of CIC stress are due to higher basal CORT secretion.
Figure 5. Corticosterone response during the time course of CIC stress.

Levels of plasma corticosterone (CORT) at the end of 6h cold stress in the animals of Experiment 2 were determined by RIA (n=10-11). *significantly different from NS control by Newman-Keuls post-hoc test at p<0.05.
We then examined whether IL-6 contributed to the sustained CORT response to repeated cold stress. For this, we examined the CORT levels at the end of either 1 day or 14 days of CIC stress in animals receiving chronic ICV infusion of an anti-rat IL-6 neutralizing antibody (see Figure 1). Control rats received isotype-matched normal serum IgG. There were significant main effects of Stress (F2,28= 7.61 p<0.01) and Antibody (F1,28= 5.23, p<0.05, n=5-7) (Figure 6A). CORT levels were significantly increased at the end of a single exposure to cold stress in both IgG and anti-IL-6 treated animals, compared to their respective No Stress (NS) controls. Although the interaction term did not achieve significance (F1,28= 1.82, p= 0.18), the main effect of the anti-IL-6 antibody treatment was clearly driven by a difference in CORT response on day 14 of cold stress. Post-hoc analysis confirmed this, revealing a significant difference between IL-6 antibody and IgG control treatment on day 14 of CIC; CORT was elevated after cold stress in animals that received control IgG treatment, whereas those receiving anti-IL-6 antibody during the course of chronic stress had CORT levels indistinguishable from NS controls (Figure 6A).
Figure 6. Effects of anti-rat IL-6 neutralizing antibody on plasma CORT, plasma ACTH and hypothalamic P-STAT3 levels in CIC stressed animals.
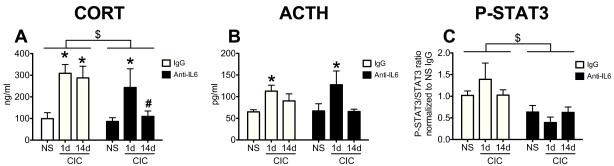
Rats received constant ICV infusions of either a rat neutralizing antibody (anti-IL-6) or a control IgG and were subjected to 1 or 14 days CIC. No Stress animals are indicated by NS. All groups received the drug treatments for the same length of time. Rats were sacrificed at the end of 6h cold exposure. CORT (panel A, n= 5-7) and ACTH (panel B, n= 5-8) were measured in the plasma and P-STAT3 levels (panel C, n= 5-6) were measured in hypothalamus lysates. * significantly different from NS within the same drug treatment, # significantly different from CIC-14d IgG, $ significant Main Effect of Antibody, p<0.05, Newman-Keuls post-hoc test.
For ACTH, there was also a significant main effect of Stress (F2,29=6.16, p<0.01, n=5-8), but no main Antibody effect and no interaction (Antibody: F1,29= 0.04, p>0.05; Interaction: F2,29= 0.87 p>0.05). ACTH was significantly elevated at the end of a single exposure to cold stress in both IgG and anti-IL-6 treated animals, compared to their respective No Stress (NS) controls (Figure 6B). In addition, in the same animals we measured the levels of P-STAT3 in the hypothalamus. As shown in Figure 6C, P-STAT3 levels were lower in animals receiving constant infusion of anti-IL-6 antibody compared to animals that did not. There was a significant main effect of Antibody (F1,27= 16.13, p<0.001), confirming the effectiveness of the anti-rat IL-6 treatment. This also suggests that basal IL-6 activity contributes to at least 50% of phosphorylated STAT3 in the hypothalamus. Further, this indicates that the lack of an IL-6 antibody effect on CORT and ACTH measures seen after a single exposure to cold was not due to a lack of IL-6 neutralization, since all anti-IL-6 groups displayed a similar reduction in STAT3 phosphorylation. There was no significant main effect of Stress on P-STAT3, nor a Stress × Antibody interaction (Stress F2,27= 0.92, p>0.05; Interaction, F2,27= 1.79, p>0.05).
3.5. Experiment 5
To further confirm that gp130 receptor activation was required for CIC stress modulation of HPA axis responses, we also tested the effects of chronic ICV infusion of 100 ng/side/day of JSI-124, an inhibitor of Janus kinase 2 (JAK2), and to a lesser extent JAK1, two effector kinases coupled to gp130 receptors. JAK2 is the main kinase responsible for STAT3 tyrosine phosphorylation. Figure 7A shows that, similar to what we observed with the neutralizing antibody, continuous inhibition of JAK activity with JSI-124 reduced the CORT response at the end of cold stress on day 14 of CIC compared to NS, but not on the first day of stress (Stress × Drug interaction: F2,29= 6.89, p<0.01; Stress main effect: F2,29=7.19, p<0.01; Drug main effect: F1,29= 0.15, p>0.05; n=5-6). This was paralleled by a similar profile in the ACTH response (Stress × Drug interaction: F2,27= 6.04, p<0.01, Stress main effect: F2,27= 7.51, p<0.01, Drug main effect, F1,27= 2.84, p>0.05; n=5-6,) (Figure 7B). A significant reduction in P-STAT3 levels in the animals receiving JSI-124 confirmed the inhibitor’s efficacy within the hypothalamus (Figure 7C) (Drug main effect: F1,33=4.58, P<0.05; Stress main effect F2,33= 0.34, P>0.05; Interaction, F2,33= 0.24, P>0.05.; n=5-8). A lower dose of JSI-124 (20ng/side/day), which did not significantly alter P-STAT3 levels, was not effective in reducing CORT levels on day 14 of CIC (data not shown). Taken together, the results indicate that inhibition of basal IL-6 signaling during repeated stress exposure attenuates the CORT response after repeated cold stress, but not to acute cold stress.
Figure 7. Effects of the STAT3 phosphorylation inhibitor JSI-124 on plasma CORT, plasma ACTH and hypothalamic P-STAT3 levels in CIC stressed animals.
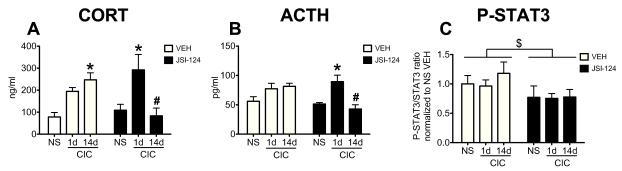
Rats received constant ICV infusions of either a STAT3 phosphorylation inhibitor, JSI-124, or vehicle and were subjected to 1 or 14 days CIC. No Stress animals are indicated by NS. All groups received the drug treatments for the same length of time. Rats were sacrificed at the end of 6h cold exposure. CORT (panel A, n=5-6) and ACTH (panel B, n=5-6) were measured in the plasma and P-STAT3 levels (panel C, n=5-8) were measured in hypothalamus lysates. * significantly different from NS within the same drug treatment, # significantly different from CIC-14d VEH, $ significant Main Effect of Drug, p<0.05,Newman-Keuls post-hoc test.
4. Discussion
In this study we explored the bidirectional relationship between stress and IL-6 signaling in the hypothalamus, in vivo. In experiments 1-3, we investigated how stress either acutely or chronically impacts IL-6 expression and signaling in the hypothalamus. In experiments 4 and 5, we investigated whether IL-6 signaling modulates the HPA axis response to acute and chronic stress.
4.1. Effects of stress on IL-6 expression and signaling
Following up on our previous observations that acute footshock significantly increased levels of IL-6 mRNA in the hypothalamus immediately after the end of footshock and 30 min into the recovery period (Girotti et al., 2011a), we now show that IL-6 protein levels are also increased in the hypothalamus with a significant peak at 3.5 h of recovery (Figure 2). In parallel, we observed increased phosphorylation of STAT3, one of the main downstream signals of the IL-6/IL-6R!/gp130 cascade, suggesting that stress-induced IL-6 in the hypothalamus is functionally coupled to the JAK/STAT3 pathway. In addition, SOCS3, a target of STAT3 transcriptional activation, is also significantly induced 30 min after the end of stress and still elevated at 3.5h of recovery, further confirming the activation of the JAK/STAT3 signaling pathway. SOCS3 is a E3 ubiquitin ligase that ubiquitinates JAK and STAT molecules targeting them to the proteasome (Yoshimura et al., 2007). It acts as a negative regulator of the JAK/STAT pathway and an anti-inflammatory signal (Croker et al., 2003). Thus, JAK/STAT3 induction of SOCS3 may be interpreted as an early negative regulatory response to an incipient inflammatory process in the hypothalamus. These data extend our previous observations and provide evidence that the IL-6 signaling pathway in the hypothalamus is activated as a consequence of footshock, complementing previous reports of increased plasma IL-6 levels following footshock (Zhou et al., 1993). In other models of footshock, IL-6 mRNA was not elevated in the hypothalamus (Hueston et al., 2010). This discrepancy may be due to differences in stressor intensity or sampling time. In those studies, IL-6 was generally measured immediately at the end of stress, whereas we have found that IL-6 induction in response to stress has a slower time course and is more pronounced during the post-stress recovery period. This pattern of expression fits with the slower time-course of IL-6 induction in the hypothalamus in response to inflammation (IL-6 is a late-term cytokine) (Laye et al., 1994). This may also be a consequence of early activation of other cytokines such as IL-1!, that often act as upstream activators of IL-6 under acute inflammatory conditions (Stylianou and Saklatvala, 1998) and that have beem shown to be induced in the hypothalamus by footshock (Blandino et al., 2006).
The effects of chronic stress on hypothalamic IL-6 signaling have not been explored. In our CIC stress model, IL-6 mRNA was elevated in the hypothalamus following stress application on days 1 and 7 of repeated stress. However, this was not complemented by detectable changes in IL-6 protein expression, STAT3 activation or SOCS3 induction. This discrepancy between IL-6 mRNA and protein levels is intriguing. One possible cause of this phenomenon, to be explored in future studies, is the inhibition of IL-6 translation by microRNAs, as has been shown under some circumstances (Kang et al., 2011). In any case, the lack of IL-6 protein increase is consistent with observations by other groups who have reported changes in hypothalamic IL-6 mRNA levels following stress without any change in protein level or number of cells expressing IL-6 (Jankord et al., 2010). Therefore, we conclude that IL-6 protein levels may not change detectably during CIC stress.
Interestingly, a more robust chronic stressor, CUS was able to induce not only a time-dependent increase in IL-6 mRNA expression, but also an increase in IL-6 protein and STAT3 phosphorylation. This phenomenon was not attributable to acute stress application on the day of sacrifice, but was a result of the preceding course of chronic unpredictable stress, demonstrating that sufficiently intense chronic stressors can also activate the JAK/STAT3 pathway in vivo. SOCS3 levels, however, did not change under these conditions. It is possible that we missed the peak of SOCS3 (and STAT3) induction in this experiment, since the time of sacrifice was at the end of a 6h cold stress, and SOCS3 mRNA has a high turnover rate (Siewert et al., 1999). This seems unlikely, however, since in pilot experiments, induction of SOCS3 was not observed after 1h, 2h or 3h of acute cold exposure (data not shown). Another possibility is that chronic stress uncouples P-STAT3 activation from SOCS3 induction. If this is the case, one could predict that failure to instate this anti-inflammatory signal may contribute to the sensitized brain responses to immune challenge following CIC stress that we have previously reported (Girotti et al., 2011a).
4.2. Effects of inhibition of basal IL-6/STAT3 signaling on HPA axis responses to cold stress
Besides being induced by acute stress, IL-6 has also been implicated in the regulation of the HPA stress response (Raber et al., 1998). For example, in hypothalamic explants IL-6 increased CRH release at the median eminence (Spinedi et al., 1992), and in rats, ICV infusion of IL-6 induced ACTH release in an AVP- and CRH-dependent manner (Kageyama et al., 1995). In the present study, we revealed a specific role for IL-6 in regulating HPA reactivity after chronic stress. Blocking IL-6 signaling by preventing binding to the IL-6R! and gp130 receptors with a neutralizing antibody directed against endogenous rat IL-6 did not inhibit CORT secretion in response to the first exposure to cold stress. However, neutralization of IL-6 during repeated cold stress completely prevented the rise in CORT seen after acute cold exposure on day 14. Importantly, this was paralleled by reduced STAT3 phosphorylation in the hypothalamus. These data indicate that basal IL-6 activity, functionally coupled to the JAK/STAT3 signaling pathway, modulates and sustains the HPA axis response to repeated stress. An effect of antibody treatment was less evident on ACTH. However, it is important to note that blood sampling occurred 6h after the beginning of stress, so it might be expected by that time that the ACTH response may have largely recovered, and thus the drug effect was less obvious. It is possible that sampling earlier in the application of stress may have revealed more robust effects of the antibody treatment on the ACTH response to cold stress.
Similar effects were observed with chronic administration of JSI-124, an inhibitor of JAK1 and JAK2 kinases coupled to gp130 and responsible for STAT3 tyrosine phosphorylation, confirming that the functional activation of gp130 signaling is required for the modulation of HPA axis response with chronic stress. These results suggest that STAT3 may be one of the downstream effectors of IL-6 function in this system, although, it is also possible that other effectors of gp130/JAK signaling may be involved (i.e., Shp/Ras/ERK and PI3k/Akt). Further, with intraventricular infusion, it is also possible that the inhibitors may have had effects in extra-hypothalamic areas known to regulate or modulate the activity of the HPA axis (e.g., bed nucleus of the stria terminalis, amygdala, lateral septum, hippocampus, prefrontal cortex, etc.). Future studies will address these possibilities. Finally, because of the emphasis on HPA output measures, this work is focused on IL-6 signaling within the hypothalamus. However, we are also currently investigating the effects of basal IL-6 and STAT3 activity in the rat orbitofrontal cortex with respect to outcomes in cognitive performance.
4.3. Mechanism of action
We envision two possible, non-mutually exclusive mechanisms of action for IL-6 signaling in regulating HPA axis outputs. One possibility is that IL-6 expressed in hypothalamic neurons is released either in the pituitary directly, or in the median eminence to reach the pituitary, acting similarly to arginine vasopressin (AVP) to enhance or prolong the effects of CRH on ACTH release. Indeed, it has been suggested that IL-6 has effects at the level of the pituitary, especially under inflammatory conditions (Lyson and McCann, 1992; Sarlis et al., 1993). IL-6 has been immunolocalized in the median eminence (Jankord et al., 2007; Jankord et al., 2010) and also in vasopressin-containing magnocellular neurons of the PVN (Ghorbel et al., 2003; Gonzalez-Hernandez et al., 2006; Jankord et al., 2010). Moreover, IL-6R! and gp130 are expressed in the anterior pituitary (Hanisch et al., 2000; Gautron et al., 2003). Acute stress preferentially activates IL-6-containing magnocellular neurons in the PVN (Jankord et al., 2010) and depletes IL-6 content in the median eminence while, concomitantly, activating STAT3 in the pituitary and increasing ACTH secretion (Jankord et al., 2007). However, not all IL-6 positive cells in the PVN localize to the magnocellular subdivision (Ghorbel et al., 2003; Gonzalez-Hernandez et al., 2006; Jankord et al., 2010). It is known that under conditions of repeated stress, neurons of the parvocellular subdivision accumulate AVP (Ma et al., 1997) and AVP released by these cells is responsible for sustained pituitary-adrenal activation (Scaccianoce et al., 1991; Ma et al., 1997) through actions on corticotroph V1b receptors to stimulate ACTH release (Aguilera and Rabadan-Diehl, 2000). Thus, it is possible that IL-6 is also expressed in these parvocellular neurons, and that IL-6 co-released with AVP during repeated stress may act on the pituitary gland to reinforce the effects of AVP, exerting paracrine effects similar to those described for another member of the gp130 cytokine family, leukemia inhibitory factor (LIF), which can activate POMC transcription via the JAK/STAT3 pathway (Auernhammer and Melmed, 2000). Alternatively, IL-6 in the pituitary may act to upregulate the enzyme pro-hormone convertase that processes POMC (Li et al., 1999).
Another possibility is that IL-6 acts in a paracrine manner within the hypothalamus to influence stress-responsive PVN neurons. Besides being present in vasopressin-containing neurons, IL-6 is found also in a small number of non-peptidergic neurons in the PVN (Jankord et al., 2010). Both IL-6R! and gp130 are also expressed in the PVN (Vallieres and Rivest, 1997). Moreover, astroglia and microglia can produce and release IL-6. In cortical slices, IL-6 reduces the amplitude of GABAA receptor currents by decreasing the number of GABAA receptor sites at the plasma membrane (Garcia-Oscos et al., 2012). It is possible that during repeated cold stress, local release of IL-6 downregulates GABA receptors on stress-responsive PVN neurons, thus reducing their sensitivity to tonic inhibition by GABAergic fibers and facilitating excitatory drive (Galic et al., 2012; Levy and Tasker, 2012). The data in Figure 5, showing that central blockade of IL-6 activity reduced HPA axis output in response to 14 d of CIC stress, would support this hypothesis. The fact that the effects of IL-6 signaling were observed after repeated but not acute stress suggests that they may involve convergence, interaction or regulation of other stress-responsive signals that are also recruited with chronic stress (e.g., repeated increases in glucocorticoids and GR activation). It is also possible that other permissive factors must first be present for basal IL-6 signaling to have these effects, and this may explain why there is no specific up-regulation of hypothalamic IL-6 mRNA or protein after two weeks of CIC stress. Interactions of STAT3 and GR in regulating expression of GR-responsive genes have been documented (Lerner et al., 2003; Langlais et al., 2012), and LIF-dependent activation of STAT3 has been shown to decrease GR expression itself (Kariagina et al., 2005). Indeed, the increased levels of IL-6 protein and P-STAT3 we observed after the more robust chronic stress treatment with CUS may represent a tonic increase in central drive responsible for the elevation of basal CORT levels (Herman et al., 1995). Thus, IL-6 signaling in the hypothalamus may contribute to plasticity in the HPA axis in response to strong or protracted stressors.
Hyperactivity of the HPA axis is observed in a subset of depressed patients, and the results of numerous clinical studies suggest that normalizing the HPA axis may be an important step for stable remission of symptoms. For example, CRH receptor 1 antagonists have held high promise in the treatment of mood disorders associated with hypersecretion of CRH, for their ability to reduce anxiety and other depressive symptoms (Refojo and Holsboer, 2009). But pharmacokinetic and hepatotoxicity issues have led to the discontinuation of clinical use of these drugs (Bosker et al., 2004). Therefore, there is an urgent need for alternatives. In this context, existing treatments that reduce IL-6 signaling, such as tocilizumab, a well-tolerated humanized anti-IL-6R! antibody used in rheumatoid arthritis patients (Nishimoto et al., 2005; Jones et al., 2011), might be effective, either alone or as an adjunct to CRH receptor inhibitors (Kehne and Cain, 2010), in reducing the HPA axis hyperactivity associated with depression.
4.4. Conclusion
In sum, we have shown that IL-6 and STAT3 in the hypothalamus are activated in response to acute and chronic stress. We have also discovered that basal IL-6 signaling through the JAK/STAT3 pathway is an important determinant of the plastic changes occurring in the HPA axis during repeated stress exposure. Elucidating the mechanisms by which constitutive IL-6 signaling exerts these effects may offer new targets for pharmacological intervention in stress-related disorders that involve dysfunction of the HPA axis.
Acknowledgements
We thank Mr. Ankur Joshi and Ms. Shiyi Geng for technical assistance. This work was supported by research grant MH053851 from the National Institute of Mental Health, which had no further role in study design, collection, analysis of interpretation of data, nor in the preparation or decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
M1ilena Girotti - designed studies, performed experiments, analyzed data, wrote the manuscript
Jennifer Donegan - contribute to experimental design, performed portions of experiments, edited manuscript
David Morilak - designed experiments, provided and allocated resources and other support to conduct the experiments, edited the paper and wrote portions, oversaw technical staff
Conflict of interest
The authors have no financial or scientific conflicts of interest to disclose.
References
- Abelson JL, Khan S, Liberzon I, Young EA. HPA axis activity in patients with panic disorder: review and synthesis of four studies. Depress Anxiety. 2007;24:66–76. doi: 10.1002/da.20220. [DOI] [PubMed] [Google Scholar]
- Aguilera G, Rabadan-Diehl C. Regulation of vasopressin V1b receptors in the anterior pituitary gland of the rat. Exp Physiol. 2000;85(Spec No):19S–26S. doi: 10.1111/j.1469-445x.2000.tb00004.x. [DOI] [PubMed] [Google Scholar]
- Auernhammer CJ, Melmed S. Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocr Rev. 2000;21:313–345. doi: 10.1210/edrv.21.3.0400. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Meaney MJ. Hypothalamic-pituitary-adrenal function in chronic intermittently cold-stressed neonatally handled and non handled rats. J Neuroendocrinol. 1995;7:97–108. doi: 10.1111/j.1365-2826.1995.tb00672.x. [DOI] [PubMed] [Google Scholar]
- Blandino P, Jr., Barnum CJ, Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol. 2006;173:87–95. doi: 10.1016/j.jneuroim.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Bondi CO, Rodriguez G, Gould GG, Frazer A, Morilak DA. Chronic unpredictable stress induces a cognitive deficit and anxiety-like behavior in rats that is prevented by chronic antidepressant drug treatment. Neuropsychopharmacology. 2008;33:320–331. doi: 10.1038/sj.npp.1301410. [DOI] [PubMed] [Google Scholar]
- Bosker FJ, Westerink BH, Cremers TI, Gerrits M, van der Hart MG, Kuipers SD, van der Pompe G, ter Horst GJ, den Boer JA, Korf J. Future antidepressants: what is in the pipeline and what is missing? CNS Drugs. 2004;18:705–732. doi: 10.2165/00023210-200418110-00002. [DOI] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Daban C, Vieta E, Mackin P, Young AH. Hypothalamic-pituitary-adrenal axis and bipolar disorder. Psychiatr Clin North Am. 2005;28:469–480. doi: 10.1016/j.psc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Capuron L, Irwin MR, Miller AH, Ollat H, Perry VH, Rousey S, Yirmiya R. Identification and treatment of symptoms associated with inflammation in medically ill patients. Psychoneuroendocrinology. 2008;33:18–29. doi: 10.1016/j.psyneuen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Jenkins BJ. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23–32. doi: 10.1016/j.tig.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Watkins LR, Maier SF. Aging sensitizes rapidly isolated hippocampal microglia to LPS ex vivo. J Neuroimmunol. 2010;226:181–184. doi: 10.1016/j.jneuroim.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33:116–125. doi: 10.1016/j.yfrne.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Oscos F, Salgado H, Hall S, Thomas F, Farmer GE, Bermeo J, Galindo LC, Ramirez RD, D’Mello S, Rose-John S, Atzori M. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. 2012;71:574–582. doi: 10.1016/j.biopsych.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautron L, Lafon P, Tramu G, Laye S. In vivo activation of the interleukin-6 receptor/gp130 signaling pathway in pituitary corticotropes of lipopolysaccharide-treated rats. Neuroendocrinology. 2003;77:32–43. doi: 10.1159/000068336. [DOI] [PubMed] [Google Scholar]
- Ghorbel MT, Sharman G, Leroux M, Barrett T, Donovan DM, Becker KG, Murphy D. Microarray analysis reveals interleukin-6 as a novel secretory product of the hypothalamo-neurohypophyseal system. J Biol Chem. 2003;278:19280–19285. doi: 10.1074/jbc.M209902200. [DOI] [PubMed] [Google Scholar]
- Gillespie CF, Nemeroff CB. Hypercortisolemia and depression. Psychosom Med. 2005;67(Suppl 1):S26–28. doi: 10.1097/01.psy.0000163456.22154.d2. [DOI] [PubMed] [Google Scholar]
- Girotti M, Donegan JJ, Morilak DA. Chronic intermittent cold stress sensitizes neuro-immune reactivity in the rat brain. Psychoneuroendocrinology. 2011a;36:1164–1174. doi: 10.1016/j.psyneuen.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti M, Donegan JJ, Morilak DA. Determining the role of hypothalamic IL-6 in sensitization of the rat HPA axis by exposure to chronic cold stress. Soc. Neuroscience Abstr. 2011b;37 online program 87.03. [Google Scholar]
- Girotti M, Pace TW, Gaylord RI, Rubin BA, Herman JP, Spencer RL. Habituation to repeated restraint stress is associated with lack of stress-induced c-fos expression in primary sensory processing areas of the rat brain. Neuroscience. 2006;138:1067–1081. doi: 10.1016/j.neuroscience.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, Spath-Schwalbe E, Jirikowski G, Scherbaum WA. Interleukin-6 messenger ribonucleic acid expression in human adrenal gland in vivo: new clue to a paracrine or autocrine regulation of adrenal function. J Clin Endocrinol Metab. 1994;79:1492–1497. doi: 10.1210/jcem.79.5.7962348. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez T, Afonso-Oramas D, Cruz-Muros I, Barroso-Chinea P, Abreu P, del Mar Perez-Delgado M, Rancel-Torres N, del Carmen Gonzalez M. Interleukin-6 and nitric oxide synthase expression in the vasopressin and corticotrophin-releasing factor systems of the rat hypothalamus. J Histochem Cytochem. 2006;54:427–441. doi: 10.1369/jhc.5A6845.2005. [DOI] [PubMed] [Google Scholar]
- Gustafsson PE, Gustafsson PA, Ivarsson T, Nelson N. Diurnal cortisol levels and cortisol response in youths with obsessive-compulsive disorder. Neuropsychobiology. 2008;57:14–21. doi: 10.1159/000123117. [DOI] [PubMed] [Google Scholar]
- Hanisch A, Dieterich KD, Dietzmann K, Ludecke K, Buchfelder M, Fahlbusch R, Lehnert H. Expression of members of the interleukin-6 family of cytokines and their receptors in human pituitary and pituitary adenomas. J Clin Endocrinol Metab. 2000;85:4411–4414. doi: 10.1210/jcem.85.11.7122. [DOI] [PubMed] [Google Scholar]
- Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. 2011;37:137–162. doi: 10.1038/npp.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61:180–190. doi: 10.1159/000126839. [DOI] [PubMed] [Google Scholar]
- Hill MN, Hellemans KG, Verma P, Gorzalka BB, Weinberg J. Neurobiology of chronic mild stress: Parallels to major depression. Neurosci Biobehav Rev. 2012;36:2085–2117. doi: 10.1016/j.neubiorev.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueston CM, Barnum CJ, Eberle JA, Ferraioli FJ, Buck HM, Deak T. Stress-dependent changes in neuroinflammatory markers observed after common laboratory stressors are not seen following acute social defeat of the Sprague Dawley rat. Physiol Behav. 2010;104:187–198. doi: 10.1016/j.physbeh.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Jankord R, Turk JR, Schadt JC, Casati J, Ganjam VK, Price EM, Keisler DH, Laughlin MH. Sex difference in link between interleukin-6 and stress. Endocrinology. 2007;148:3758–3764. doi: 10.1210/en.2006-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankord R, Zhang R, Flak JN, Solomon MB, Albertz J, Herman JP. Stress activation of IL-6 neurons in the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2010;299:R343–351. doi: 10.1152/ajpregu.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama K, Watanobe H, Takebe K. In vivo evidence that arginine vasopressin is involved in the adrenocorticotropin response induced by interleukin-6 but not by tumor necrosis factor-alpha in the rat. Neuroimmunomodulation. 1995;2:137–140. doi: 10.1159/000096883. [DOI] [PubMed] [Google Scholar]
- Kang JG, Majerciak V, Uldrick TS, Wang X, Kruhlak M, Yarchoan R, Zheng ZM. Kaposi’s sarcoma-associated herpesviral IL-6 and human IL-6 open reading frames contain miRNA binding sites and are subject to cellular miRNA regulation. J Pathol. 2011;225:378–389. doi: 10.1002/path.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariagina A, Zonis S, Afkhami M, Romanenko D, Chesnokova V. Leukemia inhibitory factor regulates glucocorticoid receptor expression in the hypothalamic-pituitary-adrenal axis. Am J Physiol Endocrinol Metab. 2005;289:E857–863. doi: 10.1152/ajpendo.00577.2004. [DOI] [PubMed] [Google Scholar]
- Kehne JH, Cain CK. Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharmacol Ther. 2010;128:460–487. doi: 10.1016/j.pharmthera.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR Interaction Code: Predictive Value of Direct/Indirect DNA Recruitment for Transcription Outcome. Mol Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Laye S, Parnet P, Goujon E, Dantzer R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27:157–162. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Lenze EJ, Mantella RC, Shi P, Goate AM, Nowotny P, Butters MA, Andreescu C, Thompson PA, Rollman BL. Elevated cortisol in older adults with generalized anxiety disorder is reduced by treatment: a placebo-controlled evaluation of escitalopram. Am J Geriatr Psychiatry. 2011;19:482–490. doi: 10.1097/JGP.0b013e3181ec806c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L, Henriksen MA, Zhang X, Darnell JE., Jr. STAT3-dependent enhanceosome assembly and disassembly: synergy with GR for full transcriptional increase of the alpha 2-macroglobulin gene. Genes Dev. 2003;17:2564–2577. doi: 10.1101/gad.1135003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy BH, Tasker JG. Synaptic regulation of the hypothalamic-pituitary-adrenal axis and its modulation by glucocorticoids and stress. Front Cell Neurosci. 2012;6:24. doi: 10.3389/fncel.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QL, Jansen E, Friedman TC. Regulation of prohormone convertase 1 (PC1) by gp130-related cytokines. Mol Cell Endocrinol. 1999;158:143–152. doi: 10.1016/s0303-7207(99)00168-9. [DOI] [PubMed] [Google Scholar]
- Lyson K, McCann SM. Induction of adrenocorticotropic hormone release by interleukin-6 in vivo and in vitro. Ann N Y Acad Sci. 1992;650:182–185. doi: 10.1111/j.1749-6632.1992.tb49118.x. [DOI] [PubMed] [Google Scholar]
- Ma S, Morilak DA. Chronic intermittent cold stress sensitises the hypothalamic-pituitary-adrenal response to a novel acute stress by enhancing noradrenergic influence in the rat paraventricular nucleus. J Neuroendocrinol. 2005;17:761–769. doi: 10.1111/j.1365-2826.2005.01372.x. [DOI] [PubMed] [Google Scholar]
- Ma XM, Levy A, Lightman SL. Emergence of an isolated arginine vasopressin (AVP) response to stress after repeated restraint: a study of both AVP and corticotropin-releasing hormone messenger ribonucleic acid (RNA) and heteronuclear RNA. Endocrinology. 1997;138:4351–4357. doi: 10.1210/endo.138.10.5446. [DOI] [PubMed] [Google Scholar]
- Marz P, Otten U, Rose-John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. Eur J Neurosci. 1999;11:2995–3004. doi: 10.1046/j.1460-9568.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- Monje FJ, Cabatic M, Divisch I, Kim EJ, Herkner KR, Binder BR, Pollak DD. Constant darkness induces IL-6-dependent depression-like behavior through the NF-kappaB signaling pathway. J Neurosci. 2011;31:9075–9083. doi: 10.1523/JNEUROSCI.1537-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106:2627–2632. doi: 10.1182/blood-2004-12-4602. [DOI] [PubMed] [Google Scholar]
- Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163:1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- Raber J, Sorg O, Horn TF, Yu N, Koob GF, Campbell IL, Bloom FE. Inflammatory cytokines: putative regulators of neuronal and neuro-endocrine function. Brain Res Brain Res Rev. 1998;26:320–326. doi: 10.1016/s0165-0173(97)00041-6. [DOI] [PubMed] [Google Scholar]
- Refojo D, Holsboer F. CRH signaling. Molecular specificity for drug targeting in the CNS. Ann N Y Acad Sci. 2009;1179:106–119. doi: 10.1111/j.1749-6632.2009.04983.x. [DOI] [PubMed] [Google Scholar]
- Sarlis NJ, Stephanou A, Knight RA, Lightman SL, Chowdrey HS. Effects of glucocorticoids and chronic inflammatory stress upon anterior pituitary interleukin-6 mRNA expression in the rat. Br J Rheumatol. 1993;32:653–657. doi: 10.1093/rheumatology/32.8.653. [DOI] [PubMed] [Google Scholar]
- Scaccianoce S, Muscolo LA, Cigliana G, Navarra D, Nicolai R, Angelucci L. Evidence for a specific role of vasopressin in sustaining pituitary-adrenocortical stress response in the rat. Endocrinology. 1991;128:3138–3143. doi: 10.1210/endo-128-6-3138. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Shelton RC, Duman RS. Functional biomarkers of depression: diagnosis, treatment, and pathophysiology. Neuropsychopharmacology. 2011;36:2375–2394. doi: 10.1038/npp.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobitz B, Pezeshki G, Pohl T, Hemmann U, Heinrich PC, Holsboer F, Reul JM. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J. 1995;9:659–664. doi: 10.1096/fasebj.9.8.7768358. [DOI] [PubMed] [Google Scholar]
- Siewert E, Muller-Esterl W, Starr R, Heinrich PC, Schaper F. Different protein turnover of interleukin-6-type cytokine signalling components. Eur J Biochem. 1999;265:251–257. doi: 10.1046/j.1432-1327.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- Spinedi E, Hadid R, Daneva T, Gaillard RC. Cytokines stimulate the CRH but not the vasopressin neuronal system: evidence for a median eminence site of interleukin-6 action. Neuroendocrinology. 1992;56:46–53. doi: 10.1159/000126207. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol. 1998;30:1075–1079. doi: 10.1016/s1357-2725(98)00081-8. [DOI] [PubMed] [Google Scholar]
- Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM. Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene. 2005;24:3236–3245. doi: 10.1038/sj.onc.1208470. [DOI] [PubMed] [Google Scholar]
- Vallieres L, Rivest S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1beta. J Neurochem. 1997;69:1668–1683. doi: 10.1046/j.1471-4159.1997.69041668.x. [DOI] [PubMed] [Google Scholar]
- Van Houdenhove B, Van Den Eede F, Luyten P. Does hypothalamic-pituitary-adrenal axis hypofunction in chronic fatigue syndrome reflect a ‘crash’ in the stress system? Med Hypotheses. 2009;72:701–705. doi: 10.1016/j.mehy.2008.11.044. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N Y Acad Sci. 2006;1071:137–166. doi: 10.1196/annals.1364.012. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133:2523–2530. doi: 10.1210/endo.133.6.8243274. [DOI] [PubMed] [Google Scholar]