

Abstract
We report here a supramolecular strategy to directly assemble the small molecular hydrophobic anticancer drug camptothecin (CPT) into discrete, stable, well-defined nanostructures with a high and quantitative drug loading. Depending on the number of CPTs in the molecular design, the resulting nanostructures can be either nanofibers or nanotubes, and have a fixed CPT loading content ranging from 23% to 38%. We found that formation of nanostructures provides protection for both the CPT drug and the biodegradable linker from the external environment and thus offers a mechanism for controlled release of CPT. Under tumor-relevant conditions, these drug nanostructures can release the bioactive form of CPT and show in vitro efficacy against a number of cancer cell lines. This strategy can be extended to construct nanostructures of other types of anticancer drugs, and thus presents new opportunities for the development of self-delivering drugs for cancer therapeutics.
The creation of vehicles for the effective delivery of hydrophobic anticancer drugs to tumor sites has garnered major attention in cancer chemotherapies for several decades1–4. A successful strategy promises immense benefits to cancer sufferers through both the reduction of side-effects and a greater treatment efficacy5,6. Current approaches focus on the use of nanocarriers, whereby the drug’s pharmacokinetic properties and biodistribution profiles are manipulated by encapsulation within liposomes, polymeric nanoparticles or micelles7–15, or by conjugation to hydrophilic polymers or inorganic nanomaterials2,16,17. Whilst these methods can be effective, there are concerns regarding the short-term and long-term toxicities arising from the synthetic nanomaterials other than the drug being delivered18,19. Furthermore, there are inherent difficulties in achieving a quantitative and high drug loading per carrier (typically less than 10%)2. Polydispersity, both in terms of polymer length and the amount of drug loaded or conjugated, is a critical issue susceptible to significant batch-to-batch variability. On the other hand, small molecule prodrugs are monodisperse but can be subject to rapid clearance and premature degradation20. Recently, the use of drug molecules to promote self-assembly into micellar structures has been reported21–24, however, these systems demonstrated limited tunability in the resulting morphologies and the drug loading content.
We report here the design of monodisperse, amphiphilic anticancer drugs – which we term drug amphiphiles (DAs) – that can spontaneously associate into discrete, stable supramolecular nanostructures with the potential for self-delivery (no additional carriers are needed). Specifically, we conjugated the hydrophobic drug camptothecin (CPT), a DNA-topoisomerase I inhibitor28,29, to a β-sheet forming peptide sequence derived from the Tau protein 30 through the reducible disulfylbutyrate (buSS) linker31 (Fig. 1a). This concept of conjugating small molecular hydrophobic units to a short peptide segment to promote new self-assembling features and to achieve new functional properties has been demonstrated by a few laboratories.24–27 The drug content was precisely controlled using the two amine functionalities of the amino acid lysine to create branching points that allow the attachment of one, two or four CPT molecules, corresponding to respective drug loadings of 23%, 31% and 38% (Fig. 1b). The drug conjugates were synthesized by reaction of the appropriate cysteine-containing Tau peptide with the activated disulfide CPT-buSS-Pyr in DMSO, followed by purification to >99% homogeneity by reversed-phase HPLC (supporting information S1, figs. S1–S4). A non-reducible CPT-containing DA, mCPT-mal-Tau, and an octanoic acid-functionalized peptide, C8-Tau, were also synthesized to act as control molecules (Figs. 1c, and S5–S6). The identity and purity for all the studied molecules were confirmed using mass spectrometry and analytical HPLC, respectively.
Figure 1.

Schematic illustration of the designed and synthesized drug amphiphiles (DAs) and control molecules. (a) The self-assembled nanostructures contain the same drug fraction as the individual DA. (b) The three key component parts of a drug amphiphile studied in this paper: the hydrophobic drug CPT, the Tau β-sheet forming peptide, and the buSS biodegradable linker. (c) The synthesized CPT DAs with quantitative drug loadings of 23%, 31% and 38%. (d) The two synthesized control molecules.
Transmission electron microscopy (TEM) and cryogenic TEM (cryo-TEM) imaging reveals that all the CPT drug amphiphiles assemble into filamentous nanostructures in water (Fig. 2). Cryo-TEM provides direct imaging of solution-state nanostructures that are preserved within a thin film of vitreous ice and thus minimizes chances of forming artifacts that typically occur during the drying and staining processes used for preparing traditional TEM samples. At 50 μM, mCPT-buSS-Tau was observed to form filaments of widths 6.7 ± 1 nm (Fig. 2a) and lengths on the scale of a few micrometers, and dCPT-buSS-Tau forms nanofibers of widths 7.2 ± 1.4 nm (Fig. 2c) that were predominantly shorter than those formed by mCPT-buSS-Tau. The diameters of these nanofibers are approximately twice that of the expected molecular length (~3.5 and 3.8 nm, respectively), indicating a cylindrical packing geometry of core-shell micelles. qCPT-buSS-Tau forms even shorter fibrous nanostructures (< 1 μm) of widths 9.5 ± 1 nm (Figs. 2e, 2g, 2h, and S7), with a dark centerline throughout all the observed nanostructures. This dark centerline is due to the deposition of the negative staining agent, uranyl acetate, and suggests the structures possess a hollow core that may have collapsed during TEM sample preparation. The image features observed here are remarkably similar to those of the tobacco mosaic virus32 and other structures in the literature that are known to have a tubular morphology33,34. The circular shape of their terminal ends further confirms the tubular morphology (Figs. 2g, 2h, S7). The comparable value of the nanotube wall thickness to the molecular length (4 nm vs. 4.1 nm) implies that the molecules are packed in a monolayered rather than bilayered fashion. These results demonstrate that the incorporation of different numbers of CPT units into the drug amphiphiles can tune both the drug loading content and the assembly morphologies.
Figure 2.

TEM characterization illustrating the effect of CPT content on the self-assembled morphology of DA molecules. TEM (a) and cryo-TEM (b) images of long filaments of widths 6.7 ± 1 nm formed by mCPT-buSS-Tau in water. TEM (c) and cryo-TEM (d) images of shorter filaments of widths 7.2 ± 1.4 nm formed by dCPT-buSS-Tau in water. TEM (e) and cryo-TEM (f) images of nanotubes of widths 9.5 ± 1 nm formed by qCPT-buSS-Tau in water. The cryo-TEM (f) resolution is insufficient to show the tubular nature. (g) and (h) High resolution TEM images of the tubular morphology formed by qCPT-buSS-Tau. The circular shape of the terminal ends (marked with white arrows) confirms the tubular structures. TEM samples for images of (a), (c), (e), (g) and (h) were stained with 2% uranyl acetate aqueous solution to enhance the image contrast. Solution concentrations: 50 μM for (a), (c), (e), (g) and (h); 1mM for (b) and (d); 100 μM for (e).
Clearly, the one dimensionality (1D) is linked to the β-sheet forming Tau peptide that provides a directional, intermolecular hydrogen bonding30. The C8-Tau control molecule without any CPT unit also forms similar filaments in water (Fig. S8), confirming the leading role of Tau peptides in these 1D assemblies. The β-sheet secondary structure was confirmed for each DA molecule by the observation of a negative signal around the 220 nm region of the circular dichroism (CD) spectra (Figs. 3a–c)26,35.
Figure 3.

Circular dichroism (CD, solid line) and UV-Vis (dashed line) spectroscopic analysis of 1 μM mCPT-buSS-Tau (a), 1 μM dCPT-buSS-Tau (b) and 1 μM qCPT-buSS-Tau (c) in 10 mM sodium phosphate at 37°C. Release study of 2 μM DA and control molecules in the presence and absence of 10 mM glutathione (GSH) in 10 mM sodium phosphate (d). Comparison of mCPT-buSS-Tau release kinetics at 2 μM and 20 μM (e). Release experiments were performed in triplicate and values are given as mean ± s.d. (Key: mono = mCPT-buSS-Tau, di = dCPT-buSS-Tau, quad = qCPT-buSS-Tau, maleimide = mCPT-mal-Tau, (+) or (−) indicates the presence or absence of 10 mM glutathione, respectively). Schematic illustration of the proposed release mechanism showing the effect of self-assembly on the susceptibility of the DAs to degradation (f).
We speculate that the morphological differences of the observed nanostructures are rooted in the number of CPT molecules within the conjugates. We therefore performed spectroscopic studies to investigate the CPT packing within the assemblies. We found that the CD spectra of both mCPT-buSS-Tau (Fig. 3a) and dCPT-buSS-Tau (Fig. 3b) aqueous solutions display strong signals in the CPT absorption regions at 250 nm and between 330 and 400 nm, in accordance with the visible absorptions in the corresponding UV-vis spectra. No CPT absorption signals, however, were observed for the CPT conjugates dissolved in DMSO where these conjugates are expected to exist in a monomeric form (Fig. S9). Since CD measures absorption differences between left and right circularly polarized light, these results clearly suggest a chiral packing of the CPT moieties in the aggregated form, and the greater amplitude of the diCPT conjugate CD signal indicates a stronger degree of chiral arrangement. Surprisingly, qCPT-buSS-Tau exhibits two bisignate CD signals centered at 265 and 366 nm and a strong positive signal at 389 nm (Fig. 3c). This bisignate Cotton effect has been frequently reported in aggregated π-conjugated systems, resulting from exciton coupling between two adjacent chromophores in a chiral orientation36,37. Hence, the observed CD signals are indicative of a strong exciton coupling between neighboring CPT quinoline rings of the same or adjacent conjugates. The positive sign of the couplet signal at the highest wavelength suggests a positive chirality and a right-handed helical arrangement of the CPT molecules within the nanotubes. The strong interactions among CPT units might play a crucial role for formation of the observed nanotube morphology, although the exact mechanism is not clear and requires further evaluation.
The short term stability of the nanostructures formed by the studied molecules towards dilution was probed using a fluorescence method based on static quenching of the CPT fluorophore (supporting information S2.3.1). At short equilibration times (2 mins), a dilution study showed dissociation occurring around 207 nM, 74 nM and 53 nM for mCPT-buSS-Tau, dCPT-buSS-Tau, and qCPT-buSS-Tau, respectively (Fig. S10). These values suggest a high stability of these self-assembled nanostructures towards dissociation. The greater stability associated with higher CPT content is expected, likely a result of an increasing hydrophobicity in combination with possible π-π associative interactions among the CPT units. Given the hydrophobic nature of CPT and the preference that the Tau peptide has for parallel β-sheet formation30, it is reasonable to assume that the CPT segments are buried in the cores of the assembled nanostructures, thereby preventing CPT from undergoing undesired conversion to the inactive acid form. Indeed, the fluorescence spectra of these DAs show an emission maximum in the 430 nm region (Fig. S11), consistent with the CPT having the closed lactone form rather than the open carboxylate which emits at 446 nm38. The lactone form is essential for CPT to interact with topoisomerase I17,28.
Given the core shell micellar nature of these assemblies, it would be expected that assembly into nanostructures would shield the degradable linker from the external environment and thus offer a mechanism for the controlled release of bioactive CPT.39 The release profiles of the DAs were assessed by incubation of 2 μM solutions at 37°C in 10 mM sodium phosphate in the presence and absence of 10 mM glutathione (GSH), respectively (Fig. 3d). Glutathione is a cancer-relevant reducing agent that can degrade the designed buSS linker to release CPT40. In the absence of GSH, hydrolytic cleavage of the CPT ester bond is responsible for any observed release. All DAs were seen to exhibit a steady rate of release, with no sudden burst kinetics. It is evident that for molecules with the reducible linkers CPT release was faster in the presence of GSH than in its absence. For the non-reducible control DA, mCPT-mal-Tau, which is susceptible to hydrolysis of the CPT ester bond only, no significant difference between the two conditions was observed. It is also evident that both dCPT-buSS-Tau and qCPT-buSS-Tau show an increased resistance to degradation relative to mCPT-buSS-Tau. We attribute this to their greater relative stability as the nanostructures formed by dCPT-buSS-Tau and qCPT-buSS-Tau will have a lesser propensity for disassembly at the concentration studied, with mCPT-buSS-Tau existing in more of a monomeric state than assembled. To confirm this effect, we performed experiments studying the degradation of the mCPT-buSS-Tau at a higher concentration (20 μM), where a significant majority of the molecule would be expected to exist in the nanostructure form. Fig. 3e supports this idea as the stability towards hydrolytic cleavage (in the absence of GSH) is greatly enhanced, with 95% of the conjugate remaining after 8 hours compared with 40% at 2 μM. A similar, but less pronounced, enhancement is observed in the presence of GSH, with 52% of the conjugate remaining after 8 hours at the higher concentration compared with 9% at the lower. These experiments suggest the CPT release mechanism shown in Fig. 3f, indicating that the self-assembled nanostructures can serve as reservoirs to provide a consistent supply of CPT conjugate monomers that can be quickly converted to bioactive CPT in the presence of GSH.
We evaluated the in vitro toxicity of the designed DAs against a number of cancer cell lines (Fig. 4). Human MCF-7 breast cancer cells and two rat gliosarcoma lines (9L and F98L) were initially tested with varying concentrations of conjugates in order to determine a dose-response relationship (Fig. 4a, 4b and Fig. S12a). For all three cell lines the general trend is clear, with the reduction-sensitive DAs exerting a greater cytotoxic effect than the non-sensitive mCPT-mal-Tau. Although we cannot rule out the possibility of extracellular hydrolysis followed by diffusion of released CPT into cells, the marked difference between the buSS and mal-linked conjugates suggests that reductive degradation by GSH is dominant and mainly responsible for the observed toxicity. The C8-Tau control experiment reveals that the Tau peptide alone presents no cytotoxic effects at the concentrations studied. Of the three DAs, dCPT-buSS-Tau was consistently observed to be the most effective at inhibiting the proliferation of cancer cells followed by qCPT-buSS-Tau and then mCPT-buSS-Tau. Since the CPT drug must enter the cell nucleus to exert its cytotoxic effect, cellular uptake presents an important step in determining the IC50 of the designed drug conjugates. One possible explanation for dCPT-buSS-Tau having the lowest IC50 may be its balanced hydrophobic-hydrophilic ratio allowing for the most efficient translocation of the conjugate into cells. Given the higher potency of dCPT-buSS-Tau, a wider range of cancer cell lines were assessed for their response to this DA (Figs. S12b–f), showing comparable to moderate activity (supporting information (S4) versus CPT.
Figure 4.
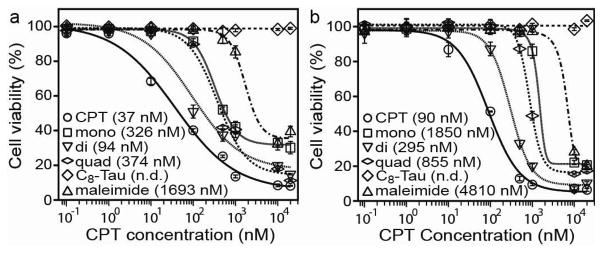
In vitro dose-response relationship study of the DA molecules against human MCF-7 breast cancer (a) and rat 9L gliosarcoma (b) cells. All cancer cells were incubated with the appropriate DA molecules for 48 hrs and cell viability was determined by SRB assay. Cytotoxicity experiments were performed in triplicate and values are given as mean ± s.d. (n = 3). The DA concentrations and the calculated IC50 values (in parentheses) were normalized with respect to the number of CPT molecules.
In this paper, we have reported a strategy to construct discrete drug nanostructures with a high and fixed drug content. We believe this strategy can be extended to fabricate nanostructures of other important anticancer drugs, such as paclitaxel and doxorubicin, and with proper molecular design and fine tuning of the self-assembly conditions it should be possible to have access to vesicular and spherical morphologies. The use of small molecular anticancer drugs as active molecular building units, not just passive cargoes to be delivered, opens up new opportunities for the development of drug nanostructures that can self-deliver.
Supplementary Material
Acknowledgments
We thank Johns Hopkins University (JHU) for the Startup fund, and the NIH for funding AC (T-32CA130840) and YL (R25CA153952). We thank the JHU Integrated Imaging Center (IIC) for the use of the TEM facility, and the JHU Dept. Chemistry Mass Spectrometry facility for MALDI-ToF analysis (NSF CHE-0840463). We also thank Prof. Kalina Hristova for the use of circular dichroism and fluorescence instrumentation.
Footnotes
Experimental details and figures can be found in online supporting material. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peer D, Karp JM, Hong S, FaroKhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Duncan R. Nat Rev Cancer. 2006;6:688. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman AS. J Control Release. 2008;132:153. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Euliss LE, DuPont JA, Gratton S, DeSimone JM. Chem Soc Rev. 2006;35:1095. doi: 10.1039/b600913c. [DOI] [PubMed] [Google Scholar]
- 5.Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Nat Rev Drug Discov. 2011;10:521. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 6.Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG. Nat Rev Cancer. 2012;12:39. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- 7.Discher DE, Eisenberg A. Science. 2002;297:967. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 8.Li SD, Huang L. Mol Pharm. 2008;5:496. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 9.Torchilin VP. Nat Rev Drug Discov. 2005;4:145. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 10.Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, Napier M, Bear JE, DeSimone JM. Nano Letters. 2012;12:5304. doi: 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang LF. Proc Natl Acad Sci USA. 2011;108:10980. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng Y, Dalhaimer P, Cai SS, Tsai R, Tewari M, Minko T, Discher DE. Nat Nanotechnol. 2007;2:249. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bae JW, Pearson RM, Patra N, Sunoqrot S, Vukovic L, Kral P, Hong S. Chem Commun. 2011;47:10302. doi: 10.1039/c1cc14331j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin LY, Karwa A, Kostelc JG, Lee NS, Dorshow RB, Wooley KL. Mol Pharm. 2012;9:2248. doi: 10.1021/mp3000887. [DOI] [PubMed] [Google Scholar]
- 15.Poon Z, Lee JB, Morton SW, Hammond PT. Nano Lett. 2011;11:2096. doi: 10.1021/nl200636r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng JJ, Khin KT, Jensen GS, Liu AJ, Davis ME. Bioconjug Chem. 2003;14:1007. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- 18.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nat Mater. 2009;8:543. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 19.Dobrovolskaia MA, McNeil SE. Nat Nanotechnol. 2007;2:469. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 20.Huttunen KM, Raunio H, Rautio J. Pharmacol Rev. 2011;63:750. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 21.MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Nat Mater. 2009;8:993. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen YQ, Jin EL, Zhang B, Murphy CJ, Sui MH, Zhao J, Wang JQ, Tang JB, Fan MH, Van Kirk E, Murdoch WJ. J Am Chem Soc. 2010;132:4259. doi: 10.1021/ja909475m. [DOI] [PubMed] [Google Scholar]
- 23.Li XQ, Wen HY, Dong HQ, Xue WM, Pauletti GM, Cai XJ, Xia WJ, Shi DL, Li YY. Chem Commun. 2011;47:8647. doi: 10.1039/c1cc12495a. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 25.Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. J Am Chem Soc. 2010;132:17707. doi: 10.1021/ja109269v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui HG, Webber MJ, Stupp SI. Biopolymers. 2010;94:1. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trent A, Marullo R, Lin B, Black M, Tirrell M. Soft Matter. 2011;7:9572. [Google Scholar]
- 28.Pommier Y. Nat Rev Cancer. 2006;6:789. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 29.Thomas CJ, Rahier NJ, Hecht SM. Bioorg Med Chem. 2004;12:1585. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 30.Goux WJ, Kopplin L, Nguyen AD, Leak K, Rutkofsky M, Shanmuganandam VD, Sharma D, Inouye H, Kirschner DA. J Biol Chem. 2004;279:26868. doi: 10.1074/jbc.M402379200. [DOI] [PubMed] [Google Scholar]
- 31.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Proc Natl Acad Sci USA. 2008;105:12128. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klug A. Philos Trans R Soc Lond B Biol Sci. 1999;354:531. doi: 10.1098/rstb.1999.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Z, Kang S, Banno M, Yamaguchi T, Lee D, Seok C, Yashima E, Lee M. Science. 2012;337:1521. doi: 10.1126/science.1224741. [DOI] [PubMed] [Google Scholar]
- 34.Pizzey CL, Pomerantz WC, Sung BJ, Yuwono VM, Gellman SH, Hartgerink JD, Yethiraj A, Abbott NL. J Chem Phys. 2008:129. doi: 10.1063/1.2955745. [DOI] [PubMed] [Google Scholar]
- 35.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proc Natl Acad Sci USA. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langeveld-Voss BMW, Beljonne D, Shuai Z, Janssen RAJ, Meskers SCJ, Meijer EW, Bredas JL. Adv Mater. 1998;10:1343. [Google Scholar]
- 37.Aida T, Meijer EW, Stupp SI. Science. 2012;335:813. doi: 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chourpa I, Millot JM, Sockalingum GD, Riou JF, Manfait M. BBA Gen Subjects. 1998;1379:353. doi: 10.1016/s0304-4165(97)00115-3. [DOI] [PubMed] [Google Scholar]
- 39.van Bommel KJC, Stuart MCA, Feringa BL, van Esch J. Org Biomol Chem. 2005;3:2917. doi: 10.1039/b507157g. [DOI] [PubMed] [Google Scholar]
- 40.Estrela JM, Ortega A, Obrador E. Crit Rev Clin Lab Sci. 2006;43:143. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.