

Abstract
Objective
To assess the role of magnetic resonance imaging (MRI) in the prenatal diagnosis of neural tube defects (NTDs).
Background
NTDs comprise a heterogeneous group of congenital anomalies that derive from the failure of the neural tube to close. Advances in ultrasonography and MRI have considerably improved the diagnosis and treatment of NTDs both before and after birth. Ultrasonography is the first technique in the morphological study of the fetus, and it often makes it possible to detect or suspect NTDs. Fetal MRI is a complementary technique that makes it possible to clear up uncertain ultrasonographic findings and to detect associated anomalies that might go undetected at ultrasonography. The progressive incorporation of intrauterine treatments makes an accurate diagnosis of NTDs essential to ensure optimal perinatal management. The ability of fetal MRI to detect complex anomalies that affect different organs has been widely reported, and it can be undertaken whenever NTDs are suspected.
Conclusion
We describe the normal appearance of fetal neural tube on MRI, and we discuss the most common anomalies involving the structures and the role of fetal MRI in their assessment.
Key Points
• To learn about the normal anatomy of the neural tube on MRI
• To recognise the MR appearance of neural tube defects
• To understand the value of MRI in assessing NTDs
Keywords: Prenatal diagnosis, Magnetic resonance imaging, Congenital abnormalities, Neural tube defects, Spinal dysraphism
Introduction
Technological developments in diagnostic imaging have improved the diagnosis and treatment of congenital anomalies immensely, and this progress is especially evident in neural tube defects (NTDs) [1].
Ultrasonography is performed to detect possible malformations at about 20 weeks into gestation. Ultrasonography allows accurate assessment of the neural tube. Fetal magnetic resonance imaging (MRI) complements ultrasonography, confirming the suspected anomaly and even detecting malformations that were not detected at ultrasonography. The additional information provided by MRI may lead to changes in the management of the pregnancy and/or of the delivery [2–5].
In this article we review the classification and clinical characteristics of the NTDs. We describe the radiological, pathological and clinical features of the most common NTDs, and finally emphasise an integrated clinical and radiological approach for accurate diagnosis.
MRI protocol and technique
In our institution, all fetal MRI studies are performed after ultrasonography and MRI never constitutes the first imaging study of the fetus. None of the women or fetuses receives any special preparation. Although it is important to be extremely careful when undertaking any procedure on a fetus, there is no evidence of associated risk to fetal development with fetal MRI [6–8].
We use a 1.5-Tesla scanner with a four-element phased-array body coil. A complete study requires between 12 and 15 sequences, so it takes about 25–30 min.
The fetal MRI images are obtained using ultrafast sequences to minimise artefacts due to fetal movements. It seems that slow, rhythmic maternal respiration reduces fetal movements, so it is not necessary to acquire images with maternal breath-holding [1]. T2-weighted single shot fast spin-echo (ssFSE) or half-Fourier single-shot turbo spin-echo (HASTE) images provide most of the information necessary for diagnosis. The spatial resolution of T1-weighted sequences improves with gestational age. Gradient echo sequences (echo-planar imaging [EPI] and true fast imaging with steady state precession [FISP]) have greater ferromagnetic susceptibility, so they make it possible to identify bony and vascular structures [1, 9].
Both the head and spine must be studied in all three planes. Even when the only indication for fetal MRI is a suspected NTD, all the fetal structures should always be studied to rule out other possible anomalies [9].
Anatomy and embryology
The development of the neural tube can be divided into three phases [10]. The first phase, gastrulation, begins in the 2nd to 3rd weeks of gestation; in this phase, the two layers of the embryonic plate divide into three definitive layers (the ectoderm, mesoderm and endoderm). In the second phase, neurulation, which takes place in the 3rd to 4th weeks of gestation, the cells that will become the notochord interact with the overlying ectodermic tissue, causing this tissue to thicken, fold over and fuse, thus forming the neural tube. The fusion begins in the medial dorsal zone of the embryo and progresses in a zipper-like fashion cranially as the rostral neuropore and caudally as the caudal neuropore. The closure of the two ends does not occur simultaneously: the cranial end closes before the caudal end. The final phase, secondary neurulation, takes place in the 5th to 6th weeks of gestation; in this phase a secondary neural tube forms from the cells of Hensen’s node, which is a mass of totipotent cells located dorsally that intervene in the formation of the notochord and later migrate caudally to the presacral region. This secondary neural tube is initially solid; it undergoes a process of retrogressive differentiation called canalisation in which it cavitates to form the medullary cone and the filum terminale. The interruption of this process at any point, whether in the cranial end or in the caudal end, will result in an NTD [11, 12].
The spinal column and spinal cord are the same length until 12–15 weeks’ gestation, after which the spinal column grows more than the spinal cord until the medullary cone is situated approximately at the level of the second lumbar vertebra, where it will be at birth and will remain without significant changes throughout life [11, 12].
Normal anatomy of the neural tube on MRI
MRI is an excellent method for evaluating the structures of the fetal head and spine. Images can be obtained in any plane, regardless of the position of the fetus in the uterus [13].
The cranial vault (Fig. 1a, b) is seen as a hypointense structure in T2-weighted images, even early in gestation. The structures in the base of the skull (Fig. 1a) can also be identified in any plane, but their visualisation improves as gestation advances.
Fig. 1.
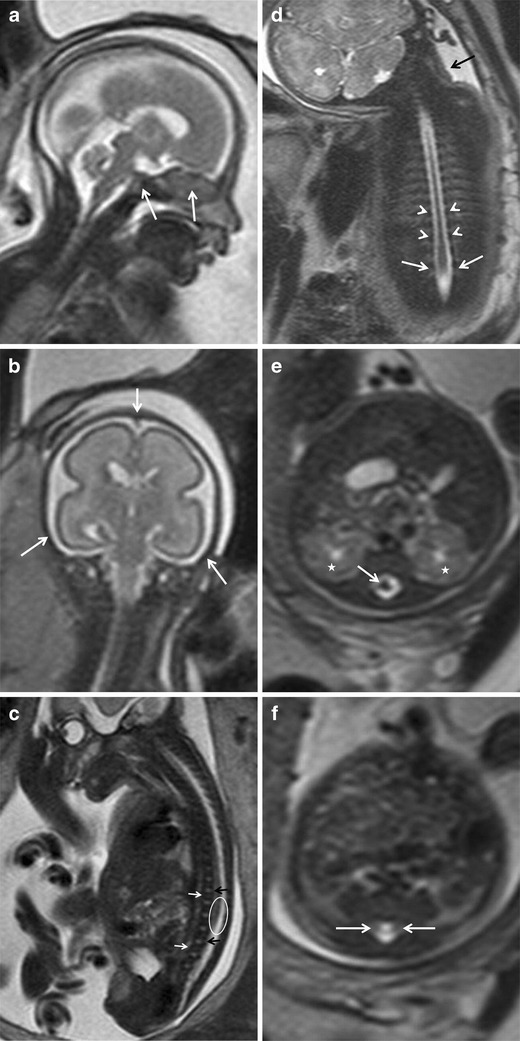
a, b Normal anatomy of the neural tube. Normal cranial bone structures. Fetus at 24 weeks’ gestation. a Sagittal and b coronal T2-weighted HASTE images of the fetal head show the osseous structures of the base of the skull (arrows in a) and bones of the cranial vault (arrows in b). c–f Normal fetal spine. Fetus at 27 weeks’ gestation. c Sagittal T2-weighted HASTE image of the spinal canal. The vertebral bodies are hypointense with a band of higher intensity in the centre (white arrows), the intervertebral spaces are hyperintense (black arrows). The ellipse marks the medullary cone. d Coronal T2-weighted HASTE image shows the vertebral pedicles as hypointense rounded structures on both sides of the spinal canal (arrowheads). The interpedicular distance in the lumbar spine is slightly increased (arrows); this is a normal finding that should not be confused with spina bifida. A false cervicothoracic scoliosis is also seen (black arrow). e Axial image at the level of L1–L2 shows the medullary cone (white arrow) inside the spinal canal (the asterisks mark the kidneys). f The medullary cone is no longer seen in this axial image of the spinal canal below the kidneys; sometimes the nerve roots can be identified (white arrows)
The vertebral bodies have low signal intensity on T2-weighted sequences and are clearly seen on sagittal images. True FISP sequences provide good information about bony structures. The intervertebral spaces are hyperintense and are clearly depicted in the sagittal plane (Fig. 1c). The vertebral pedicles are better visualised in the coronal plane (Fig. 1d). The interpedicular distance should be constant throughout the spinal column, although it does increase slightly in the lumbar region and this widening should not be confused with an NTD (Fig. 1d). The posterior elements of the vertebrae are best seen in the axial plane (Fig. 1e, f).
Caution is warranted in the assessment of possible scoliosis or kyphosis, because the spinal column can be altered by the position of the fetus in the uterus; these pseudoalterations are more common as gestation advances, resulting in proportionately less space within the uterine cavity (Fig. 1d).
The spinal canal is hyperintense in T2-weighted sequences because it is filled with cerebrospinal fluid. The spinal cord can be identified as a hypointense structure inside the spinal canal. Although the spinal cord is well depicted in all three planes, it can probably be evaluated best in the axial plane (Fig. 1e). The position of the fetus within the uterus results in a certain degree of thoracolumbar kyphosis that displaces the spinal cord anteriorly in the thoracic spine, making it difficult to see because it is adjacent to the vertebral bodies (Fig. 1c). Precaution should be taken to ensure that the spinal cord disappears below the fetal kidneys (Fig. 1e, f); sometimes, small hypointense structures that correspond to nerve roots are seen distal to the medullary cone (Fig. 1f).
It is important to evaluate the paravertebral soft tissues in MRI studies, especially in fetuses with spina bifida or destructive processes or tumours that could affect these tissues. The skin and the paravertebral muscular structures are hypointense in T2-weighted sequences. The spinal canal should be closed posteriorly by soft tissues (Fig. 1c, e, f).
Neural tube defects
NTDs are a heterogeneous group of anomalies that result from the partial or total failure of the neural tube to close. NTDs can affect the cranial region (anencephaly) or the spinal region (spina bifida); spinal NTDs can be divided into open or closed defects, depending on whether they are covered by a layer of skin [11] (Fig. 2).
Fig. 2.
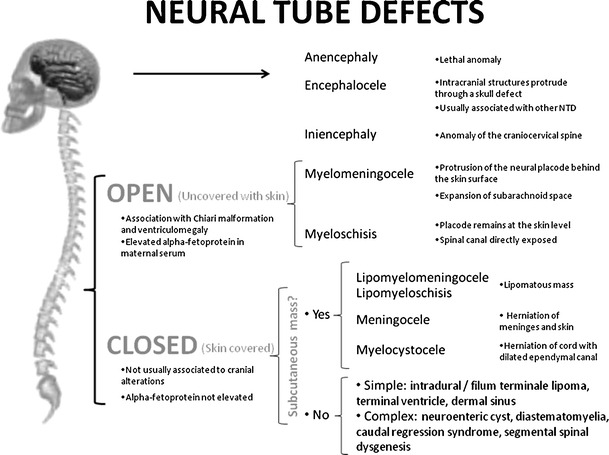
Neural tube defects
The overall worldwide incidence of NTDs is between one and ten per 1,000 live births [14, 15]. The rate of NTDs has varied widely over time and has a clear geographical distribution. The highest incidences have been reported in Mexico, Ireland, India and northern China [16, 17]. Most cases have a sporadic and multifactorial origin. A family history of NTDs is absent in 90–95% of cases, and NTDs are reported in 3% of miscarriages [11]. Some genetic factors like single gene mutations (Meckel-Gruber and Roberts syndromes) and chromosomopathies (trisomies 13 and 18, triploidy) have been identified. Some maternal factors (hyperthermia, diabetes, hyperinsulinaemia, obesity, psychosocial or emotional stress [18–23]) and some environmental factors (maternal medication with anticonvulsants like valproic acid and carbamazepine [24]) can increase the incidence of NTDs [25]. Nutritional factors that can predispose to NTDs have also been identified. Administering folic acid during pregnancy reduces the risk of NTDs [26–28].
Prenatal screening for NTDs includes the measurement of diverse markers like acetylcholinesterase and alpha-fetoprotein in amniotic fluid and in maternal serum (alpha-fetoprotein is a glycoprotein secreted by the liver and by the fetal yolk sac) [9]. A recent meta-analysis [29] confirmed that measuring alpha-fetoprotein in maternal serum during the second trimester is useful in screening for NTDs. Nevertheless, this marker is only useful for detecting open NTDs; furthermore, alpha-fetoprotein levels are also elevated in a long list of fetal anomalies, including omphalocele, Turner’s syndrome, and sacrococcygeal teratoma, amongst others [29].
Anencephaly/exencephaly
It is the absence of a major portion of the brain, skull and scalp above the orbits. There are two subtypes: anencephaly, in which the cranial vault, cerebral hemispheres and diencephalic structures are replaced by amorphous neurovascular tissue, and exencephaly, in which the cranial vault is absent but relatively normal amounts of brain tissue, albeit abnormally developed, is present.
Some authors consider exencephaly an early stage of anencephaly, because the unprotected anomalous brain tissue located outside the cranial vault will eventually take on the appearance of anencephaly after being destroyed by the trauma induced by constant collision against the walls of the uterus [11]. In both exencephaly and anencephaly, the osseous structures above the orbits are absent. The base of the skull, the facial structures, and the orbits are present and are usually normal.
MRI is usually not used to evaluate fetuses with these conditions because ultrasonography usually detects this anomaly very early and MRI is not required to confirm this diagnosis. Fetal MRI studies show the absence of osseous structures above the orbits and the absence of brain tissue or alterations in brain tissue when it is present. It is considered a lethal anomaly, associated anomalies usually have no significance.
Encephalocele
In encephalocele, intracranial structures protrude through a defect in the skull. The specific name of the anomaly depends on the structures contained in the herniated sac; for example, a meningocele contains meninges and cerebrospinal fluid, an encephalomeningocele contains meninges and brain tissue and an encephalomeningocystocele or encephalomeningohydrocele contains meninges, brain tissue, and part of the ventricular system (Fig. 3). Most encephaloceles occur along the midline in the occipital or frontal region, but they can also involve the anterior cranial fossa (into the nasal sinuses) or even the base of the skull (into the sphenoid or ethmoid sinus).
Fig. 3.
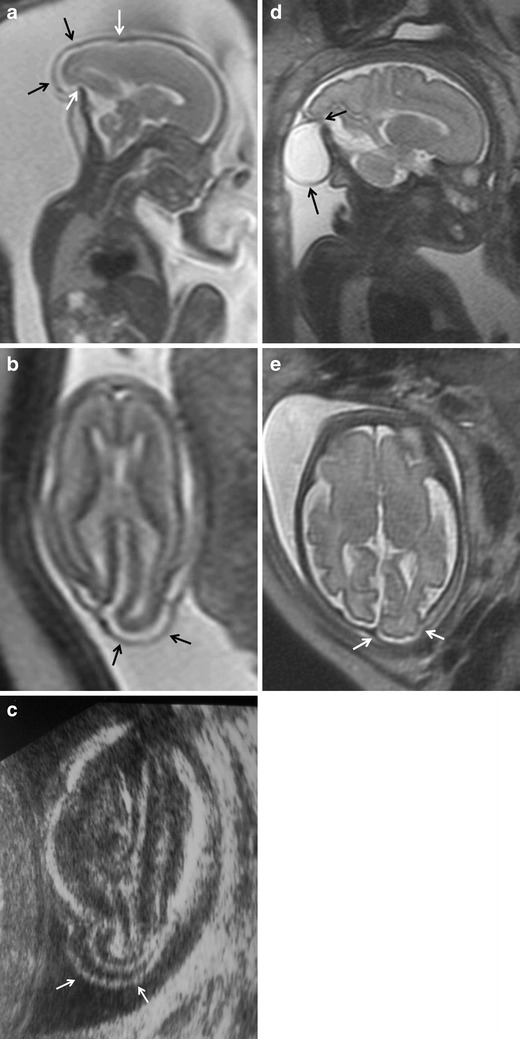
Encephalocele. Fetus at 21 weeks’ gestation. a Sagittal and b axial T2-weighted HASTE images show the protrusion of the occipital lobe and occipital horn of the ventricular system (black arrows in a and b) through a defect in the bone and soft tissues (white arrows in a). c Ultrasound image also shows the protrusion of the occipital lobe (white arrows). d, e Images of the same fetus at 31 weeks’ gestation show a decrease in the extruded encephalic component (arrow in e) and an increase in the associated meningeal component (arrow in d)
Encephaloceles are usually diagnosed by ultrasonography. MRI’s role is to specify the type of lesion, which is more important in smaller lesions and in those that affect the anterior cranial fossa or base of the skull. It is essential to evaluate the rest of the structures in the head and the neural tube, fundamentally the posterior fossa because it can be difficult to evaluate with ultrasonography.
The MRI semiology varies with the type of lesion. MRI shows a homogeneously hyperintense signal in cases of meningocele (Fig. 3d) as well as the presence of brain tissue inside the herniated sac in cases of encephalomeningocele (Fig. 3a, b, e). Other anomalies like open spina bifida or ventriculomegaly that are often associated with encephaloceles are easy to evaluate with MRI.
The differential diagnosis for encephalocele is based on the size, content and location of the lesion. The main differential diagnosis for encephaloceles located in the occipital region is with cystic hygroma, whereas for those located in the frontal region, the differential diagnosis is with teratoma and haemangioma of the scalp. Both teratomas and haemangiomas of the scalp are shown as slightly heterogeneous but well-defined solid masses with variable signal intensity; haemangiomas may have areas of signal voiding due to their intense vascularisation [30].
An encephalocele may form part of a syndrome like Meckel-Gruber syndrome and it can be associated with other anomalies like ventriculomegaly, spina bifida (7–15% of all encephaloceles) [11], microcephaly, or cleft lip and/or cleft palate.
The prognosis depends on the size, location and extent of the lesion as well as on whether other anomalies are present [31].
Iniencephaly
Iniencephaly is an NTD affecting the occipital bone combined with rachischisis of the cervicothoracic spine and retroflexion of the head (Fig. 4). Fetuses with iniencephaly have a variable defect in the occipital bone with an enlarged foramen magnum, the partial or total absence of cervicothoracic vertebrae with irregular fusion of those that are present and incomplete closure of the vertebral bodies and arches. These alterations result in significant shortening of the spine and of the fetus due to marked lordosis with hyperextension of the head. Iniencephaly can be open (associated with encephalocele) or closed (spinal anomaly without encephalocele) [11].
Fig. 4.
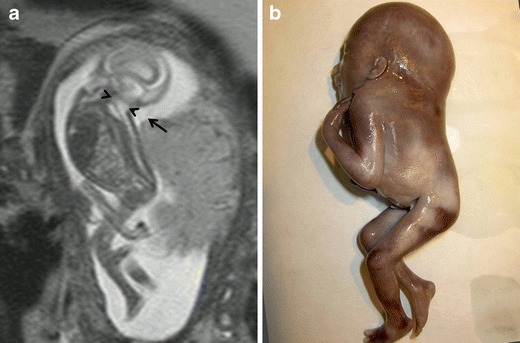
Closed iniencephaly. Fetus at 17 weeks’ gestation. a Sagittal T2-weighted HASTE images show a severe spinal anomaly. The spine is short, the head is hyperextended, and there is a nuchal fold (arrow). The foramen magnum is enlarged (arrowheads). b Pathological specimen
The differential diagnosis is with Klippel-Feil syndrome (short neck and fusion of the cervical vertebrae). Some authors believe that Klippel-Feil syndrome is a milder form of iniencephaly [11].
The prognosis of iniencephaly in the neonatal period is dismal, so whether other anomalies are present is unimportant.
Spinal dysraphism or spina bifida
Spinal dysraphism is the result of a congenital vertebral defect due to the failure of the caudal neuropore to close that leads to the exposure of the contents of the medullary canal to the exterior. The defect usually involves the posterior vertebral elements, although it can also involve the vertebral body itself. The defect is usually located in the lumbosacral region, but it can occur at any level and it can even involve the entire spine. The absence of skin and muscle in these areas is due to the failure to induct the overlying ectodermal and mesodermal tissues [11].
Other theories postulate the existence of an unbalance in the production and reabsorption of cerebrospinal fluid during the embryonic period that leads to an excessive accumulation in the already closed neural tube (hydromyelia), which in turn causes a secondary separation from the dorsal wall [11].
Spinal dysraphism comprises a wide spectrum of anomalies; these can be divided into open and closed defects. Open defects include rachischisis and myelomeningocele. Rachischisis is the most severe form of spina bifida; in this lethal condition, an extensive segment of the rachis is absent, resulting in extensive exposure of neural tissue [11]. Myelomeningocele and myeloschisis (myelocele) are the most common spinal dysraphism, accounting for 85% of all cases [12, 32]. In these lesions, neural contents are exposed to the exterior through midline defects in the bone and skin [12, 33]. When the spinal canal is directly exposed, the anomaly is called myeloschisis or myelocele [1] (Fig. 5), and when intramedullary structures lined with meninges protrude or herniate due to the expansion of the underlying subarachnoid space [34], it is called myelomeningocele (Fig. 6). Myelomeningocele is the most common open spinal dysraphism [32, 35]. By definition, there must be a vertebral defect, the most common of which is the absence of the posterior vertebral arches (Figs. 5d and 6a, b). The interpedicular distance is increased (Fig. 6c). The main characteristic that allows myelomeningoceles to be differentiated from myeloschisis is the position of the neural placode, a remnant of the unfolded (non-neurulated) neural tube, with respect to the surface of the skin. In myelomeningocele, the neural placode protrudes behind the surface of the skin (Fig. 6a, b, f), whereas in myeloschisis it is at the same level (Fig. 5c, d, f) [32]. Another, extremely rare type of open dysraphism is the hemimyelomeningocele or hemimyelocele: in this anomaly, diastematomyelia is associated with one of the above-mentioned anomalies in which one of the hemicords does not complete neurulation [32, 36]. This finding is important because if diastematomyelia is not suspected, the surgical repair of the myelomeningocele will not include the repair of the tethered cord that is nearly always present in diastematomyelia [1, 9, 37].
Fig. 5.
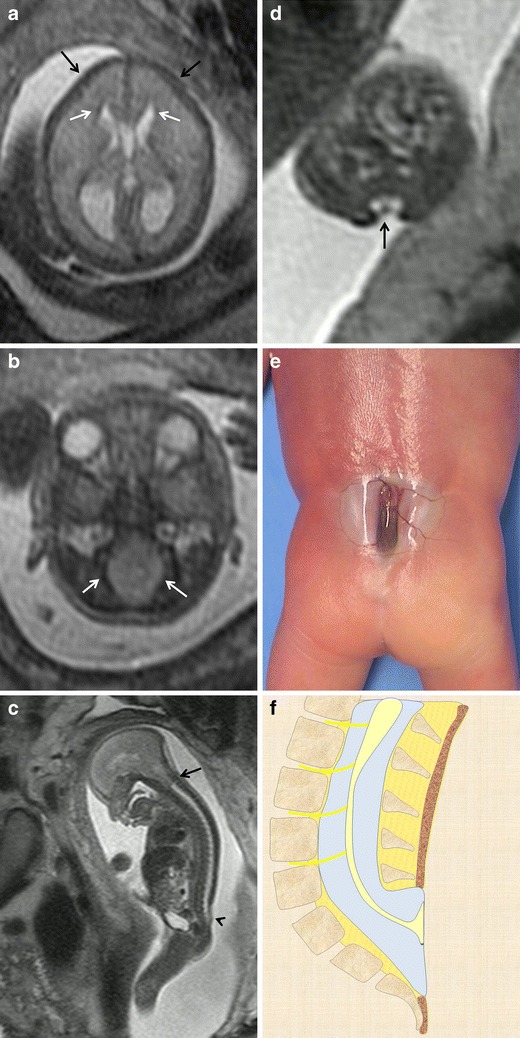
Myeloschisis. Fetus at 23 weeks’ gestation. a, b Axial T2-weighted HASTE image of the fetal cranium shows the deformity of the frontal bones (black arrows in a) and the characteristic beaklike shape of the ventricular horns (arrows in a). The posterior fossa deformity can also be seen: the cisterna magnum is absent and the foramen magnum is enlarged due to the descent of the structures of the posterior fossa toward the spinal canal (arrows in b). c Sagittal T2-weighted HASTE image shows a cutaneous defect or thinning in the lumbosacral region (arrowhead) with no mass protruding through the defect as well as an associated Chiari malformation (black arrow). d Axial image at the level of the cutaneous defect shows the abnormally low neural placode within the spinal canal without expansion of the subarachnoid spaces. These findings are characteristic of myeloschisis. e Pathological specimen. f Sagittal diagrams of the anomaly
Fig. 6.
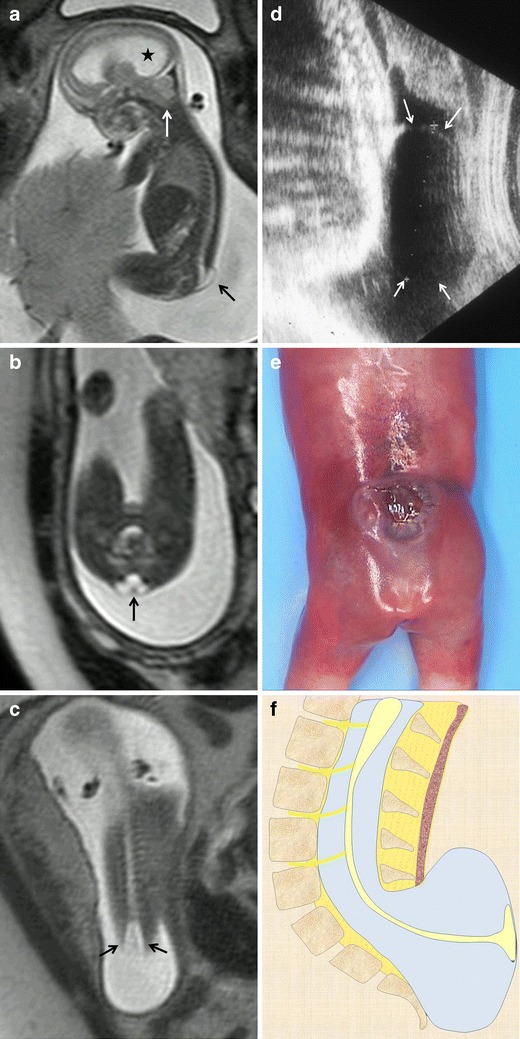
Lumbosacral myelomeningocele. Fetus at 22 weeks’ gestation. a Sagittal T2-weighted HASTE image shows a small cystic mass protruding at the thoracolumbar level (black arrow). Associated hydrocephalus (asterisk) and a Chiari malformation (white arrow) are also seen. b Axial T2-weighted HASTE image at the lumbosacral level shows the neural placode protruding outside the surface of the skin due to expansion of the adjacent subarachnoid space (black arrow). c Coronal image at the spinal level shows increased interpedicular distance at the level of the neural defect. d Ultrasound image of a different patient shows a cystic mass protruding at lumbar level (white arrows). e Pathological specimen. f Sagittal diagrams of the anomaly
Open spina bifida is nearly always found together with Chiari type II malformation [34], which is an anomaly of the posterior fossa consisting of herniation of part of the cerebellum (vermis and fourth ventricle) and of the brainstem through the foramen magnum (Figs. 5c and 6a). This association is thought to be due to the collapse of the primitive fetal ventricular system (brought about by the loss of cerebrospinal fluid through the spinal defect), which prevents the rhombencephalic vesicle (from which the brainstem, fourth ventricle, and cerebellum are derived) from expanding and results in deficient development of the posterior fossa and herniation [38]. Recent studies have reported worse prognoses for fetuses with greater cerebellar herniation, related to an increased incidence of seizures, vesical dysfunction, and dependency for ambulation [39].
Open spina bifida is accompanied by a collapse of the extra-axial subarachnoid spaces (Fig. 5a) and/or ventriculomegaly (Fig. 6a) secondary to mechanical obstruction from the Chiari malformation or to dysfunctional reabsorption of cerebrospinal fluid [38, 40] in 75% of cases [11]. This is why it is essential to evaluate the spine carefully to rule out spinal dysraphism when ventriculomegaly is detected at any gestational age. Likewise, the shape of the ventricles is abnormal owing to the acute angle of the ventricular horns [41] (Fig. 5a). As in ultrasonography, in MRI both direct and the indirect signs of spina bifida at the level of the fetal cranium (the lemon sign: special shape of the cranium due to deformity of the frontal bones [Fig. 5a] and the banana sign: special shape of the posterior fossa secondary to displacement of the cerebellar hemispheres toward the cervical canal, which results in the obliteration of the cisterna magnum [Fig. 5b]) are of the utmost importance for the diagnosis. Other cranial anomalies, like the collapse of the subarachnoid space, a beak-shaped mesencephalic tectum, dysgenesis of the corpus callosum and subependymal heterotopias are all better seen on MRI [9].
The differential diagnosis will depend fundamentally on the contents and location of the spinal defect. The main differential diagnosis with myelomeningocele will be sacrococcygeal teratoma. The intracranial anomalies that accompany NTDs can help in differentiating between these two entities, because teratomas are not usually accompanied by intracranial anomalies.
Distinguishing between open and closed spinal dysraphism is of the utmost importance because the long-term functional and neurological prognoses are notably better in cases of closed dysraphism than in cases of open dysraphism [39, 41].
It is of the utmost importance to obtain an accurate and specific assessment of the anomaly due to the progressive incorporation of in utero treatments. Latest trials in fetal surgery for myelomeningocele have shown a significant reduction of VP shunt placement, an improvement in the overall neuromotor function and a significantly reversed hindbrain herniation in those patients who underwent fetal repair compared with those who underwent postnatal surgery. This trial has also revealed that in utero therapy increases the risk of spontaneous rupture of membranes, oligohydramnios and preterm delivery [42]. Fetal MRI provides a better overview for the neurosurgeon who is familiar with MRI and not as much with ultrasound. Specific details with respect to the level and magnitude of the defect can help the intra-uterine management of the spinal anomaly. The resolution of MRI is less than ultrasound, but the diagnostic value of MRI is less dependent on the fetal position.
Closed dysraphism accounts for about 15% of cases [11]. These tend to be small lesions and they are completely covered with skin. The term “closed” is more appropriate than “occult”, because cutaneous signs that raise suspicion of these anomalies are present in up to 50% of cases [34, 43]. The soft tissues that cover the spine dorsally can be identified as a hypointense line on sagittal and axial T2-weighted images (Figs. 7a, b). In cases of open spina bifida, this line is interrupted at the level of the lesion; this finding is essential for differentiating between open and closed dysraphism. Closed dysraphism is not usually associated to cranial alterations like Chiari malformation or ventriculomegaly [1, 9] and does not manifest with elevated alpha-fetoprotein [9]. Closed spinal dysraphism can be subdivided into lesions with subcutaneous masses and those without.
Fig. 7.
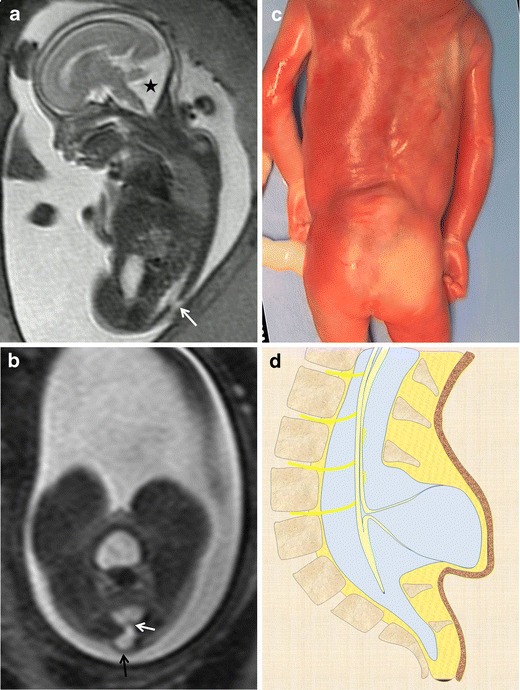
Myelocystocele. Fetus at 23 weeks’ gestation. a Sagittal T2-weighted HASTE image shows an interruption in the posterior vertebral arches in the lumbar region (arrow). A posterior fossa anomaly with an elevated vermis and dilation of the IV ventricle is also seen (asterisk). b Axial image at the lumbar level showing the cystic mass protruding through the spinal defect (black arrow). Two nerve roots (white arrow) can be seen arising from the non-neurulated neural placode inside the spinal defect. c Pathological specimen. d Sagittal diagram of the anomaly
There are three main types of closed spinal dysraphisms with masses. In the first, further subdivided into lipomyelomeningocele and lipomyeloschisis (lipomyelocele) (Fig. 8), direct contact between the mesenchymal tissue and neural tube affects the differentiation of fatty tissue [12]. Clinically, these lesions are characterised by the presence of a lipomatous subcutaneous mass above the gluteal cleft [32]. In these cases, it is important to note the point where the lipoma joins the neural placode to differentiate between a lipomyelomeningocele and lipomyeloschisis [34]. The point of union in lipomyelomeningoceles is outside the neural canal due to the expansion of the subarachnoid space, whereas in lipomyeloschisis the lipoma joins the neural placode inside the neural canal after entering through a defect in the bone (Fig. 8g) [34].
Fig. 8.
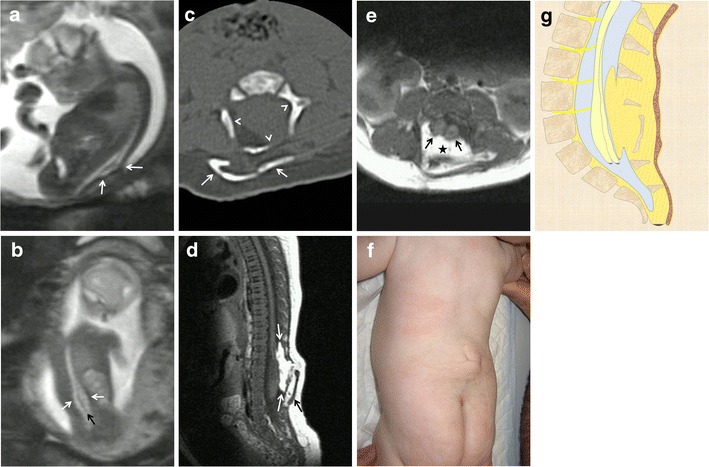
Complex closed spinal dysraphism: lipomyelocele and diastematomyelia. Fetus at 22 weeks’ gestation. a Sagittal T2-weighted HASTE image shows a deformity of the spinal canal completely covered by soft tissues (arrows). b Coronal image showing the widening of the spinal canal at the lumbar level (white arrows) and abnormally low termination of the medullary cone (black arrow) below the kidneys. c CT study after birth shows the deformed, widened spinal canal (arrowheads) and soft-tissue calcification behind the bone defect (white arrows). d Sagittal T1-weighted MRI image obtained after birth shows the abnormally low medullary cone, a lipomatous component within the spine (white arrows), and soft-tissue calcification (black arrow). e Axial T1-weighted image showing spinal cord duplication (black arrows) and the lipomatous component within the spine (asterisk). f Photograph of the patient after birth. g Sagittal diagrams of the anomaly
The second type is a meningocele with cutaneous lining. These lesions are herniated sacs of cerebrospinal fluid lined with meninges and skin. They are usually posterior herniations and are most commonly located in the thoracic spine [32]. Anterior meningoceles are much less common and tend to be located in the presacral region [44]. The third type, myelocystocele (Fig. 7), consists of the herniation of a segment of the spinal cord with a dilated ependymal canal within a meningocele [32]; these lesions can occur at any point in the spine [45]. Myelocystoceles are similar to myelomeningoceles; the two entities are differentiated by the thickness of the wall of the herniated sac (Fig. 7b), the absence of the Chiari malformation, and the absence of elevated alpha-fetoprotein levels [1, 9]. Myelocystoceles can be associated with cloacal anomalies [46].
Closed spinal dysraphisms without subcutaneous masses can be divided into simple and complex anomalies. Simple lesions include intradural lipomas, filum terminale lipomas, tight filum terminale syndrome, persistent terminal ventricle, and dermal sinus [29]. Intradural lipomas are lipomas located within the spine that are contained within the dural sac. These lesions generally affect the lumbosacral region and are usually associated with the tethered cord syndrome. Filum terminale lipomas or fibrolipomatous thickening of the filum terminale can be considered normal variants unless there is clinical evidence of a tethered cord. Filum terminale lipomas are identified as linear thickening of the filum terminale that is hyperintense on T1-weighted sequences. The tight filum terminale syndrome is characterised by shortening and hypertrophy of the filum terminale and consequent cord tethering syndrome. Persistent terminal ventricle consists of small linear cavities inside the medullary cone immediately above the filum terminale. A dermal sinus is an epithelial fistula that connects the neural tissue or the meninges with the surface of the skin. Dermal sinuses are usually located in the lumbosacral region and are usually found together with an intraspinal dermoid cyst. Clinically, they present with a small midline pit, a cutaneous nevus, an area of hyperpigmentation, or a capillary haemangioma. Detecting all these findings prenatally is extremely difficult; however, the fistulous tract can be identified using high resolution MRI techniques [1]. These patients require surgical repair after birth because the fistulous tract that communicates the skin with the neural tissue can predispose to infections of the neural contents.
Complex closed spinal dysraphisms can be divided into disorders of midline notochord integration and disorders of notochord malformation [32]. Disorders of midline notochord integration include enteric fistula, which is an abnormal connection between the digestive tract and the surface of the skin that may or may not be accompanied by a neurenteric cyst. Neurenteric cysts are lined with epithelial mucosa similar to that of the gastrointestinal tract and are typically located in the anterior cervicothoracic region. Another disorder of midline notochord integration is diastematomyelia or diplomyelia (Fig. 8), which consists of a longitudinal splitting of the spinal cord into two hemicords [47]. There are two types of diastematomyelia: in type 1, both hemicords are wrapped in independent meningeal linings and are separated by an osseous or cartilaginous septum; whereas in type 2, the two hemicords share a common meningeal lining [32, 48]. It is difficult to diagnose diastematomyelia prenatally. On MRI, the septum in type 1 diastematomyelia can be seen as a hypointense linear structure within the spinal canal on T2-weighted sequences. Gradient echo sequences can be useful for detecting these structures [1]. Caution should be taken not to confuse this septum with the spinal cord, as both structures have the same signal intensity. The soft tissues and the skin that cover these anomalies are usually intact unless they form part of a more complex anomaly. Diastematomyelia is normally accompanied by vertebral anomalies like hemivertebrae or butterfly vertebrae, tethered cord, or open spina bifida (hemimyelomeningocele or hemimyelocele) [49]. It is most commonly located in the lower thoracic or upper lumbar spine.
Disorders of notochord malformation include caudal regression syndrome [46], which consists of the total or partial agenesis of the lumbar and sacral spine, probably secondary to a disturbance in the formation of the secondary neural tube. Caudal regression syndrome is divided into two types: in type 1, the medullary cone is high and ends abruptly; in type 2, the spinal cord is tethered and the medullary cone is low-lying [32, 46]. Caudal regression syndrome comprises a wide spectrum of anomalies ranging from sacral agenesis or dysgenesis to sirenomelia (fusion of the lower limbs). Some authors believe that type 1 and type 2 caudal regression actually represent two distinct entities [11]. Caudal regression can be found in association with other anomalies like omphalocele, bladder exstrophy, imperforate anus and spinal defects (OEIS complex); vertebral anomalies, anal anomalies, cardiac anomalies, tracheoesophageal fistula, limb malformations, hydrocephalus (VACTERL-H); and sacral agenesis, anal atresia and presacral teratoma or meningocele (Currarino triad) [9].
Another disorder of notochord malformation is segmental spinal dysgenesis [50], which includes agenesis or dysgenesis of the thoracic or lumbar spine, alterations of the spinal cord and nerve roots, congenital paraparesis or paraplegia, and deformities in the lower limbs.
Conclusion
Anomalies of the spine and neural tube are uncommon. Their prenatal diagnosis can predict the viability of the fetus, has implications for family counselling about the pregnancy, and orients the prognosis and management of the newborn. Ultrasonography remains the primary imaging test for the prenatal diagnosis of NTDs, but sometimes it does not enable an accurate diagnosis so other diagnostic techniques are necessary. Fetal MRI has advanced rapidly in the last 25 years, developing from an experimental technique to become a fundamental tool in normal clinical practice in many centres around the world. MRI’s ability to detect complex anomalies that involve different organs has been widely reported. Fetal MRI should be restricted to centres with experienced staff and should never be considered a replacement for ultrasonography, which continues to be the technique of choice to evaluate fetal morphology.
Acknowledgments
The authors thank John Giba for linguistic aid.
References
- 1.Simon EM. MRI of the fetal spine. Pediatr Radiol. 2004;34:712–719. doi: 10.1007/s00247-004-1245-1. [DOI] [PubMed] [Google Scholar]
- 2.Simon EM, Goldstein RB, Coakley FV, et al. Fast MR imaging of fetal CNS anomalies in utero. AJNR Am J Neuroradiol. 2000;21:1688–1698. [PMC free article] [PubMed] [Google Scholar]
- 3.Duczkowska A, Bekiesinska-Figatowska M, Herman-Sucharska I, et al. Magnetic resonance imaging in the evaluation of the fetal spinal canal contents. Brain Dev. 2011;33:10–20. doi: 10.1016/j.braindev.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths PD, Widjaja E, Paley MNJ, et al. Imaging the fetal spine using in utero MR: diagnostic accuracy and impact on management. Pediatr Radiol. 2006;36:927–933. doi: 10.1007/s00247-006-0234-y. [DOI] [PubMed] [Google Scholar]
- 5.Glenn OA, Barkovich J. Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis: part 2. AJNR Am J Neuroradiol. 2006;27:1807–1814. [PMC free article] [PubMed] [Google Scholar]
- 6.International commission onnon-ionizing radiation protection Guidelines on limits of exposure to static magnetic fields. Health Phys. 2009;96:504–514. doi: 10.1097/01.HP.0000343164.27920.4a. [DOI] [PubMed] [Google Scholar]
- 7.Kok RD, de Vries MM, Heerschap A, et al. Absence of harmful effects of magnetic resonance exposure at 1.5 T in utero during the third trimester of pregnancy: a follow-up study. Magn Reson Imaging. 2004;22:851–854. doi: 10.1016/j.mri.2004.01.047. [DOI] [PubMed] [Google Scholar]
- 8.Juutilainen J. Developmental effects of electromagnetic fields. Bioelectromagnetics Suppl. 2005;7:107–115. doi: 10.1002/bem.20125. [DOI] [PubMed] [Google Scholar]
- 9.Bulas D. Fetal evaluation of spine dysraphism. Pediatr Radiol. 2010;40:1029–1037. doi: 10.1007/s00247-010-1583-0. [DOI] [PubMed] [Google Scholar]
- 10.Tortori-Donati P, Rossi A (2005) Pediatric neuroradiology, 1st edn. Springer, Berlin Heidelberg New York
- 11.Nyberg DA, McGahan JP, Pretorius DH et al (2002) Diagnostic imaging of fetal anomalies. Lippincott Williams & Wilkins, Philadelphia
- 12.Atlas SW (2008) Magnetic resonance imaging of the brain and spine, 4th edn. Lippincott Williams & Wilkins, Philadelphia
- 13.Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg. 1976;4:69–92. doi: 10.1016/S0301-0503(76)80013-6. [DOI] [PubMed] [Google Scholar]
- 14.Au KS, Ashley-Koch A, Northrup H. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev Disabil Res Rev. 2010;16:6–15. doi: 10.1002/ddrr.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oi S. Current status of prenatal management of fetal spina bifida in the world: worldwide cooperative survey on the medico-ethical issue. Childs Nerv Syst. 2003;19:596–599. doi: 10.1007/s00381-003-0778-1. [DOI] [PubMed] [Google Scholar]
- 16.Lemire RJ. Neural tube defects. JAMA. 1988;259:558–562. doi: 10.1001/jama.1988.03720040050026. [DOI] [PubMed] [Google Scholar]
- 17.Moore CA, Li S, Li Z, et al. Elevated rates of severe neural tube defects in a high-prevalence area in northern China. Am J Med Genet. 1997;73:113–118. doi: 10.1002/(SICI)1096-8628(19971212)73:2<113::AID-AJMG2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 18.Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. Am J Med Genet C Semin Med Genet. 2005;135C:77–87. doi: 10.1002/ajmg.c.30056. [DOI] [PubMed] [Google Scholar]
- 19.Moretti ME, Bar-Oz B, Fried S, et al. Maternal hyperthermia and the risk for neural tube defects in offspring: systematic review and meta-analysis. Epidemiology. 2005;16:216–219. doi: 10.1097/01.ede.0000152903.55579.15. [DOI] [PubMed] [Google Scholar]
- 20.Shaw GM, Velie EM, Morland KB. Parental recreational drug use and risk for neural tube defects. Am J Epidemiol. 1996;144:1155–1160. doi: 10.1093/oxfordjournals.aje.a008894. [DOI] [PubMed] [Google Scholar]
- 21.Shaw GM, Quach T, Nelson V, et al. Neural tube defects associated with maternal periconceptional dietary intake of simple sugars and glycemic index. Am J Clin Nutr. 2003;78:972–978. doi: 10.1093/ajcn/78.5.972. [DOI] [PubMed] [Google Scholar]
- 22.Wasserman CR, Shaw GM, Selvin S, et al. Socioeconomic status, neighborhood social conditions, and neural tube defects. Am J Public Health. 1998;88:1674–1680. doi: 10.2105/AJPH.88.11.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernández-Díaz S, Werler MM, Walker AM, et al. Neural tube defects in relation to use of folic acid antagonists during pregnanc. Am J Epidemiol. 2001;153:961–968. doi: 10.1093/aje/153.10.961. [DOI] [PubMed] [Google Scholar]
- 24.Lindhout D, Schmidt D. In-utero exposure to valproate and neural tube defects. Lancet. 1986;1:1392–1393. doi: 10.1016/S0140-6736(86)91711-3. [DOI] [PubMed] [Google Scholar]
- 25.De Marco P, Merello E, Calevo MG, et al. Maternal periconceptional factors affect the risk of spina bifida-affected pregnancies: an Italian case–control study. Childs Nerv Syst. 2011;27:1073–1081. doi: 10.1007/s00381-010-1372-y. [DOI] [PubMed] [Google Scholar]
- 26.MRC Vitamin Study Research Group (1991) Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 338:131–137 [PubMed]
- 27.Czeizel AE, Dudás I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832–1835. doi: 10.1056/NEJM199212243272602. [DOI] [PubMed] [Google Scholar]
- 28.Kirke PN, Molloy AM, Daly LE, et al. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q J Med. 1993;86:703–708. [PubMed] [Google Scholar]
- 29.Wang ZP, Li H, Hao LZ, et al. The effectiveness of prenatal serum biomarker screening for neural tube defects in second trimester pregnant women: a meta-analysis. Prenat Diagn. 2009;29:960–965. doi: 10.1002/pd.2325. [DOI] [PubMed] [Google Scholar]
- 30.Robson CD, Barnewolt CE. MR imaging of fetal head and neck anomalies. Neuroimaging Clin N Am. 2004;14:273–291. doi: 10.1016/j.nic.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Saleem SN, Said AH, Abdel-Raouf M, et al. Fetal MRI in the evaluation of fetuses referred for sonographically suspected neural tube defects (NTDs): impact on diagnosis and management decision. Neuroradiology. 2009;51:761–772. doi: 10.1007/s00234-009-0549-0. [DOI] [PubMed] [Google Scholar]
- 32.Rufener SL, Ibrahim M, Raybaud CA, et al. Congenital spine and spinal cord malformations—pictorial review. AJR Am J Roentgenol. 2010;194:S26–S37. doi: 10.2214/AJR.07.7141. [DOI] [PubMed] [Google Scholar]
- 33.Barkovich AJ (2005) Pediatric neuroimaging, 4th edn. Lippincott Williams & Wilkins, Philadelphia
- 34.Rossi A, Biancheri R, Cama A, et al. Imaging in spine and spinal cord malformations. Eur J Radiol. 2004;50:177–200. doi: 10.1016/j.ejrad.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 35.Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42:471–491. doi: 10.1007/s002340000325. [DOI] [PubMed] [Google Scholar]
- 36.Parmar H, Patkar D, Shah J, et al. Diastematomyelia with terminal lipomyelocystocele arising from one hemicord: case report. Clin Imaging. 2003;27:41–43. doi: 10.1016/S0899-7071(02)00522-3. [DOI] [PubMed] [Google Scholar]
- 37.Proctor MR, Bauer SB, Scott RM. The effect of surgery for split spinal cord malformation on neurologic and urologic function. Pediatr Neurosurg. 2000;32:13–19. doi: 10.1159/000028891. [DOI] [PubMed] [Google Scholar]
- 38.McLone DG, Dias MS. The Chiari II malformation: cause and impact. Childs Nerv Syst. 2003;19:540–550. doi: 10.1007/s00381-003-0792-3. [DOI] [PubMed] [Google Scholar]
- 39.Chao TT, Dashe JS, Adams RC, et al. Central nervous system findings on fetal magnetic resonance imaging and outcomes in children with spina bifida. Obstet Gynecol. 2010;116:323–329. doi: 10.1097/AOG.0b013e3181e666e8. [DOI] [PubMed] [Google Scholar]
- 40.Simon EM, Pollock AN. Prenatal and postnatal imaging of spinal dysraphism. Semin Roentgenol. 2004;39:182–196. doi: 10.1053/j.ro.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Levine D (2005) Atlas of fetal MRI, 1st edn. Informa Healthcare, New York
- 42.Adzick NS. Fetal surgery for myelomeningocele: trials and tribulations. Isabella Forshall Lecture. J Pediatr Surg. 2012;47:273–281. doi: 10.1016/j.jpedsurg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drolet B. Birthmarks to worry about. Cutaneous markers of dysraphism. Dermatol Clin. 1998;16:447–453. doi: 10.1016/S0733-8635(05)70245-X. [DOI] [PubMed] [Google Scholar]
- 44.Turgut M, Akyüz O, Unsal A. Occult intrasacral meningocele: case report and review of the literature. Zentralbl Neurochir. 2007;68:34–37. doi: 10.1055/s-2007-968169. [DOI] [PubMed] [Google Scholar]
- 45.Muthukumar N. Terminal and nonterminal myelocystoceles. J Neurosurg. 2007;107:87–97. doi: 10.3171/PED-07/08/087. [DOI] [PubMed] [Google Scholar]
- 46.Nievelstein RA, Valk J, Smit LM, et al. MR of the caudal regression syndrome: embryologic implications. AJNR Am J Neuroradiol. 1994;15:1021–1029. [PMC free article] [PubMed] [Google Scholar]
- 47.Sonigo-Cohen P, Schmit P, Zerah M, et al. Prenatal diagnosis of diastematomyelia. Childs Nerv Syst. 2003;19:555–560. doi: 10.1007/s00381-003-0771-8. [DOI] [PubMed] [Google Scholar]
- 48.Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451–480. doi: 10.1227/00006123-199209000-00010. [DOI] [PubMed] [Google Scholar]
- 49.Struben H, Visca E, Holzgreve W, et al. Prenatal diagnosis of diastematomyelia and tethered cord—a case report and review of the literature. Ultraschall Med. 2008;29:72–76. doi: 10.1055/s-2007-963208. [DOI] [PubMed] [Google Scholar]
- 50.Tortori-Donati P, Fondelli MP, Rossi A, et al. Segmental spinal dysgenesis: neuroradiologic findings with clinical and embryologic correlation. AJNR Am J Neuroradiol. 1999;20:445–456. [PMC free article] [PubMed] [Google Scholar]