

Abstract
We outline a set of strategies to infer protein function from structure. The overall approach depends on extensive use of homology modeling, the exploitation of a wide range of global and local geometric relationships between protein structures and the use of machine learning techniques. The combination of modeling with broad searches of protein structure space defines a “structural BLAST” approach to infer function with high genomic coverage. Applications are described to the prediction of protein–protein and protein–ligand interactions. In the context of protein–protein interactions, our structure-based prediction algorithm, PrePPI, has comparable accuracy to high-throughput experiments. An essential feature of PrePPI involves the use of Bayesian methods to combine structure-derived information with non-structural evidence (e.g. co-expression) to assign a likelihood for each predicted interaction. This, combined with a structural BLAST approach significantly expands the range of applications of protein structure in the annotation of protein function, including systems level biological applications where it has previously played little role.
Keywords: protein interaction prediction, protein interface prediction, structure/function relationship, machine learning
Introduction
The protein structures that populate the PDB have provided crucial insights at the atomic level as to the molecular mechanisms that underlie protein function. Indeed, structural studies have had, and continue to have, a significant and sometimes revolutionary impact in all areas of biology. However, structural biology has tended to focus on single proteins or biological systems and, despite significant advances in the general area of structural bioinformatics, the horizontal integration of the vast quantity of structural information available in the PDB has had little or no impact in the larger biological community. This is in contrast to protein sequence information, which is more routinely, automatically and broadly used. Given that structure is more conserved than sequence, structural similarity has the potential to yield a great deal of functional information that sequence relationships cannot provide and to identify relationships between many more pairs of proteins. In this article we argue that the exploitation of statistical and machine learning techniques combined with the vast amount of high-throughput experimental data constantly being generated enable a significant expansion in the scale and diversity of application of structural information to biological problems.
The exploitation of structural relationships has often been based on similarity in the overall shape, or “fold”, of proteins. For example, the largely manually curated SCOP1 and partially curated CATH2 databases classify proteins into discrete groups. It is frequently possible to deduce function for a given protein based on membership in such predefined categories which can also be determined with tools such as DALI which provide structural similarity scores that have been shown to correlate well with database classifications.3
The use of global structural similarity to infer function, despite its value, has limitations as well. As we and others have extensively discussed, there are numerous relationships between proteins that are based on local structural similarity.4–9 Indeed, mechanisms for how proteins with diverse shapes but common functional cores have emerged from common ancestors in the course of evolution10–12 have been proposed. For example, homologous proteins that contain different compositions and relative orientations of secondary structure elements (SSEs) and thus belong to different protein folds or even classes (all-α vs. α/β) may still preserve a local functional core (e.g., a helix-turn-helix DNA binding motif that appears in evolutionarily related transcription factors that have different global folds13). Moreover, local sequence relationships between ungapped sequence fragments have also been observed between proteins with different SSE compositions.14,15 Proteins with such local structural similarities have been shown to share a range of function relationships, including common protein–ligand, protein–protein, and drug binding sites,5,16,17 protein–protein interaction (PPI) sites and approximate binding mode.18,19
The ultimate potential impact of both global and local structural relationships in inferring function is highlighted by the observation that, given a suitably “loose” definition of structural similarity, the repertoire of structures currently in the structural databases is nearly complete at the domain level.20–23 Thus, it can be expected that most newly solved protein structures will have both near and remote structural neighbors which can provide clues as to their function. Programs such as BLAST use local sequence relationships to quickly scan sequence databases. Since structure-based scans of protein structural databases can be carried out very quickly with current technology (typically minutes for a database of tens of thousands of structures), a similar strategy can be used for structural relationships as well, essentially defining a “structural BLAST”. Here we discuss a number of issues associated with the use of structural relationships to assign function and review applications to the prediction of protein–protein and protein–ligand binding sites.
Issues in the exploitation of local structural similarity
Much work has been done relating sequence similarity to function similarity.24,25 Annotation schemes such as GO and EC number can be used to define function and there are widely accepted quantitative measures of sequence similarity (although a great deal of functional diversity is expected even within groups of proteins that are obviously sequence-related26). Quantitative measures of structural similarity are also available4,27–29 but these focus on global rather than local structural relationships.
One solution is simply to circumvent the problem and, instead of using an unreliable score to dictate what functional hypotheses should be considered, provide a researcher interested in a particular problem with enough information to make an informed decision as to the relevance of a structure/function relationship. This is the philosophy behind the design of our MarkUs server5,30 which applies a structural BLAST approach to identify a large number of structural neighbors with a wide range of similarities for a given query. These can then be visualized and filtered using a number of different properties (e.g., GO annotation or an interaction with a certain type of ligand) based on user-provided hypotheses. Properties of the structural neighbor that determine function (catalytic residues, biophysical properties, conservation) can be displayed and compared to the same properties in the query structure to determine if they are consistent with the hypothesized function. For example, if a protein is thought to be a transcription factor, it might be expected to have an electrostatic pattern on its surface that is similar to other DNA binding proteins.
The MarkUs server was designed with the expectation that users interested in a particular protein or set of proteins will not want automatically generated answers but rather will benefit from access to a list of structurally related proteins and the ability to query that list for relevant functional information. MarkUs' interactive capabilities are designed to enable the testing of hypotheses and the generation of new ones “on the fly” based on the assumption that an answer is likely to be found somewhere in protein structure space.
Predicting protein–protein and protein–ligand interfaces
The expectation underlying the use of MarkUs is that proteins that have a similar function, even if only remotely related structurally, will be likely to share common features at structurally related locations. We have shown that this is often the case in the specific context of protein–ligand and PPIs. Our general strategy is based on the exploration of protein structure space with structural BLAST (Fig. 1). The figure represents the protein structure universe where some proteins are shown forming complexes with other proteins or ligands (which could in principle include DNA and RNA). Given a query protein its structural neighbors are identified by considering global and local geometric relationships. Each structural neighbor is placed in the coordinate system of the query. Any binding partners of structural neighbors are then located in the same coordinate system by applying the same transformation to the binding partner that relates the query to the structural neighbor.16,31–33
Figure 1.
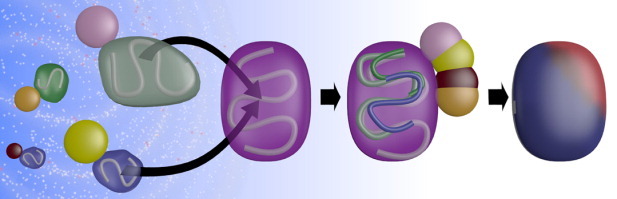
Exploring the protein universe using structural BLAST. To infer function of a given query protein (purple), the universe of structures (left) is searched broadly for proteins that are structurally similar to the query and each one is placed in the query coordinate system (curved arrows) by superposing the protein backbones (shown schematically within the surface of each protein). In the specific application shown in the figure, binding partners (small molecules, other proteins, nucleic acids, etc.) of the structural neighbors (shown as pink, yellow, magenta, and orange circles) are also transformed into the coordinate system of the query (middle panel). The query is then analyzed to find surface residues that show a statistical propensity to contact ligands from structural neighbors. This information is represented as a heat map on the surface of the query (right panel). Regions where the query is likely to bind other molecules are shown in red and unlikely regions are shown in blue.
Using this procedure, interactions made by a collection of structurally similar proteins with their binding partners are “mapped” to a single representative structure (the query) and one can then ask whether the geometric locations to which interactions are mapped are “conserved”. Russell et al.31 defined a rigorous measure of the conservation of the geometric location of protein–ligand binding sites by statistically quantifying the degree of conservation with a Z-score, which reflects whether there are locations on the surface of the representative protein that preferentially have ligand interactions mapped to them, as compared to a random mapping.
We adapted this method to analyze the geometric conservation of PPI sites.16 In this analysis, we used the program SKA34,35 to find structural neighbors, applying a “protein structural distance” (PSD35) cutoff of 0.6, which is large enough to detect relationships across categories such as SCOP fold, superfamily or family. We found that nearly all proteins in a structurally diverse benchmark36 had Z-scores >3, indicating significant conservation of the location on the surface of protein–protein binding sites among structural neighbors. Importantly, for most proteins, this was still true when the analysis was repeated excluding proteins classified in the same SCOP category (family, superfamily or fold) so that, for example, even proteins in different folds that share local structural similarity tend to use the same regions of their surface to bind other proteins. We used these observations as the basis of an algorithm, PredUs, that predicts residues on a protein surface involved in a protein–protein interfaces and showed that the use of remote structural relationships substantially improved overall prediction performance, increasing recall with little or no cost in precision.37
We have used the same approach to analyze the geometric conservation of protein ligand interfaces (Fig. 2). We obtained a representative set of query proteins from LigASite,38 a curated database of amino acid–ligand interactions. As with protein–protein interfaces, more than 80% of the proteins in the benchmark set had Z-scores > 3 suggesting significant conservation in the location of the protein–ligand binding sites on their surfaces. Moreover, there is still significant conservation in binding site location when proteins which share SCOP annotations with the query at different levels of the hierarchy are excluded. Indeed, even proteins in different SCOP folds exhibit this tendency. (In our analysis of ligands, we increased the PSD cutoff from 0.6 to 0.8 implying a broader search for remote structural neighbors than used for PPIs. Even using this looser criterion for structural similarity we found statistically meaningful relationships.)
Figure 2.

Conservation of protein–ligand interfaces. The figure shows the fraction of query proteins in the LigASite benchmark with a Z-score above the value shown on the x-axis. The blue line corresponds to results where structural neighbors of each query are chosen from a 60% non-redundant (with respect to sequence) pool of ligand-containing proteins which we compiled. Structural neighbors are obtained using a PSD cutoff of 0.8 that is large enough to detect geometric similarities even between proteins in different SCOP folds. The remaining lines show results obtained when structural neighbors from the same SCOP family (green), superfamily (purple), and fold (red) as the query were excluded from the set of structural neighbors (proteins without SCOP annotations were also excluded). Since some queries do not have many structural neighbors, Z-scores in this figure are calculated only for 146 proteins (out of a total of 337 in the LigASite benchmark) that had ≥5 neighbors in each set.
Figure 3 provides an example of how remote similarities can reflect not only conservation in the geometric location of the protein–ligand interface, but similar ligand shapes as well. The figure shows two nucleotide binding protein domains that are classified in different SCOP folds that have a pairwise sequence identity in the “twilight zone” (∼15%). However, the sequence identity in the active site is much larger (48.2%) and the global coverage of one by the other in the structural alignment is 67.5%. As evident from the figure, the ADP molecule and the ADP substructure in FAD overlap very closely, with a heavy atom RMSD of 0.62Å. Based on a broader analysis of how geometric conservation of protein–ligand binding sites correlates with ligand shape, relationships such as those described in Figure 3 appear to be common and provide a basis for the development of a new algorithm to predict what type of ligands bind to a site on a given query protein.39
Figure 3.

Structural alignment can reveal ligand shape similarity. NAD(P)-binding domain (purple, SCOP domain d1hyua1 and fold classification c.3) and nucleotide-binding domain (yellow, SCOP domain d1djna3, and fold classification c.4) and are structurally superimposed and shown in ribbon representation as a stereo pair. An FAD molecule from d1hyua1 (red) and ADP from d1djna3 (cyan) are shown in stick representation. There is a clear similarity in ligand shape and binding mode even though only some secondary structure elements overlap (regions of both structures without structurally equivalent regions in the other are shown as transparent).
Using Bayesian approaches to quantify structure/function relationships
Despite the valuable information that can be found in remote structural relationships, there are significant uncertainties associated with defining and measuring shape similarity. Remote relationships might be expected to provide only weak evidence of a functional relationship given the large functional diversity that can be expected in any set of structurally similar proteins. This general problem arises in many systems biology applications where functional relationships are often inferred from evidence such as co-expression which will often occur even in the absence of direct physical contact between two proteins. To address such issues Bayesian40 and other statistical approaches have been used to quantify functional relationships between proteins.41 In a typical application, a Bayesian network is used to classify objects into binary categories (e.g., two proteins do or do not share a GO annotation, they do or do not physically interact, etc.). Membership in a category is quantified using a “likelihood ratio” (LR), which reflects the increase/decrease in the probability that an object belongs to a category after learning that it has a particular characteristic, or “clue” (Fig. 4).
Figure 4.
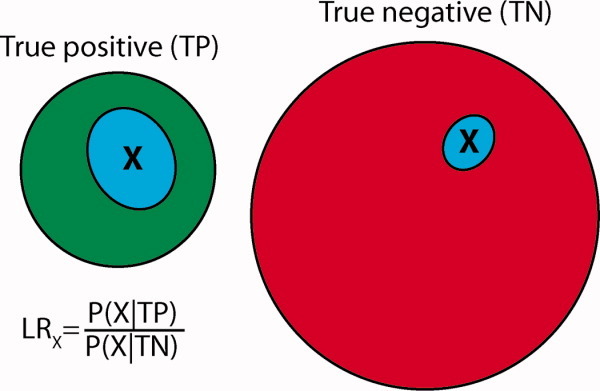
Learning with Bayesian statistics. Learning in the Bayesian approach is carried out by examining reliable true positive (TP) and true negative (TN) reference sets. For example, TP might be a set of protein–protein interactions which have been experimentally validated multiple times and TN might be a set of protein pairs that are known not to interact. The degree to which having property X indicates membership in TP is quantified by the likelihood ratio (LR), calculated as the percentage of objects in the TP set that have the property X (P(X|TP)) divided by the percentage in the TN set with property X (P(X|TN)). A LR >1 indicates that an object with property X is more likely to be in the TP set than the TN set.
A distinct advantage of Bayesian approaches is that even if a particular clue is not a strong indicator of membership in a category on its own (e.g., remote structural similarity), different clues can be combined to yield a reliable classifier (in some implementations this can be accomplished in practice by simply multiplying the LRs generated by different clues). For example, Bayesian approaches have been used to predict PPIs in different species42–44 using multiple clues that, on their own, might be expected to be quite weak indicators of a potential interaction between two proteins but, when combined, result in high confidence predictions.44,45 More importantly in the context of the current discussion, Bayesian approaches can circumvent the uncertainties associated with defining geometric similarity and inherently ambiguous structure–function relationships by relating different measures of structure and function in a statistical sense.
Large scale prediction of protein–protein interactions
We have recently reported a new algorithm, called PrePPI, that exploits both global and local structural similarity and uses Bayesian statistics to combine structural and non-structural evidence (e.g., co-expresssion) to predict PPIs.46 Similar to other approaches,33,47,48 we construct approximate models of potentially interacting protein pairs by simply superposing each, individually, on structurally similar proteins that interact in the PDB. In PrePPI, we take a structural BLAST approach to the identification of structurally similar proteins, allowing for the detection of remote relationships that may contain valuable clues as to an interaction. Given the large numbers of interaction models that are generated in this way (tens of millions for yeast and billions for human), an important component of the PrePPI algorithm is the way in which these models are evaluated. Rather than trying to assign a score to a three-dimensional model, we define a set of empirical properties using only the structure-based sequence alignment between the query and template proteins (see Fig. 5). We then use a Bayesian approach to ask how well these properties correlate with being a true interaction based on reference sets of true positive and true negative interactions that we compiled.
Figure 5.
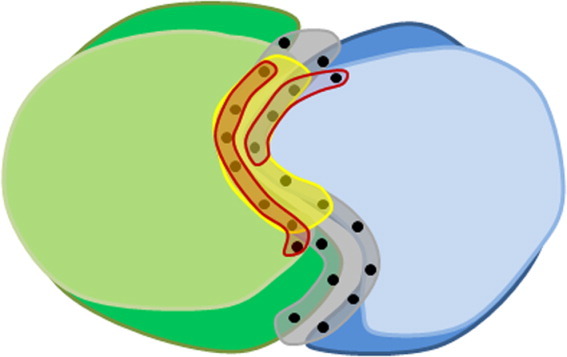
Evaluating protein interaction models. The figure shows an interaction model of a complex formed between two query proteins A and B (light green and light blue), that is derived from a known template complex present in the PDB (dark green and dark blue) using PrePPI's structural BLAST algorithm.46 The interaction model is obtained by superimposing the structure of each query protein on its respective structural neighbor in the template complex. The model is evaluated with a set of simple empirical scores that depend on (a) The quality of the structural alignment of the queries with their templates, (b) How well residues in the modeled interface (highlighted in yellow) overlap the template interface (highlighted in gray), and (c) How well predicted interfacial residues in both query proteins (highlighted in red) overlap with interfacial residues in the template.46
PrePPI can reliably predict PPIs on a genome-wide scale. Although the models PrePPI generates using a structural BLAST approach may be crude, they often contain enough information to provide an important clue as to the possibility of a given PPI. Moreover, the availability of structural clues for a large set of potential interactions results in an even larger number of reliable predictions when combined with non-structural information. This point is evident in Figure 6 where, as can be seen in the left panel which uses structural information alone, about 78,000 high-confidence predictions (false positive rate <0.001 corresponding to an LR > 60046) are made for the human genome and, of these, only about 400 are derived from remote structural homologs. In contrast, over 300,000 high confidence predictions are made when structural and non-structural information are combined (right panel) and of these, 77,000 are derived from remote structural homologs. To understand the basis of this effect, consider an example where an interaction model based on a remote relationship has a structure-based LR of 100 (low confidence). If the same interaction model has an LR based on co-expression of 10 (even lower confidence), the combination results in a final LR of 1000 (high confidence). As shown in the right panel of Figure 6, combining different sources of evidence increases the number of high-confidence predictions in general and, moreover, there is a dramatic expansion in predictions that use intermediate and remote relationships.
Figure 6.

Expanding the number of reliable predictions using remote relationships. The graphs show the numbers of high-confidence (LR>600) protein–protein interaction predictions that use close (blue, PSD<0.2), intermediate (red, 0.2<PSD<0.4), and remote (green, 0.4<PSD<0.6) structural relationships. For reference, at a PSD cutoff of 0.2, about two thirds of the structural neighbors are expected to be in the same fold, while at PSD cutoff of 0.6 about half of the structural neighbors would be in the same fold. The results on the left are based on structural information alone while those on the right are based on both structural and non-structural information.
Overall, PrePPI performs comparably to high-throughput experimental methods.46 Moreover, out of 19 protein–protein predictions selected for validation by four different groups, fifteen were confirmed using co-immunoprecipitation assays. The validated interactions most were mostly unexpected, i.e. could not be identified from sequence or function-based approaches. They were based on a wide range of structural relationships and suggested novel participants in specific functional pathways: transcription factor interactions with nuclear hormone receptors, novel interactions between cell surface receptors, kinases and adaptor proteins and novel components large multi-subunit protein complexes (an extended discussion of predictions and how they were chosen for validation is provided in46). We have created a publically available database that provides a probability that two proteins will interact. The database now contains over 30,000 high confidence interactions for yeast and over 300,000 for human and is being extended to other genomes. Distinct from high-throughput experiments, PrePPI also provides a structural model for the interaction that can be tested and refined. We expect the accuracy of these models to vary greatly with some providing nearly correct docking geometries while others may just identify a number of interfacial residues even though the details of the model are inaccurate. Nevertheless, such models offer a starting point for further refinement using computational approaches and for experimental tests based on site-directed-mutagenesis.
Concluding discussion
There are two pre-requisites for the application of a structural BLAST approach to biological problems. One, of course, is that protein structures must be available for a particular problem and, for genome-wide applications, for most proteins in a genome. Given the difficulty of obtaining structures experimentally, it is essential that homology models be used. Although homology models may not always be accurate, the same is true for sequence alignments. Our results strongly suggest that, in many applications, homology models can play a major role in bridging the sequence–structure gap. This was an important rationale for structural genomics initiatives and we believe that the success of the PrePPI algorithm, which bases most of its high likelihood predictions on homology models, validates the underlying logic of these large-scale efforts. (We do not address the controversial issue of cost/benefit analysis).
A second requirement for a more complete integration of structural information into biology involves the full exploration of protein structure space as embodied in the structural BLAST approach. The use of structural alignments is not new but the use of local structural relationships that may not be evident from the overall protein fold puts structural alignments on more of an equivalent footing with sequence alignments than has been common in the past. Of course computational tools need to be developed that make structure-based searches as accessible to the community as sequence alignments. This is not a trivial task and above we have outlined some early steps we have taken in this direction. Ultimately, the investment of time required to master structure-based tools will have to be justified in terms of what can be learned. It is our contention that we have just begun to extract the wealth of functional information that exists in the “universe” of protein structures.
Finally, given the enormous amount of genomic/systems biology information becoming available it is essential that structural information be fully integrated with other sources of evidence to infer protein function. As we have discussed here, the structural BLAST approach and the use of homology models can, with the help of machine learning techniques, such as the Bayesian methods used in PrePPI, pave the way for the seamless integration of experimental and computational information.49
Acknowledgments
This work is supported by NIH grants GM030518, GM094597, CA121852 (QZ, DP, BH), and Swiss National Science Foundation grant PBZHP3-123276 and the Novartis Foundation (FD).
References
- 1.Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucl Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearl F, Todd A, Sillitoe I, Dibley M, Redfern O, Lewis T, Bennett C, Marsden R, Grant A, Lee D, Akpor A, Maibaum M, Harrison A, Dallman T, Reeves G, Diboun I, Addou S, Lise S, Johnston C, Sillero A, Thornton J, Orengo C. The CATH Domain Structure Database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucleic Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolodny R, Petrey D, Honig B. Protein structure comparison: implications for the nature of ‘fold space’, and structure and function prediction. Curr Opin Struct Biol. 2006;16:393–398. doi: 10.1016/j.sbi.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Petrey D, Fischer M, Honig B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proc Natl Acad Sci USA. 2009;106:17377–17382. doi: 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrey D, Honig B. Is protein classification necessary? Toward alternative approaches to function annotation. Curr Opin Struct Biol. 2009;19:363–368. doi: 10.1016/j.sbi.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao M, Skolnick J. Structural space of protein–protein interfaces is degenerate, close to complete, and highly connected. Proc Natl Acad Sci. 2010;107:22517–22522. doi: 10.1073/pnas.1012820107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shindyalov IN, Bourne PE. An alternative view of protein fold space. Proteins. 2000;38:247–260. [PubMed] [Google Scholar]
- 9.Krishna SS, Sadreyev RI, Grishin NV. A tale of two ferredoxins: sequence similarity and structural differences. BMC Struct Biol. 2006;6:8. doi: 10.1186/1472-6807-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grishin NV. Fold change in evolution of protein structures. J Struct Biol. 2001;134:167–185. doi: 10.1006/jsbi.2001.4335. [DOI] [PubMed] [Google Scholar]
- 11.Alva V, Koretke KK, Coles M, Lupas AN. Cradle-loop barrels and the concept of metafolds in protein classification by natural descent. Curr Opin Struct Biol. 2008;18:358–365. doi: 10.1016/j.sbi.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Dokholyan NV, Shakhnovich B, Shakhnovich E. Expanding protein universe and its origin from the biological Big Bang. Proc Natl Acad Sci USA. 2002;99:14132–14136. doi: 10.1073/pnas.202497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newlove T, Konieczka JH, Cordes MH. Secondary structure switching in Cro protein evolution. Structure. 2004;12:569–581. doi: 10.1016/j.str.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 14.Friedberg I, Harder T, Kolodny R, Sitbon E, Li Z, Godzik A. Using an alignment of fragment strings for comparing protein structures. Bioinformatics. 2007;23:e219–e224. doi: 10.1093/bioinformatics/btl310. [DOI] [PubMed] [Google Scholar]
- 15.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci U S A. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang QC, Petrey D, Norel R, Honig BH. Protein interface conservation across structure space. Proc Natl Acad Sci U S A. 2010;107:10896–10901. doi: 10.1073/pnas.1005894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie L, Wang J, Bourne PE. In silico elucidation of the molecular mechanism defining the adverse effect of selective estrogen receptor modulators. PLoS Comp Biol. 2007;3:e217. doi: 10.1371/journal.pcbi.0030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O. Architectures and functional coverage of protein-protein interfaces. J Mol Biol. 2008;381:785–802. doi: 10.1016/j.jmb.2008.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protocols. 2011;6:1341–1354. doi: 10.1038/nprot.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Skolnick J. The protein structure prediction problem could be solved using the current PDB library. Proc Natl Acad Sci USA. 2005;102:1029–1034. doi: 10.1073/pnas.0407152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Hubner IA, Arakaki AK, Shakhnovich E, Skolnick J. On the origin and highly likely completeness of single-domain protein structures. Proc Nat Acad Sci USA. 2006;103:2605–2610. doi: 10.1073/pnas.0509379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kihara D, Skolnick J. The PDB is a covering set of small protein structures. J Mol Biol. 2003;334:793–802. doi: 10.1016/j.jmb.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 23.Skolnick J, Zhou H, Brylinski M. Further evidence for the likely completeness of the library of solved single domain protein structures. J Phys Chem B. 2012;116:6654–6664. doi: 10.1021/jp211052j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333:863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 25.Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y. Automatic prediction of protein function. Cell Mol Life Sci. 2003;60:2637–2650. doi: 10.1007/s00018-003-3114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aravind L, Anantharaman V, Koonin EV. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implications for protein evolution in the RNA world. Proteins. 2002;48:1–14. doi: 10.1002/prot.10064. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 29.Kolodny R, Koehl P, Levitt M. Comprehensive evaluation of protein structure alignment methods: Scoring by geometric measures. J Mol Biol. 2005;346:1173–1188. doi: 10.1016/j.jmb.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer M, Zhang QC, Dey F, Chen BY, Honig B, Petrey D. MarkUs: a server to navigate sequence–structure–function space. Nucleic Acids Res. 2011;39:W357–W361. doi: 10.1093/nar/gkr468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell RB, Sasieni PD, Sternberg MJ. Supersites within superfolds. Binding site similarity in the absence of homology. J Mol Biol. 1998;282:903–918. doi: 10.1006/jmbi.1998.2043. [DOI] [PubMed] [Google Scholar]
- 32.Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA. 2008;105:129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu L, Lu H, Skolnick J. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 2002;49:350–364. doi: 10.1002/prot.10222. [DOI] [PubMed] [Google Scholar]
- 34.Petrey D, Honig B. GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- 35.Yang AS, Honig B. An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and a quantitative measure for protein structural distance. J Mol Biol. 2000;301:665–678. doi: 10.1006/jmbi.2000.3973. [DOI] [PubMed] [Google Scholar]
- 36.Hwang H, Vreven T, Janin J, Weng Z. Protein–protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang QC, Deng L, Fisher M, Guan J, Honig B, Petrey D. PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 2011;39:W283–W287. doi: 10.1093/nar/gkr311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dessailly BH, Lensink MF, Orengo CA, Wodak SJ. LigASite--a database of biologically relevant binding sites in proteins with known apo-structures. Nucleic Acids Res. 2008;36:D667–D673. doi: 10.1093/nar/gkm839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dey F, Petrey D, Honig B. Prediction of ligand-binding properties. In preparation [Google Scholar]
- 40.Pearl J. Probabilistic reasoning in intelligent systems: networks of plausible inference. San Mateo, CA: Morgan Kaufmann; 1988. [Google Scholar]
- 41.Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- 42.Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 43.Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, Basso K, Beltrao P, Krogan N, Gautier J, Dalla-Favera R, Califano A. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010:6. doi: 10.1038/msb.2010.31. art. num 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mani KM, Lefebvre C, Wang K, Lim WK, Basso K, Dalla-Favera R, Califano A. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B-cell lymphomas. Mol Systems Biol. 2008;4:1–9. doi: 10.1038/msb.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Troyanskaya OG, Dolinski K, Owen AB, Altman RB, Botstein D. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae) Proc Natl Acad Sci USA. 2003;100:8348–8353. doi: 10.1073/pnas.0832373100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, Maniatis T, Califano A, Honig B. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mukherjee S, Zhang Y. Protein-protein complex structure predictions by multimeric threading and template recombination. Structure. 2011;19:955–966. doi: 10.1016/j.str.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis FP, Braberg H, Shen MY, Pieper U, Sali A, Madhusudhan MS. Protein complex compositions predicted by structural similarity. Nucleic Acids Res. 2006;34:2943–2952. doi: 10.1093/nar/gkl353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang QC, Petrey D, Garzón JI, Deng L, Honig B. PrePPI: a structure-informed database of protein–protein interactions. Nucleic Acids Res. 2013;41:D828–D833. doi: 10.1093/nar/gks1231. [DOI] [PMC free article] [PubMed] [Google Scholar]