

Abstract
Abnormalities in dopaminergic activity have been implicated in psychiatric diseases, such as attention deficit hyperactivity disorder (ADHD), and are treated with therapeutic stimulants, commonly methylphenidate or amphetamine. Amphetamine administration increases glycogen synthase kinase-3 (GSK3) activation, which is necessary for certain acute behavioral responses to amphetamine, including increased locomotor activity and impaired sensorimotor gating. Here, we tested if modulating GSK3 by administration of the GSK3 inhibitor lithium or expression of constitutively active GSK3 altered behavioral responses to methylphenidate administered to mice acutely or daily for 8 days. Methylphenidate or amphetamine was administered to mice intraperitoneally for 1 or 8 days. Open-field activity and pre-pulse inhibition (PPI) were measured. In contrast to lithium’s blockade of acute amphetamine-induced locomotor hyperactivity, lithium treatment did not significantly reduce methylphenidate-induced locomotor hyperactivity in wild-type mice after acute or 8 days of repeated methylphenidate administration. Lithium treatment significantly increased the impairment in PPI caused by methylphenidate, but significantly reduced the amphetamine-induced PPI deficit. In GSK3 knockin mice, expression of constitutively active GSK3β, but not GSK3α, significantly increased locomotor hyperactivity after acute methylphenidate treatment, and significantly impaired PPI, preventing further methylphenidate-induced impairment of PPI that was evident in wild-type mice and GSK3α knockin mice. Lithium does not counteract locomotor activity and PPI responses to methylphenidate as it does these responses to amphetamine, indicating that different mechanisms mediate these behavioral responses to methylphenidate and amphetamine. Only active GSK3β, not GSK3α, modulates behavioral responses to MPH, indicating selectivity in the actions of GSK3 isoforms.
Keywords: Amphetamine, Attention deficit hyperactivity disorder, Glycogen synthase kinase-3, Methylphenidate, Pre-pulse inhibition
1. Introduction
Attention deficit hyperactivity disorder (ADHD) is a neurobehavioral disorder characterized by inattentiveness, aggression, irritability, hyperactivity and increased impulsivity (Waldman et al., 1998). The causes of ADHD are unknown, but patients often respond to a combination of behavioral therapy and pharmacological intervention. Psychostimulants, such as methylphenidate and amphetamine, are commonly used therapeutic drugs, which often reduce the symptoms of ADHD (Cantwell, 1996; Solanto, 2002; Olfson et al., 2003). Therapeutic effects of both stimulants are due to enhancing dopaminergic activity. Methylphenidate binds the dopamine transporter, and to a lesser extent the norepinephrine transporter, thereby inhibiting reuptake of dopamine and norepinephrine into presynaptic terminals, allowing greater stimulation of their respective receptors (Sulzer et al., 1995; Gatley et al., 1996). Amphetamine decreases uptake and increases efflux of dopamine by the dopamine transporter, decreases dopamine content in synaptic vesicles (Sulzer et al., 1995), and inhibits norepinephrine and serotonin reuptake, which increases activation of adrenergic and serotonergic receptors. Neurotransmitter release is also promoted by amphetamine, and more modestly by methylphenidate (Gatley et al., 1996; Kuczenski and Segal, 1997; Nestler et al., 2001).
Acute treatment with amphetamine causes a rapid increase in locomotor activity in rodents. Beaulieu reported that this locomotor-activating effect of amphetamine is mediated by activation of glycogen synthase kinase-3 (GSK3) (Beaulieu et al., 2004, 2007). The activity of the two GSK3 isoforms, GSK3α and GSK3β, is predominantly regulated by inhibitory phosphorylation on Ser21-GSK3α and Ser9-GSK3β, a modification that is mediated by Akt (Doble and Woddgett, 2003; Jope and Johnson, 2004). We recently reported that 8 days of amphetamine or methylphenidate administration activated GSK3 in mouse striatum (Mines and Jope, 2012), similarly to acute amphetamine responses.
The importance of inhibitory control of GSK3 can be studied using GSK3α21A/21A/β9A/9A knockin mice, with the regulatory serines of one or both GSK3 isoforms mutated to alanines (McManus et al., 2005), thereby maintaining GSK3 maximally active. Both GSK3 isoforms are expressed at normal levels. Administration of amphetamine activates GSK3 by decreasing the activating phosphorylation of Akt on Thr-308, which decreases the inhibitory serine-phosphorylation of both isoforms of GSK3 (Beaulieu et al., 2004, 2007). Beaulieu et al. (2004) found that dopamine transporter-knockout mice exhibit hyperactivity in a novel open field that was attenuated by administration of lithium and other GSK3 inhibitors, and amphetamine-induced hyperactivity was diminished by reduced GSK3 expression in GSK3β−/+ mice. Conversely, GSK3 knockin mice are hypersensitive to amphetamine-induced locomotor hyperactivity, further linking amphetamine and GSK3 activation in promoting locomotor hyperactivity (Polter et al., 2010).
In the present study, we tested if GSK3 mediates responses of mice to methylphenidate similarly to previous findings with amphetamine (Beaulieu et al., 2004, 2007; Mines and Jope, 2012), and if the behavioral effects are altered following longer term stimulant administration. The results indicate that in contrast to lithium’s counteraction of locomotor and PPI responses to amphetamine, lithium administration did not alter these behavioral responses to methylphenidate. Studies in GSK3 knockin mice demonstrated that only active GSK3β, not active GSK3α, promoted locomotor activation induced by methylphenidate.
2. Materials and methods
2.1. Animals
This study used adult, male C57Bl/6J littermate mice, ~3 months of age, expressing wild-type GSK3 or GSK3α21A/21A (hereafter referred to as GSK3α knockin mice), or GSK3β9A/9A (GSK3β knockin mice), originally kindly provided with matched controls by Dr. D. Alessi (University of Dundee). These mutations disable the inhibitory serine phosphorylation of GSK3, but both isoforms are expressed at normal levels so GSK3 retains maximal activities within the normal physiological range (McManus et al., 2005). GSK3 knockin mice reproduce and develop normally and no overt phenotype has been reported. For chronic lithium treatment, mice were given ad libitum water and saline (to prevent hyponatremia caused by lithium-induced increased excretion of sodium) and were fed pelleted chow containing 0.2% lithium carbonate (Teklad, Madison, WI) for three weeks, as previously described (De Sarno et al., 2002; Eom and Jope, 2009). All mice were housed and treated in accordance with National Institutes of Health guidelines and procedures with mice were approved by the University of Alabama at Birmingham and University of Miami Institutional Animal Care and Use Committees.
2.2. Drug administration
Methylphenidate and amphetamine (Sigma, St. Louis, MO) were dissolved in saline and injected intraperitoneally (i.p.). Control mice received equal amounts of saline vehicle. Acute injections were given in the testing chambers. For the 8 days of repeated injections, drugs were administered in the home-cage, except for the last injection, which was administered in the testing chamber.
2.3. Open-field behavior
Locomotion was measured in an open-field arena (43.2 cm × 43.2 cm × 30.5 cm) fitted with 16 evenly spaced infrared sources and sensors juxtaposed around the periphery of the chamber (Med Associates, St. Albans, VT). The outer walls were wrapped with white paper to limit external stimuli and light gradients. A white noise generator was used (55 dB) during all tests. Each chamber was connected to a computer that recorded beam breaks (50 ms sampling rate). Three consecutive beam breaks represented an ambulatory episode (horizontal activity) and the total distance of all ambulatory episodes was recorded in 60 s intervals. Each mouse was placed in the center of the box to begin the test. Testing sessions were 45 min for methylphenidate treatment and 80 min for amphetamine treatment. Habituation to the apparatus was carried out for the first 15 min before methylphenidate injection and for the first 30 min before amphetamine injection. Stereotypic behaviors, measured as repetitive breaks of the same beams during slow, small movement periods, were also recorded. Separate experimental groups were used for measurements after acute administration and after 8 days of repeated administration, as pre-exposure to the open-field apparatus could confound results.
2.4. Pre-pulse inhibition behavior
Pre-pulse inhibition was measured using the SR-Lab System (San Diego Instruments, San Diego, CA). Mice were placed in the Plexiglas cylinder and left undisturbed with background white noise (65 dB) for 5 min prior to 7 trials. During the first trial no stimulus was presented to measure baseline movement in the cylinder. Another trial consisted of a 40 ms, 120 dB sound burst used as the startle stimulus. Three different 20 ms pre-pulse sounds (69, 73, and 81 dB) were presented either alone (pulse alone) or 100 ms before the startle stimulus (pre-pulse trials). Each trial type was repeated six times, once per block of seven trials in pseudorandom order. The inter-trial interval was 15 s. The startle response was recorded every 1 ms during a 65 ms period that followed the onset of the startle stimulus. The maximum startle amplitude during this period was used as the dependent variable. Percent pre-pulse inhibition of the startle response was calculated as 100—[(startle response on acoustic pre-pulse plus startle stimulus trials/startle response alone trials)−100].
2.5. Statistical analysis
Data were analyzed by unpaired t test and two-way ANOVA with Bonferroni post tests. Values are expressed as mean ± S.E.M.
3. Results
3.1. Locomotor hyperactivity is dose-dependently induced by methylphenidate in wild-type mice
Administration of methylphenidate dose-dependently induced increases in open-field locomotor activity in wild-type mice (Fig. 1A; Fdose(3,54)=18.87, P<0.0001; Ftime(8,432)=20.51, P< 0.0001; Finteraction(24,432)=13.83, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to vehicle-treated control mice). Administration of 5 mg/kg methylphenidate did not significantly d increase locomotor activity, whereas 10 mg/kg methylphenidate caused a rapid and prolonged hyperactivity that was significantly increased within 10 min of methylphenidate administration and remained significantly elevated for 30 min. Interestingly, after administration of the highest dose tested, 20 mg/kg methylphenidate, wild-type mice displayed a rapid onset of hyperactivity that reached a peak at 10 min post treatment, followed quickly by a return towards basal activity. This pattern of locomotor activation is similar to a previous report assessing the effects of acute administration of 30 mg/kg methylphenidate in mice (Beaulieu et al., 2006).
Fig. 1.

Psychostimulant responses in wild-type mice. (A) Open-field activity responses to 0 (saline control), 5, 10, or 20 mg/kg methylphenidate in wild-type mice after 15 min acclimation to the test chamber. *P≤0.05, post-test, as compared to vehicle-treated control mice. (B) Effects of 8 days of repeated administration of 10 mg/kg methylphenidate on open-field activity. (C) Effects of 8 days of repeated administration of 20 mg/kg methylphenidate on open-field activity. *P≤0.05, post-test, as compared to Day 1. (D) Effects of 1 or 8 days of repeated administration of 20 mg/kg methylphenidate on stereotypic behavior. *P≤0.05, post-test, as compared to vehicle-treated control mice. (E) Effects of 2 mg/kg amphetamine on open-field activity. (F) Effects of 1 or 8 days of repeated administration of 2 mg/kg amphetamine on stereotypic behavior *P≤0.05, post-test, as compared to vehicle-treated control mice. Values are expressed as means±S.E.M.
The effects of 8 days of repeated daily administration of methylphenidate on locomotor activity were compared with a single treatment. Locomotor activity following repeated administration of 10 mg/kg methylphenidate was similar to acute administration (Fig. 1B; Fdose(1,16)=0.05, P=0.8244; Ftime(8,128)=71.48, P<0.0001; Finteraction(8,128)=2.80, P=0.0068, two-way ANOVA). Compared to acute treatment, repeated administration of 20 mg/kg methylphenidate did not alter the initial rate of onset of locomotor hyperactivity, but significantly reduced the maximum hyperactivity response (Fig. 1C; Fdose(1,24)=4.48, P=0.0448; Ftime(8,192)=5.40, P<0.0001; Finteraction(8,192)=4.65, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to Day 1).
The pattern of methylphenidate-induced hyperactivity at the highest dose (20 mg/kg) suggested the possible development of stereotypic behavior. However, assessment of stereotypic behavior indicated no significant increase in stereotypic counts in mice treated with 20 mg/kg methylphenidate for 1 or 8 days, as compared to vehicle-treated control mice (Fig. 1D; Fdose(2,45)=1.05, P=0.3592; Ftime(8,360)=2.66, P=0.0075; Finteraction(16,360)=5.15, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to vehicle-treated control mice). Nevertheless, it should be noted that since automatic measurements of stereotypy may not accurately analyze the expression of various forms of stereotypy, a possibility of contribution of some type of stereotypic movements in the reduction of locomotor activity following repeated methylphenidate treatment cannot be fully excluded. However, a previous study also reported a rapid increase in locomotor hyperactivity after administration of 30 mg/kg methylphenidate that was followed by a secondary reduction in ambulatory activity (Beaulieu et al., 2006).
In comparison, administration of 2 mg/kg amphetamine caused a prolonged increase in locomotor activity (Fig. 1E; Fdose(1,18) =0.004, P=0.9467; Ftime(15,270) =5.11, P<0.0001; Finteraction(15,270) =1.47, P=0.1177, two-way ANOVA), as previously reported (Beaulieu et al., 2007; Polter et al., 2010). Repeated treatment with amphetamine for 8 days did not significantly alter hyperactivity from that induced by acute amphetamine administration (Fig. 1E). There was no significant increase in stereotypic behavior in mice treated with 2 mg/kg amphetamine, as compared to control mice (Fig. 1F; Fdose(2,27) = 36.81, P<0.0001; Ftime(15,405) =17.11, P <0.0001; Finteraction(30, 405)=4.89, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to vehicle-treated control mice).
3.2. Lithium treatment does not reduce methylphenidate-induced hyperactivity in wild-type mice
Lithium was administered to mice for three weeks to assess its effect on methylphenidate-induced locomotor hyperactivity, a lithium treatment paradigm that does not alter basal locomotor activity in this strain of mice (Yuskaitis et al., 2010). As compared to methylphenidate treatment alone, lithium treatment did not significantly alter 20 mg/kg methylphenidate-induced hyperactivity (Fig. 2A; Fdose(1,23)=2.85, P=0.1049; Ftime(8,184)=10.44, P< 0.0001; Finteraction(8,184)=0.96, P=0.4724, two-way ANOVA), or decrease the maximum or duration of hyperactivity following repeated administration of 20 mg/kg methylphenidate (Fig. 2B; Fdose(1,15)=0.72, P=0.4091; Ftime(8,120)=5.94, P<0.0001; Finteraction (8,120)=1.40, P=0.2028, two-way ANOVA). In contrast to the lack of statistically significant effects of lithium on the response to methylphenidate, lithium treatment completely blocked amphetamine-induced hyperactivity (Fig. 2C; Fdose(1,18)=3.71, P=0.0700; Ftime(15,270)=9.15, P<0.0001; Finteraction(15,270)=3.45, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to the response to amphetamine in the absence of lithium), as reported previously (Beaulieu et al., 2004). There was no significant difference between the locomotor activities of mice treated with lithium and administered repeated doses of amphetamine from the response of mice treated with repeated doses of amphetamine alone (Fig. 2D; Fdose (1,18)=0.74, P=0.4019; Ftime(15,270)=2.32, P=0.0040; Finteraction (15,270)=3.23, P<0.0001, two-way ANOVA). Thus, chronic lithium treatment had no effect on methylphenidate-induced locomotor hyperactivity, but lithium effectively counteracted the locomotor response to acute amphetamine administration, whereas this effect of lithium on the response to amphetamine was absent after prolonged amphetamine treatment.
Fig. 2.
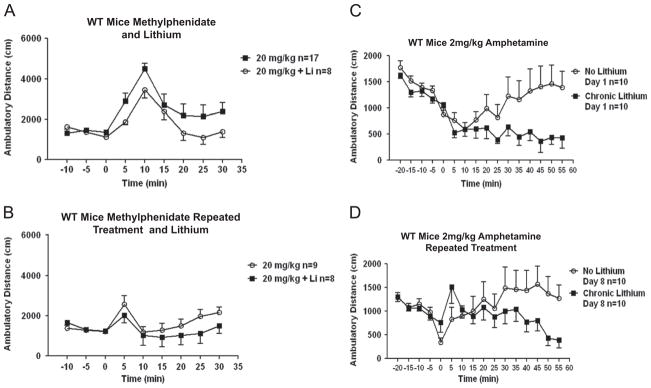
Effects of lithium treatment on psychostimulant-induced responses in wild-type mice. (A) Effect of lithium treatment on open-field activity after acute administration of 20 mg/kg methylphenidate in wild-type mice. (B) Effect of lithium treatment on open-field activity after 8 days of administration of 20 mg/kg methylphenidate. (C) Effect of lithium treatment on open-field activity after acute administration of 2 mg/kg amphetamine. *P≤0.05, post-test, as compared to the response to amphetamine in the absence of lithium. (D) Effect of lithium treatment on open-field activity after 8 days of administration of 2 mg/kg amphetamine. Values are expressed as means±S.E.M.
3.3. MPH affects sensorimotor gating behavior
Methylphenidate (20 mg/kg) administration significantly decreased PPI compared to PPI in vehicle-treated control mice (Fig. 3A; *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to vehicle-treated control mice, t test). Lithium treatment, which alone does not alter PPI (Umeda et al., 2006), facilitated the methylphenidate-induced decrease in PPI (Fig. 3A; #P≤0.05, as compared to methylphenidate treatment alone, t test). Amphetamine (2 mg/kg) significantly reduced PPI at the highest tone (81 dB) (Fig. 3B; *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to vehicle-treated control mice, t test). There was also no significant increase in PPI from mid (73 dB) to high (81 dB) tones in amphetamine treated mice, as observed in control mice. Lithium treatment modestly restored the PPI response in amphetamine-treated mice, as a significant increase in PPI was observed with increasing pre-pulse tones from 73 dB to 81 dB in lithium-treated mice given amphetamine, as occurred in control mice. Thus, chronic lithium treatment significantly promoted PPI deficits induced by methylphenidate, but significantly reduced the amphetamine-induced PPI deficit.
Fig. 3.

Effects of lithium on sensorimotor gating in wild-type mice. (A) Effect of acute administration of 20 mg/kg methylphenidate on PPI with or without lithium pre-treatment in wild-type mice. *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to vehicle-treated control mice, t test; #P≤0.05, as compared to methylphenidate treatment alone, t test. (B) Effect of acute administration of 2 mg/kg amphetamine on PPI with or without lithium pretreatment in wild-type mice. *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to vehicle-treated control mice, t test. Values are expressed as means±S.E.M.
3.4. GSK3β, but not GSK3α, regulates methylphenidate-induced behavioral responses
Since amphetamine-induced locomotor hyperactivity was heightened in GSK3α/β knockin mice that express constitutively active GSK3α and GSK3β (Polter et al., 2010), we tested if that occurred following methylphenidate administration separately in GSK3α or GSK3β knockin mice. Acute administration of 20 mg/kg methylphenidate induced hyperactivity in GSK3α knockin mice that was not significantly different from the response to methylphenidate in wild-type mice (Fig. 4A; Fgenotype(1,21) =0.002, P=0.9636; Ftime(8,168) =14.05, P<0.0001; Finteraction(8,168) = 0.73, P=0.6613, two-way ANOVA). The attenuated locomotor ) activation in response to 8 days of repeated administration of 20 mg/kg methylphenidate in wild-type mice (Fig. 1C) also was equivalent in GSK3α mice (Fig. 4B; Fdose(1,9) =14.83, P =0.0039; Ftime(8,72) =10.40, 0 P <0.0001; Finteraction(8,72) =11.39, P <0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to Day 1). In D contrast to the lack of effect of active GSK3α, in GSK3β knockin mice the maximal locomotor hyperactivity induced by acute 20 mg/kg methylphenidate administration was significantly higher and the duration of hyperactivity was significantly prolonged compared with wild-type mice (Fig. 4C; Fgenotype(1,23)=6.34, P=0.0192; Ftime(8,184)=22.99, P<0.0001; Finteraction(8,184)=5.03, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to wild-type mice). However, GSK3β knockin mice (Fig. 4D; Fdose(1,14)=44.02, P<0.0001; Ftime(8,112)=13.76, P<0.0001; Finteraction(8,112)= 19.49, P<0.0001, two-way ANOVA. *P≤0.05, post-test, as compared to Day 1) responded similarly to wild-type mice (Fig. 1C) with blunted locomotor responses after 8 days of 20 mg/kg methylphenidate administration. Thus, activated GSK3β significantly increased the hyperactive locomotor response to acute methylphenidate administration, but active GSK3α did not, and prolonged methylphenidate administration resulted in diminished locomotor activation compared with acute administration regardless of the phosphorylation status of either GSK3 isoform.
Fig. 4.

GSK3 is involved in methylphenidate-induced open-field behavior. (A) Effect of acute administration of 20 mg/kg methylphenidate on open-field activity in GSK3α knockin mice. (B) Effect of 8 days of repeated administration of 20 mg/kg methylphenidate on open-field activity in GSK3α knockin mice. *P≤0.05, post-test, as compared d to Day 1. (C) Effect of acute administration of 20 mg/kg methylphenidate on open-field activity in GSK3β knockin mice. *P≤0.05, post-test, as compared to wild-type mice. (D) Effect of 8 days of repeated administration of 20 mg/kg methylphenidate on open-field activity in GSK3β knockin mice. *P≤0.05, post-test, as compared to Day 1. Values are expressed as means±S.E.M.
PPI in GSK3α knockin mice and its inhibition by methylphenidate treatment (Fig. 5A; *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to control, t test) were similar to PPI in wild-type mice (Fig. 3A). In contrast, PPI was impaired in GSK3β knockin mice, as there was no significant difference in response to increasing tones (Fig. 5B), and methylphenidate had no significant effect on PPI in GSK3β knockin mice, although there was a trend suggesting an effect (P=0.06 at lowest tone and P=0.08 at highest tone).
Fig. 5.

GSK3 affects methylphenidate-induced sensorimotor behaviors. (A) Effect of 20 mg/kg methylphenidate on PPI in GSK3α knockin mice. *P≤0.05, as compared to previous tone intensity; &P≤0.05, as compared to control, t test. (B) Effect of 20 mg/kg methylphenidate on PPI in GSK3β knockin mice. Values are expressed as means±S.E.M.
Taken together, these findings indicate that constitutively active GSK3β, but not GSK3α, in the knockin mice significantly alters responses to methylphenidate, revealing differing roles for the two GSK3 isoforms in methylphenidate-induced behavioral responses.
4. Discussion
Abnormalities in dopaminergic activity and signaling are linked to several neurobehavioral disorders. The increasing incidence of neurobehavioral disorders, such as ADHD (Center for Disease Control, 2010), and increases in the prescription of stimulants, such as methylphenidate, emphasizes the critical need for further understanding of drug-induced behavioral responses. The results of this study indicate that lithium treatment differentially modifies locomotor activity and PPI behavioral responses to methylphenidate and amphetamine, and that there are distinct differences between the impact of the two isoforms of GSK3 on these behavioral responses to methylphenidate.
Inhibition and activation have been used to decipher the role of GSK3 in stimulant-induced behaviors. Beaulieu et al. (2004) clearly demonstrated that GSK3 promotes amphetamine-mediated actions in vivo, such as locomotor activity. They showed that reduced expression of GSK3β, using GSK3β−/+ mice, or inhibition of GSK3 with lithium treatment significantly reduced amphetamine-induced locomotor hyperactivity (Beaulieu et al., 2004). Using hyperactive dopamine transporter-knockout mice, they also showed that inhibition of GSK3, using SB216763, alsterpaullone, indirubin-3-monoxime, valproate, and TDZD, reduced dopamine-mediated open-field locomotor activity (Beaulieu et al., 2004). The influence of GSK3 on other psychostimulant-induced behaviors has also been reported. GSK3 inhibition by administration of SB216763 or valproate in mice or AR-A014418 in rats attenuated cocaine- and amphetamine-induced locomotor hyperactivity (Miller et al., 2009; Gould et al., 2004). Lithium and SB216763 also reduced methamphetamine-induced hyperactivity and behavioral sensitization in mice and rats paradigms (Ago et al., 2012; Xu et al., 2011). Clozapine, an atypical antipsychotic that reduces GSK3 activity in mouse and rat brain (Kang et al., 2004; Li et al., 2006), reduced amphetamine-mediated hyperactivity (Tang et al., 1997) and cocaine-induced sensitization (Park et al., 2010). Reciprocally, over-expression of constitutively active GSK3βS9A was sufficient to increase locomotor activity of mice in a novel environment (Prickaerts et al., 2006), and GSK3α/β knockin mice displayed increased locomotor activation in response to amphetamine administration (Polter et al., 2010). In contrast to the reports that lithium treatment blocks amphetamine-stimulated locomotor activity, as also demonstrated in this study, the current study found that lithium treatment does not significantly alter locomotor activation in response to methylphenidate. GSK3α and GSK3β knockin mice were used to determine if constitutive activation of either, or both, isoforms altered responses to methylphenidate. Whereas GSK3α knockin mice displayed no difference from wild-type mice in methylphenidate-induced locomotor hyperactivity, GSK3β knockin mice displayed greater locomotor activation in response to methylphenidate compared to wild-type mice. These findings support the unexpected conclusion that lithium does not influence methylphenidate-induced locomotor activity, and that only active GSK3β, not active GSK3α, promotes this response to methylphenidate, although further studies using mice with selectively reduced expression of each of the GSK3 isoforms could be used to further examine these interactions. These findings extend a growing appreciation for the differential actions of the two isoforms of GSK3 (Liang and Chuang, 2007; Force and Woodgett, 2009).
Differential regulation by lithium of acute methylphenidate-and amphetamine-induced behavioral responses and different effects of the two GSK3 isoforms provoked examination of their effects after repeated administration. Administration of 20 mg/kg methylphenidate for 8 days did not increase locomotor activity to the same extent as acute methylphenidate in wild-type mice, which matches previous reports of diminished locomotor responses to methylphenidate after repeated treatments of a lower dose of methylphenidate administered orally to mice (Kuczenski and Segal, 2002). This contrasts with 8 days of treatment with 2 mg/kg amphetamine, after which the locomotor response was indistinguishable from the acute response. Interestingly, lithium did not alter locomotor responses after repeated administration of either stimulant and the promotion by constitutively active GSK3β of locomotor activation was absent after repeated administration of methylphenidate.
Deficits in PPI have often been observed in psychiatric disorders, such as schizophrenia (Geyer et al., 2001; Swerdlow et al., 2008). These deficits have been modeled in rodents, for example by administration of amphetamine, which is well-known to impair PPI (Flood et al., 2010; Geyer et al., 2001; Swerdlow et al., 2008). Impaired PPI has also been reported after methylphenidate administration to wild-type mice (Flood et al., 2010). Inhibition of GSK3 has been reported to restore PPI after amphetamine administration (Ong et al., 2005). In the present study, methylphenidate induced significant decreases in PPI in wild-type mice and this impairment was increased by lithium treatment. In contrast, lithium treatment reduced amphetamine-induced PPI deficits in wild-type mice. Thus, as with stimulant-induced locomotor hyperactivity, lithium treatment had different effects on the PPI responses to methylphenidate and amphetamine. Also, as with locomotor activity, there were differential actions of the two GSK3 isoforms on stimulant-induced impairment of PPI. Constitutively active GSK3α had no effect on PPI or the impairment caused by methylphenidate administration, whereas constitutively active GSK3β impaired PPI, which negated any potential impairment of PPI following methylphenidate administration. Collectively, these PPI measurements demonstrate differential actions of lithium following administration of methylphenidate compared with amphetamine, and a selective modulatory effect of GSK3β rather than GSK3α.
With dopamine-mediated signaling and behaviors being linked to more neurological disease and disorders, it’s becoming increasingly important to understand all facets of regulation. In this study, we identify differential stimulant-induced responses after chronic administration. We also confirm previous reports identifying GSK3 regulation as a key contributor to dopamine-mediated therapeutic efficacy and responsiveness.
Acknowledgments
We thank Dr. Dario Alessi for providing the GSK3 knockin mice. This research was funded by grants from the NIH (MH092970 and MH038752). The content is solely the responsibility of the authors and does not represent the official views of the NIH.
References
- Ago Y, Tanaka T, Kita Y, Tokumoto H, Takuma K, Matsuda T. Lithium attenuates methamphetamine-induced hyperlocomotion and behavioral sensitization via modulation of prefrontal monoamine release. Neuropharmacol. 2012;62:1634–1639. doi: 10.1016/j.neuropharm.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Beaulieu M, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, Borrelli E, Caron MG. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. 2007;27:881–885. doi: 10.1523/JNEUROSCI.5074-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Paradoxical striatal cellular signaling responses to psychostimulants in hyperactive mice. J Biol Chem. 2006;281:32072–32080. doi: 10.1074/jbc.M606062200. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium anatagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc N Nat Acad Sci, USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantwell DP. Attention deficit disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 1996;35:978–987. doi: 10.1097/00004583-199608000-00008. [DOI] [PubMed] [Google Scholar]
- Center for Disease Control. Increasing Prevalence of Parent-Reported Attention-Deficit/Hyperactivity Disorder Among C Children—United States, 2003 and 2007. Morbidity and Mortality Weekly Report. 2010;59:1439–1443. [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woddgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom TY, Jope RS. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3α/β impairs in vivo neural precursor cell proliferation. Biol Psychiatry. 2009;66:494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood DG, Zuvich E, Marino MJ, Gasior M. The effects of d-amphetamine, methylphenidate, syndocarb, and caffeine on pre-pulse inhibition of the startle reflex in DBA/2 mice. Psychopharmacol. 2010;211:325–336. doi: 10.1007/s00213-010-1901-0. [DOI] [PubMed] [Google Scholar]
- Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284:9643–9647. doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatley SJ, Pan D, Chen R, Chaturvedi G, Ding YS. Affinities of methylphenidate derivatives for dopamine, norepinephrine and serotonin transporters. Life Sci. 1996;58:231–239. doi: 10.1016/0024-3205(96)00052-5. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of pre-pulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Neuropsychopharmacol. 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GBW. The glamour and gloom of glycogen synthase kinase-3 (GSK3) Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kang UG, Seo MS, Roh MS, Kim Y, Yoon SC, Kim YS. The effects of K clozapine on the GSK-3-mediated signaling pathway. FEBS Lett. 2004;560:115–119. doi: 10.1016/S0014-5793(04)00082-1. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Exposure of adolescent rats to oral methylphenidate: preferential effects on extracellular norepinephrine and absence of sensitization and cross-sensitization to methamphetamine. J Neurosci. 2002;16:7264–7271. doi: 10.1523/JNEUROSCI.22-16-07264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem. 1997;68:2032–2037. doi: 10.1046/j.1471-4159.1997.68052032.x. [DOI] [PubMed] [Google Scholar]
- Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2006;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JS, Tallarida RJ, Unterwald EM. Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacol. 2009;56:1116–1123. doi: 10.1016/j.neuropharm.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines MA, Jope RS. Brain region differences in regulation of Akt and GSK3 by chronic stimulant administration in mice. Cell Signal. 2012;24:1398–1405. doi: 10.1016/j.cellsig.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE, Malenka RC. Molecular Neuropharmacology: a foundation for Clinical Neuroscience. 1. The McGraw-Hill Companies; New York: 2001. [Google Scholar]
- Olfson M, Gameroff MJ, Marcus SC, Jensen PS. National trends in the treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2003;160:1071–1077. doi: 10.1176/appi.ajp.160.6.1071. [DOI] [PubMed] [Google Scholar]
- Ong JC, Brody SA, Large CH, Geyer MA. An investigation of the efficacy of mood stabilizers in rodent models of pre-pulse inhibition. J Pharmacol Exp Ther. 2005;315:1163–1171. doi: 10.1124/jpet.105.090845. [DOI] [PubMed] [Google Scholar]
- Park HJ, Cui FJ, Hwang JY, Kang UG. Effects of clozapine on behavioral sensitization induced by cocaine. Psychiatry Res. 2010;175:165–170. doi: 10.1016/j.psychres.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, Sweatt JD, McMahon L, Bartolucci AA, Li X, Jope RS. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacol. 2010;8:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J, Moerchars D, Cryns K, Lanaerts I, van Craenndonk H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice over-expressing glycogen synthase kinase β: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanto MV. Dopamine dysfunction in AD/HD: integrating clinical and basic neuroscience research. Behav Brain Res. 2002;130:65–71. doi: 10.1016/s0166-4328(01)00431-4. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of pre-pulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199:331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Fanklin SR, Himes CS, Smith MW, Tenbrink RE. PNU-96415E, a potential antipsychotic agent with clozapine-like pharmacological properties. J Pharmacol Exp Ther. 1997;281:440–447. [PubMed] [Google Scholar]
- Umeda K, Suemaru K, Todo N, Egashira N, Mishima K, Iwasaki K, Fujiwara M, Araki H. Effects of mood stabilizers on the disruption of prepulse inhibition induced by apomorphine or dizocilpine in mice. Eur J Pharmacol. 2006;553:157–162. doi: 10.1016/j.ejphar.2006.09.050. [DOI] [PubMed] [Google Scholar]
- Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH, Sherman SL, Cleveland HH, Sanders ML, Gard JM, Stever C. Association and linkage of the dopamine transporter gene and attention-deficit hyperactivity disorder in children: heterogeneity owing to diagnostic subtype and severity. Am J Hum Genet. 1998;63:1767–1776. doi: 10.1086/302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang J, Wu P, Xue Y, Zhu W, Li Q, Zhai H, Shi J, Lu L. Glycogen synthase kinase 3β in the nucleus accumbens core is critical for methamphetamine-inducedi behavioral sensitization. J Neurochem. 2011;118:126–139. doi: 10.1111/j.1471-4159.2011.07281.x. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JC, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol. 2010;79:632–646. doi: 10.1016/j.bcp.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]