

Abstract
Two known cleistriosides and six known cleistetrosides were synthesized and evaluated for anticancer and antibacterial activities. This study, for the first time, reports anticancer activity and comprehensively the antibacterial activity for these oligosaccharide natural products. In addition, two new unnatural cleistetroside analogues were synthesized and tested. Biological activities for the 10 oligosaccharides against B. subtilis were found to range between 4 and >64 μM and for NCI-H460 human lung cancer epithelial cells between 7.5 and 90.9 μM. Similar activities were found for seven of the oligosaccharides against the NCI panel of 60 cell lines. The degree of acylation and location of the specific acetate groups had significant effects on the anticancer and antibacterial activity of both the cleistriosides and the cleistetrosides.
Keywords: cleistetroside, cleistrioside, antibacterial, anticancer, palladium-catalyzed glycosylation
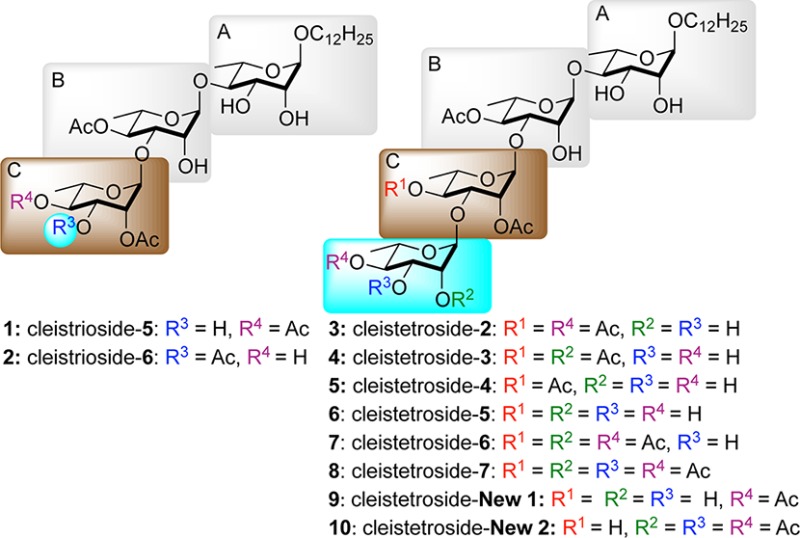
As part of a high-throughput-based search for new natural products with unique structures and interesting biological activity, two partially acetylated trisaccharides (cleistrioside-5/-6, 1 and 2) and six partially acetylated tetrasaccharides (cleistetroside-2/-3/-4/-5/-6/-7, 3–8) natural products were discovered (Figure 1) from the leaves and bark of trees with a folk medicinal tradition.1−3 These dodecanyl tri- and tetrarhamnoside structures with various degrees of acylation were isolated from Cleistopholis patens and glauca.1,2 The structures of the cleistriosides and cleistetrosides were assigned by detailed NMR analysis and later confirmed by total synthesis. The absolute and relative stereochemistry for one member of the cleistetrosides (3) was confirmed by two total synthetic efforts by Zhang4 and Chen5 in 2007 via a traditional carbohydrate approach. In 2010, we reported a divergent de novo asymmetric synthesis of both cleistriosides (1 and 2) as well as the six cleistetrosides (3–8), thus confirming the structures of the remaining members.6
Figure 1.
Targeted cleistriosides and cleistetrosides.
Because of the limited amounts of natural isolates (1–3 mg), only two [cleistrioside-5 (1) and cleistetroside-2 (3)] of these eight oligo-rhamno-natural products were extensively evaluated for antimicrobial activity.3 These antibacterial activities are outlined in Table 1,3 which shows quite potent activity across a broad range of organisms. Of particular note is that both cleistrioside-5 (1) and cleistetroside-2 (3) showed significant activity against several oxacillin-resistant organisms (e.g., Staphylococcus aureus ATCC 33591, Table 1).3 Because of the rhamnan structural motif of the cleistriosides and cleistetrosides, their antibacterial activity is assumed to occur via inhibition of mycobacterial cell wall synthesis. Similarly, the fact that the oligo-rhamnose motif is not found in mammalian cells makes them even more interesting as a potential antibiotic.7
Table 1. Antibacterial Activity of Cleistroside-5 (1) and Cleistetroside-2 (3)3.
| MIC (μg/mL) |
||
|---|---|---|
| organism | 1 | 3 |
| Staphylococcus aureus ATCC33591 (oxacillin-resistant) | 2 | 2–4 |
| Coagulase-negative staphylococci | 2–4 | >32 |
| Enterococcus faecalis | 2 | 2–4 |
| Bacillus spp. | 2 | 2 |
| Streptococcus pneumoniae | 4–16 | 4 |
| Corynebacterium spp. | 8 | 16 |
| viridans group streptococci | 4 | 4 |
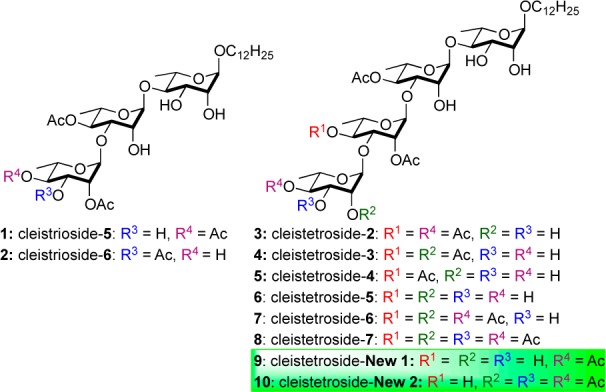
Over the years, we have been interested in the de novo synthesis and study of partially acylated rhamnoside natural products.8,9 Accordingly, the structurally novel cleistrioside and cleistetroside potential antibiotics inspired us to develop a divergent de novo asymmeric approach, which resulted in a reasonably efficient approach to eight of the members of these two classes of natural products (Scheme 1). While our de novo route to any particular member was not notably more efficient than the two traditional carbohydrate approaches to cleistetroside-2 (3),4,5 the approach allows for the divergent synthesis of all eight oligo-rhamnosides (1–8) with the minimal use of protecting groups. The overall efficiency of the route is best exemplified by the fact that it affords sufficient quantities of material for detailed biological investigations. Herein, we describe our successful use of this divergent de novo approach to the eight natural products (1–8) along with two unnatural cleistetroside analogues (9 and 10) as well as their evaluation in antibacterial and cytotoxicity assays.
Scheme 1. De Novo Retrosynthetic Approach to the Cleistriosides and Cleistetrosides.
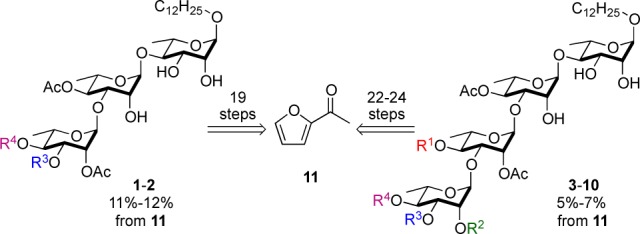
Previously, we demonstrated that the eight oligosaccharides (1–8) could be efficiently prepared via a divergent de novo route that derives all the sugar carbons from the achiral acylfuran 11.6 Key to the approach was the efficient synthesis of a common trisaccharide intermediate 17 (Scheme 2). In a three-step asymmetric protocol, acylfuran was converted into pyranone glycosyl donor 12,10,11 which in turn is converted to pyranone 13 via a Pd-catalyzed glycosylation [1:4/Pd2(dba)3·CHCl3 to PPh3].12−14 A three-step postglycosylation protocol was used to stereoselectively install the desired rhamno-stereochemistry and acetonide protecting groups. A subsequent glycosylation, reduction, acylation, and dihydroxylation sequence provided disaccharide 15 followed by a regioselective glycosylation, and chloroacylation gives the orthoganally protected trisaccharide 17. This 14-step synthesis was accomplished with only two traditional (acetonide and chloroacetate) and two nontraditional (ketone and alkene) alcohol protecting groups.15
Scheme 2. Approach to the Intermediate 17.

Reagents and conditions: (a) (i) Noyori (S,S), HCO2Na(aq), 10 mol % CTAB, rt, 24 h; (ii) NBS, THF/H2O (3:1), NaHCO3, NaOAc·3H2O, 0 °C, 1 h; (iii) Boc2O, DMAP, CH2Cl2, −78 °C, 12 h. (b) HOCH2(CH2)10CH3, 5 mol % Pd(PPh3)2, CH2Cl2, 0 °C. (c) (i) NaBH4, CeCl3, CH2Cl2/MeOH, −78 °C, 2 h; (ii) OsO4, NMO, t-BuOH/acetone, rt, 12 h; (iii) TsOH, 2,2-DMP, acetone, 0 °C, 4 h. (d) (i) 12, 5 mol % Pd(PPh3)2, CH2Cl2, 0 °C, 2 h; (ii) NaBH4, CH2Cl2/MeOH, −78 °C, 2 h; (iii) Ac2O, Py, rt, 3 h; (iv) OsO4, NMO, t-BuOH/acetone, rt, 12 h. (e) (i) n-Bu2SnO, MeOH, reflux, 1h; (ii) 12, 5 mol % Pd(PPh3)2, CH2Cl2, 0 °C, 2 h. (f) (ClAc)2O, Py, 3 h. Pd(PPh3)2 = Pd2(dba)3·CHCl3/4PPh3.
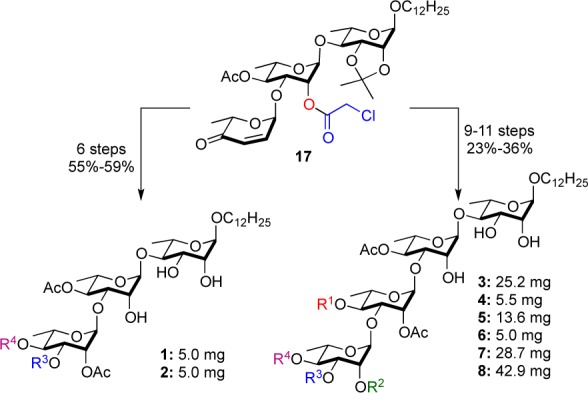
The enone portion of trisaccharide 17 was ready for further elaboration into the final rhamno-sugar required for 1 and 2; in only six steps, 17 was selectively converted into either trisaccharides 1 or 2. By simply incorporating an additional glycosylation/postglycosylation reaction sequence, along with traditional two deprotections, the final two rhamno-sugars of the cleistetrosides were installed onto 17.16 Thus, in 9–11 steps, 17 was selectively converted into the six cleistetroside natural products 3–8. In contrast to the isolation work, this synthetic approach provided significantly enough pure material for detailed biological evaluation (vide infra), providing 5 mg of both cleistriosides and between 5 and 43 mg of the cleistetrosides (Scheme 3).
Scheme 3. Divergent Approach to the Cleistriosides and Cleistetrosides 1–8.
In addition to the six naturally occurring cleistetrosides, this route was amenable to the synthesis of two unnatural cleistetroside analogues 9 and 10 (Scheme 4). The new route begins with the same trisaccharide intermediate 17, which was reduced, chloroacylated, and dihydroxylated to form diol 18. A regioselective acylation, glycosylation, reduction, acylation, and dihydroxylation converted 18 into tetrasaccharide 19 in 48% yield.17,18 Finally the two chloroacetates [(NH2)2CS/NaHCO3/Bu4NI] and acetonide (AcOH/H2O) were removed to give cleistetroside analogue 9 in 83% yield. Similarly, 18 can be converted into 20 via a regioselective acylation, glycosylation, reduction, and dihydroxylation (58% yield for four steps). Finally, a per-acylation followed by a chloroacetate and acetonide deprotection gave cleistetroside analogue 10 in 93% yield.
Scheme 4. Approach to the Cleistetrosides 9 and 10.

Reagents and conditions: (a) NaBH4, CeCl3, CH2Cl2/MeOH, −78 °C, 2 h. (b) (ClAc)2O, DMAP, Py, rt, 3 h. (c) OsO4, NMO, t-BuOH/acetone, rt, 12 h. (d) CH3C(OMe)3, p-TsOH, CH2Cl2, 0 °C, 20 min, then AcOH(aq), 20 min. (e) 12, 5 mol % Pd(PPh3)2, CH2Cl2, 0 °C, 2 h. (f) Ac2O, DMAP, Py, rt, 3 h. (g) (NH2)2CS, NaHCO3, Bu4NI, THF, 55 °C, 2 h. (h) 80% AcOH, 80 °C, 2 h. Pd(PPh3)2 = Pd2(dba)3·CHCl3/4PPh3.
With the 10 target oligosaccharides in hand, we next set out to evaluate the structure–activity relationship (SAR) with respect to their activity against three different bacteria strains. We chose two Escherichia coli strains (MG1655 and imp)19 and one Bacillus subtilis strain (JH642). Of the two E. coli strains, the first (K-12 MG1655) is the wild-type Gram-(−) organism and the second E. coliimp (K-12 BAS901) is a Gram-(+)-like strain, which has been genetically modified (imp-4213) to increases the permeability of the outer membrane.20 The B. subtilis strain (JH642) is a Gram-(+) organism. Consistent with the findings of Hu et al.,3 we found both of the cleistriosides and all of the cleistetrosides were inactive against wild-type E. coli, which suggests that the oligosaccharides are not getting past the Gram-(−) organism cell wall. Further evidence along these lines came from the screening against the Gram-(+) like strain (imp), with three of the oligosaccharides showing improved activity. Improved MIC values were found for one of the natural trisaccharides (2, 32 μM) and one tetrasaccharide (6, 16 μM). Interestingly, of the two unnatural analogues (9 and 10), cleistetroside-new 1 (9, 16 μM) showed similar activity to 6, which differs by only one acetate (R4). As was suggested from the E. coli study, we found significantly greater activity against the B. subtilis strain, where the activities ranged from 4 to >64 μM.
Once again, cleistrioside-6 (2) was the more active of the two trisaccharide natural products, whereas significantly different trends were found for the tetrasaccharides 3–10. Of the cleistetrosides, only two were inactive against B. subtilis (8 and 10, >64 μM), with the rest having MICs ≤ 8 μM. In particular, cleistetroside-2 and -6 (3 and 7) were found to be the most active (4 μM). As with the Gram-(+)-like E. coliimp, the unnatural cleistetroside 9 was more active than 10 against B. subtilis.
Because the related acylated oligo-rhamnoside natural products were reported to possess anticancer activity,21 we decided to test the set of 10 oligosaccharides for their cytotoxicity against the human lung cancer cell line (NCI-H460). We found IC50 values for this class of natural products at similar concentrations (7.5–90.9 μM, Table 3) as the MIC for the Gram-(+) B. subtilis strain (4–>64 μM, Table 2). Once again, of the two trisaccharides, cleistrioside-6 (2) was the most active. In contrast to the antibacterial activity, we found all of the cleistetrosides to have significant cytotoxicity (<20 μM). As we have found for anti-B. subtilis activity, we found cleistetrosides-6 (7) to be the most active (7.5 μM), with cleistetrosides-2 (3) showing similar activity (9.1 μM).
Table 3. Cancer Cytotoxicity of the 10 Oligosaccharides on NCI-H460 Human Lung Cancer Epithelial Cellsa.
| compd | IC50 (μM) | compd | IC50 (μM) |
|---|---|---|---|
| 1 | 90.9 | 6 | 15.4 |
| 2 | 12.5 | 7 | 7.5 |
| 3 | 9.1 | 8 | 16.4 |
| 4 | 12.8 | 9 | 16.5 |
| 5 | 12.2 | 10 | 9.8 |
The results are an average of at least three independent experiments.
Table 2. MIC Values for Cleistrio/Cleistetroside Derivativesa.
| MIC (μM) |
|||||||
|---|---|---|---|---|---|---|---|
|
E. coli |
B. subtilis | ||||||
| R1 | R2 | R3 | R4 | MG1655 | imp | JH642 | |
| 1 | H | Ac | >64 | >64 | 32 | ||
| 2 | Ac | H | >64 | 32 | 8 | ||
| 3 | Ac | H | H | Ac | >64 | >64 | 4 |
| 4 | Ac | Ac | H | H | >64 | >64 | 8 |
| 5 | Ac | H | H | H | >64 | >64 | 8 |
| 6 | H | H | H | H | >64 | 16 | 8 |
| 7 | Ac | Ac | H | Ac | >64 | >64 | 4 |
| 8 | Ac | Ac | Ac | Ac | >64 | >64 | >64 |
| 9 | H | H | H | Ac | >64 | 16 | 8 |
| 10 | H | Ac | Ac | Ac | >64 | >64 | >64 |
Antibacterial activity was assessed by MIC method, which was performed by the broth dilution method described by the National Committee for Clinical Laboratory Standard Methods (M7-A6, 2003) (see the Supporting Information).
The fact that cleistetrosides-2 and -6 (3 and 7) are the most active and cleistetroside-7 (8) is the least active in both the anti-B. subtilis and the anticancer screens suggests the possibility that both the anti-B. subtilis and the anticancer activities for this class of natural products result from the same molecular target or mode of action. To challenge this trend, we were inspired to prepare the two unnatural cleistetrosides (9 and 10), which had the C-4 acetate (R1, Ac to H) in the third rhamnose sugar removed. The removal of the same acetate in the cleistriosides (R4) was found to improve activity. Thus, cleistetroside-new 1 (9) is analogous to the quite potent cleistetroside-2 (3), and cleistetroside-new 2 (10) is analogous to least potent cleistetrosides 7 (8) with the C-4 acetates having been removed. The testing of compounds 9 and 10 clearly demonstrates the importance of the R1 group to cleistetroside SAR. For instance, the removal of the acetate in cleistetroside-2 (3) had a small but negative effect on its potency in both screens. In contrast, cleistetroside-new 2 (10) possessed dramatically different effects on its anti-B. subtilis activity (>64 μM) as compared to anticancer activity (<10 μM). Thus, with the synthesis and testing of only two new structures, we were able to find a compound that contradicted the correlation between anti-B. subtilis and anticancer activity. This, of course, could result from different modes of action and/or different esterase activities in bacterial versus human cancer cells.
While there is not enough information to fully understand the effects of the acetate groups, clearly, the location of the individual acetate groups has a significant effect on both the anticancer and the anti-B. subtilis activity. In contrast to the tetrasaccharides, this effect can easily be seen in cleistriosides-5 and -6 (1 and 2), where the simple migration of a C-4 acetate group on the terminal sugar in 1 to the C-3 position in 2 dramatically increases its cytotoxicity to both B. subtilis and cancer cells. Interestingly, the most active tetrasaccharides (3, 7, and 10) have a C-4 acetate and are glycosylated at the C-3 position.
To gain a more complete understanding of the anticancer activities, the set of 10 acylated oligo-rhamnosides (1–10) were screened against the NCI panel of 60 cell lines. Of the 10 compounds screened, three (5, 6, and 9) did not pass the initial single dose assay. A representative subset (two from the nine tissue types) of these results is outlined in Table 4 (for the results of the full 60 cell lines, see the Supporting Information). Because these experiments were run under different conditions (e.g., time) and measure different parameters (e.g., protein concentrations vs mitochondrial activity), it is difficult to directly compare the activities. However, some interesting trends can be seen as well as differences. For instance, the data from the NCI screen and our H460 MTT assay both show compounds 3, 7, and 10 to have similar anticancer activity, with 8 being an outlier. Similarly, the unnatural tetrasaccharide 10 was significantly more active than 9. In contrast to our data for NCI-H460, the NCI screen found tetrasaccharide 8 to be one of the most active against the full panel of cell lines, including the NCI-H460. This discrepancy presumably arises from the subtle differences in the two protocols.
Table 4. Growth Inhibition GI50 Evaluation of Cleistrioside and Cleistetroside Analogues (μM)a.
| cell type | cell line | 1 | 2 | 3 | 4 | 7 | 8 | 10 |
|---|---|---|---|---|---|---|---|---|
| leukemia | RPMI-8226 | 4.12 | 1.00 | 1.86 | 2.84 | 0.96 | 1.04 | 0.92 |
| SR | 3.20 | 1.20 | 1.45 | 3.83 | 0.86 | 0.92 | 0.85 | |
| nonsmall cell lung cancer | A549 | 6.69 | 2.08 | 9.47 | 8.18 | 1.18 | 0.80 | 0.97 |
| NCI-H460 | 6.32 | 1.54 | 7.58 | 8.43 | 0.94 | 0.93 | 0.96 | |
| colon cancer | HCT-15 | 8.39 | 7.38 | 7.54 | 8.33 | 1.01 | 0.82 | 1.52 |
| HT-29 | 4.35 | 1.21 | 8.02 | 8.13 | 0.97 | 0.91 | 0.91 | |
| CNS cancer | SF-539 | 7.11 | 3.10 | 4.70 | 7.17 | 0.83 | 0.80 | 0.91 |
| SNB-75 | 3.42 | 0.90 | 1.36 | 2.18 | 0.50 | 0.54 | 0.62 | |
| melanoma | MDA-MB-435 | 7.68 | 1.49 | 6.00 | 7.25 | 0.83 | 0.88 | 0.84 |
| SK-MEL-5 | 6.21 | 1.23 | 1.76 | 6.27 | 0.74 | 0.76 | 0.82 | |
| ovarian cancer | OVCAR-3 | 5.91 | 1.49 | 4.84 | 6.07 | 0.77 | 0.84 | 0.80 |
| ADR-R | 9.29 | 7.92 | 9.15 | 8.90 | 3.38 | 0.92 | 2.24 | |
| renal cancer | A498 | 6.73 | 5.13 | 7.57 | 5.64 | 0.73 | 0.67 | 0.02 |
| CAKI-1 | 8.78 | 6.57 | 8.45 | 9.24 | 1.42 | 0.86 | 0.60 | |
| prostate cancer | PC-3 | 2.37 | 0.89 | 5.30 | 6.55 | 0.82 | 0.71 | 0.75 |
| DU-145 | 7.94 | 2.48 | 7.92 | 7.47 | 0.80 | 0.78 | 0.79 | |
| breast cancer | MCF7 | 4.84 | 1.62 | 1.74 | 5.29 | 0.87 | 0.89 | 0.94 |
| BT-549 | 7.90 | 4.54 | 8.46 | 8.39 | 0.94 | 1.05 | 0.88 |
Growth inhibition of 50% (GI50) is calculated from [(Ti – Tz)/(C – Tz)] × 100 = 50, which is the compound concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the compound incubation. Compounds 5, 6, and 9 were excluded because they did not pass the initial signal dose screen (10 μM).
In conclusion, we have demonstrated the utility of a de novo asymmetric synthesis for providing sufficient quantities of material for comprehensive SAR type study. The route was amenable to the synthesis of unnatural oligosaccharide analogues for further SAR studies for both anticancer and antibacterial activities. It is worth noting that these efforts discover previously unreported anticancer activities for this class of acylated oligo-rhamnoside natural products. Further SAR studies along these lines are ongoing and will be reported in due course.
Acknowledgments
We are grateful to Prof. Yon Rojanasakul (West Virginia University) and Dr. Todd A. Stueckle (West Virginia University) for providing NCI-H460 cancer cells and Prof. Kim Lewis (Northeastern University) for the wild-type E. coli and imp strains and Prof. Alan D. Grossman (Massachusetts Institute of Technology) for the B. subtilis strain.
Supporting Information Available
Assay protocols, statistical analysis data, synthetic procedures, characterization data, and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ The order of these authors is alphabetical. The manuscript was written with contributions from all authors. B.W. and M.L. were responsible for the synthesis. P.S. was responsible for compound evaluations with help from M.C.S. and H.-Y.L.W. N.G.A. was responsible for compound characterization.
We thank NIH (GM090259), NSF (CHE-1213596 to G.A.O. and MCB-0845033 to P.J.B.), the American Cancer Society (RSG-12-161-01-DMC to P.J.B.), and the Research Corporation for Science Advancement (Cottrell Scholar for P.J.B.) for their support of our research.
The authors declare no competing financial interest.
Dedication
This work is dedicated to Dr. Josephine O'Doherty on the occasion of her 75th birthday.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tané P; Ayafor J. P.; Sondengam B. L.; Lavaud C.; Massiot G.; Connolly J. D.; Rycroft D. S.; Woods N. Partially-acetylated dodecanyl tri- and tetra-rhamnoside derivatives from Cleistopholis glauca (annonaceae). Tetrahedron Lett. 1988, 29, 1837–1840. [Google Scholar]
- Seidel V.; Baileul F.; Waterman P. G. J. Partially acetylated tri- and tetrarhamnoside dodecanyl ether derivatives from Cleistopholis patens. Phytochemistry 1999, 52, 465–472. [DOI] [PubMed] [Google Scholar]
- Hu J.-F.; Garo E.; Hough G. W.; Goering M. G.; O'Neil-Johnson M.; Eldridge G. R. Antibacterial, Partially Acetylated Oligorhamnosides from Cleistopholis patens. J. Nat. Prod. 2006, 69, 585–590. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Wang P; Ding N.; Song G.; Li Y. Total synthesis of cleistetroside-2, partially acetylated dodecanyl tetrarhamnoside derivative isolated from Cleistopholis patens and Cleistopholis glauca. Carbohydr. Res. 2007, 342, 1159–1168. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Chen Q.; Du Y. Facile synthesis of cleistetroside-2, a partially acetylated oligorhamnoside from Cleistopholis glauca and patens. Carbohydr. Res. 2007, 342, 1496–1501. [DOI] [PubMed] [Google Scholar]
- Wu B.; Li M.; O'Doherty G. A. Synthesis of Several Cleistrioside and Cleistetroside Natural Products via a Divergent De Novo Asymmetric Approach. Org. Lett. 2010, 12, 5466–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parakkottil Chothi M.; Duncan G. A.; Armirotti A.; Abergel C.; Gurnon J. R.; Van Etten J. L.; Bernardi C.; Damonte G.; Tonetti M. Identification of an L-Rhamnose Synthetic Pathway in Two Nucleocytoplasmic Large DNA Viruses. J. Virol. 2010, 84, 8829–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O'Doherty G. A. Synthesis of SL0101 Carbasugar Analogues: Carbasugars via Pd-Catalyzed Cyclitolization and Post Cyclitolization Transformations. Org. Lett. 2010, 12, 2986–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O'Doherty G. A. De Novo Asymmetric Synthesis of SL0101 and its Analogues via a Palladium-Catalyzed Glycosylation. Org. Lett. 2006, 8, 5149–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; O'Doherty G. A. De Novo Asymmetric Synthesis of Digitoxin via a Palladium Catalyzed Glycosylation Reaction. Org. Lett. 2006, 8, 4339–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; O'Doherty G. A. De Novo Approach to 2-Deoxy-β-glycosides: Asymmetric Syntheses of Digoxose and Digitoxin. J. Org. Chem. 2007, 72, 2485–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu R. S.; O'Doherty G. A. A Palladium-Catalyzed Glycosylation Reaction: The De Novo Synthesis of Natural and Unnatural Glycosides. J. Am. Chem. Soc. 2003, 125, 12406–12407. [DOI] [PubMed] [Google Scholar]
- Babu R. S.; Zhou M.; O'Doherty G. A. De-Novo Synthesis of Oligosaccharides Using a Palladium Catalyzed Glycosylation Reaction. J. Am. Chem. Soc. 2004, 126, 3428–3429. [DOI] [PubMed] [Google Scholar]
- Babu R. S.; O'Doherty G. A. Palladium Catalyzed Glycosylation Reaction: De-Novo Synthesis of Trehalose Analogues. J. Carbohydr. Chem. 2005, 24, 169–177. [Google Scholar]
- Babu R. S.; Chen Q.; Kang S.-W.; Zhou M.; O’Doherty G. A. De Novo Synthesis of Oligosaccharides Using Green Chemistry Principles. J. Am. Chem. Soc. 2012, 134, 11952–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; Xing Y.; O'Doherty G. A. De Novo Asymmetric Synthesis of an α-6-Deoxyaltropyranoside as Well as its 2-/3-Deoxy and 2,3-Dideoxy Congeners. J. Org. Chem. 2009, 74, 5961–5966. [DOI] [PubMed] [Google Scholar]
- Guo H.; O'Doherty G. A. De Novo Asymmetric Synthesis of the Anthrax Tetrasaccharide via a Palladium Catalyzed Glycosylation Reaction. Angew. Chem., Int. Ed. 2007, 46, 5206–5208. [DOI] [PubMed] [Google Scholar]
- Guo H.; O'Doherty G. A. De Novo Asymmetric Synthesis of Anthrax Tetrasaccharide and Analogue. J. Org. Chem. 2008, 73, 5211–5220. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Mulzer M.; Shi P.; Beuning P. J.; Coates G. W.; O’Doherty G. A. De Novo Asymmetric Synthesis of Fridamycin E. Org. Lett. 2011, 13, 6592–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson B. A.; Misr R.; Benson S. A. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 1989, 122, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B.; Chai H.; Santisuk T.; Reutrakul V.; Farnsworth N. R.; Cordell G. A.; Pezzuto J. M.; Kinghorn A. D. Novel Cytotoxic Acylated Oligorhamnosides from Mezzettia leptopoda. J. Nat. Prod. 1998, 61, 1535–1538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.