

Abstract
Background
Mutations in SACS, leading to autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), have been identified as a frequent cause of recessive early-onset ataxia around the world. Here we aimed to enlarge the spectrum of SACS mutations outside Quebec, to establish the pathogenicity of novel variants, and to expand the clinical and imaging phenotype.
Methods
Sequencing of SACS in 22 patients with unexplained early-onset ataxia, assessment of novel SACS variants in 3.500 European control chromosomes and extensive phenotypic investigations of all SACS carriers.
Results
We identified 11 index patients harbouring 17 novel SACS variants. 9/11 patients harboured two variants of at least probable pathogenicity which were not observed in controls and, in case of missense mutations, were located in highly conserved domains. These 9 patients accounted for at least 11% (9/83) in our series of unexplained early onset ataxia subjects. While most patients (7/9) showed the classical ARSACS triad, the presenting phenotype reached from pure neuropathy (leading to the initial diagnosis of Charcot-Marie-Tooth disease) in one subject to the absence of any signs of neuropathy in another. In contrast to its name “spastic ataxia”, neither spasticity (absent in 2/9=22%) nor extensor plantar response (absent in 3/9=33%) nor cerebellar ataxia (absent in 1/9=11%) were obligate features. Autonomic features included urine urge incontinence and erectile dysfunction. Apart from the well-established MRI finding of pontine hypointensities, all patients (100%) showed hyperintensities of the lateral pons merging into the (thickened) middle cerebellar peduncles. In addition, 63% exhibited bilateral parietal cerebral atrophy, and 63% a short circumscribed thinning of the posterior midbody of the corpus callosum. In 2 further patients with differences in important clinical features, VUS class 3 variants (c.1373C>T [p.Thr458Ile] and c.2983 G>T [p.Val995Phe]) were identified. These variants were, however, also observed in controls, thus questioning their pathogenic relevance.
Conclusions
We here demonstrate that each feature of the classical ARSACS triad (cerebellar ataxia, spasticity and peripheral neuropathy) might be missing in ARSACS. Nevertheless, characteristic MRI features – which also extend to supratentorial regions and involve the cerebral cortex – will help to establish the diagnosis in most cases.
Keywords: Ataxia, Recessive ataxia, Spastic ataxia, Early onset ataxia, Spasticity, Genetics, Magnetic resonance imaging, Electrophysiology, Thin corpus callosum, Charcot Marie Tooth, Hereditary spastic paraplegia
Background
Autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) was for long regarded as a subtype of early onset ataxia restricted to the Quebec region due to founder mutations from a French ancestor. After cloning of the responsible gene, SACS, it became evident that ARSACS is not limited to Quebec, and more than 100 different pathogenic mutations have now been identified worldwide [1]. Most ARSACS patients show a typical triad of early-onset cerebellar ataxia, lower limb spasticity and peripheral neuropathy. Together with characteristic T2-hypointensities in the pons [2,3], these phenotypic features help to identify ARSACS in patients with unexplained ataxia. However, three mutation-proven individuals with an unusual phenotype (lacking either spasticity or peripheral neuropathy) have been described so far [1,4,5], indicating that the classical triad might not be the presenting feature in all ARSACS patients, and that ARSACS might be underdiagnosed in patients with atypical phenotypes. Moreover, the presumed high frequency of allele carriers around the world [6] suggests that many subjects will be identified in the future with SACS variants of unknown significance, thus warranting the identification of further phenotypic features that help to establish the clinical diagnosis.
Here we aimed (i) to expand the spectrum of SACS mutations outside Quebec and to establish the pathogenicity of novel variants, (ii) to identify phenotypic characteristics that are shared by most ARSACS patients and that further help to identify ARSACS in patients with unexplained ataxia, spasticity or neuropathy, and (iii) to assess non-classical presenting phenotypes that indicate additional disease groups, in which mutational screening of SACS should be considered in the molecular genetic work-up.
Methods
From 2005 until 2012 we recruited a consecutive series of 172 index patients in the ataxia outpatient clinics of the Departments of Neurology and Pediatric Neurology at the University Clinic in Tübingen (Germany) with (i) progressive degenerative ataxia with age of onset ≤30 years, (ii) absence of ataxia in the parental generation, and (iii) genetic exclusion of common subtypes of dominant ataxias (SCA 1, 2, 3, 6, 7, and 17). Mutations causing Friedreich-, POLG- and C10orf2 (PEO1/Twinkle)-ataxia were screened in the whole cohort. A molecular diagnosis could not be established in 83/172 patients (48%). 22 of these 83 patients with unexplained ataxia were screened for ARSACS by direct sequencing of the SACS gene (for details, see Additional file 1). These 22 patients were selected for direct sequencing by 2 criteria: (1) pyramidal tract signs to the lower extremity in addition to the degenerative ataxia (20/22 patients); or (2) variant abnormalities including the SACS region detected by preceding homozygosity mapping and/or whole-exome sequencing approaches (2/22 patients). In all index patients with only one heterozygous or apparently homozygous SACS variants, macro-deletions were excluded by MLPA (see Additional file 1 for details). Following earlier recommendations on variant classification and reporting [7], the likelihood of pathogenicity of novel sequence variants was graded according to a 5-class-system: class 1 = not pathogenic; class 2 = variant of uncertain clinical significance (VUS), likely not pathogenic; class 3 = VUS, uncertain pathogenicity; class 4 = VUS, likely pathogenic; class 5 = definitely pathogenic [7]. Only patients harbouring two variants of class 3 or higher are reported here (see Additional file 1 for details on variant classes and assignment of variants). The frequency of the identified SACS variants was determined in 3.500 European control chromosomes from individuals with unrelated phenotypes analyzed by exome sequencing (for methodological details, see [8]) in Munich, Germany. In addition, the frequency was assessed in the exome variant server (EVS) of the National Heart, Lung, and Blood Institute GO Exome Sequencing Project (Seattle, WA, USA; URL: evs.gs.washington.edu/EVS/) with the corresponding nucleotide positions being analyzed in >8.000 European and >4.000 African American alleles and in the Database of Single Nucleotide Polymorphisms (dbSNP; Bethesda: National Center for Biotechnology Information, National Library of Medicine [dbSNP Build ID: 137]; available from: http://www.ncbi.nlm.nih.gov/SNP/). In-silico predictions of the pathogenicity of point mutations were performed using PolyPhen-2 [9], SIFT [10] and MutationTaster [11]. Cosegregation of novel variants was confirmed in all families with a second affected family member. Compound heterozygosity was confirmed by segregation analysis where parental DNA was available.
Each mutation carrier was clinically characterized by a movement disorder specialist and investigated by nerve conduction studies (NCS), motor evoked potentials (MEP), standard funduscopy, and routine magnetic resonance imaging (MRI) including T1-, T2-, diffusion-weighted images (DWI) and fluid attenuated inversion recovery T2 (FLAIR) images. The presence vs. absence of thinning of the corpus callosum and of atrophy of the cerebellar and parietal cortex were evaluated independently by two examiners (M.S.; B.B.), and the inter-rater reliability was determined by Cohen’s kappa coefficient. Its value was interpreted following Landis and Koch [12]. In case of disagreement, the more conservative assessment (i.e. the absence of thinning and atrophy, respectively) was adopted for final judgement.
Results
Genetic spectrum
In total, 19 different SACS variants were identified across different exons (Figure 1). 17 out of 19 variants are novel, 2 have already been identified earlier (p. Arg728*; p.Gln1709*; [6]) (see Table 1). The identified variants comprised 6 nonsense mutations, 5 missense mutations, 5 frameshift mutations, 5 in-frame deletions, and 1 in-frame duplication. These variants were observed in 11 index patients. In three families with known parental consanguinity, homozygous SACS variants were observed (Table 1). Four index patients had a positive family history with affected siblings. Cosegregation of the respective variants was demonstrated in all of them. No macro-deletion or duplications were identified by MLPA.
Figure 1.

SACS mutations. Graphical overview of mutations found in this and other studies (Human Gene Mutation database, except gross deletions). Different mutation types are marked with different symbols: missense mutations = squares; insertions/duplications = triangles with downward orientation; deletions = triangles with upward orientation. Sequence variations identified in this study are coloured, with the colour indicating the pathogenicity class: red = pathogenic, orange = likely pathogenic, yellow = uncertain. Known protein domains of the sacsin protein are highlighted in green. AA = amino acid; HGMD = Human Gene Mutation Database.
Table 1.
SACS mutations
| Family No. | Nucleotide change1 | Amino acid change | Mutation type | Exon | Allele frequency in 1846 controls | dbSNP | EVS | PhyloP2 [-14,1;6,4] | Grantham distance3 [0–215] | In-silico predictions4 | Novel variant | Pathogenicity class5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Patients with 2 pathogenic or likely pathogenic SACS variants |
|
|
|
|
|
|
|
|
|
|
|
|
|
#1 index |
c.9305_9306insT |
p.Leu3102Phefs*8 |
frame shift |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Pathogenic (class 5) |
| |
c.9305_9306insT |
p.Leu3102Phefs*8 |
|
|
|
|
|
|
|
|
|
|
| #1 sister |
see above |
|
|
|
|
|
|
|
|
|
|
|
|
#2 |
c.2182C>T |
p.Arg728* |
nonsense |
9 |
0 |
- |
- |
ND |
NA |
NA |
no6 |
Pathogenic (class 5) |
| |
c.13056delT |
p.Phe4352Leufs*11 |
frame shift |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Pathogenic (class 5) |
|
#3 index |
c.3769G>T |
p.Gly1257* |
nonsense |
10 |
0 |
- |
- |
ND |
NA |
NA |
yes |
Pathogenic (class 5) |
| |
c. 8584A>T |
p.Lys2862* |
nonsense |
10 |
0 |
- |
- |
ND |
NA |
NA |
yes |
Pathogenic (class 5) |
| #3 sister |
see above |
|
|
|
|
|
|
|
|
|
|
|
|
#4 index |
c.11542_11544del |
p.Ile3848del |
in-frame deletion |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Likely pathogenic (VUS class 4) |
| |
c.11542_11544del |
p.Ile3848del |
|
|
|
|
|
|
|
|
|
|
| #4 brother |
see above |
|
|
|
|
|
|
|
|
|
|
|
|
#5 index |
c.2387delT |
p.Leu796Tyrfs*13 |
frame shift |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Pathogenic (class 5) |
| |
c.11984_11986 dupTGT |
p.L3995dup |
in-frame duplication |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Likely pathogenic (VUS class 4) |
| #5 sister |
see above |
|
|
|
|
|
|
|
|
|
|
|
| #6 |
c.11624 G>A |
p.Arg3875His |
missense |
10 |
0 |
- |
- |
6.34 |
29 |
Deleterious |
yes |
Likely pathogenic (VUS class 4) |
| |
c.1647_1658del |
p.Leu549_Leu552del |
In-frame deletion |
8 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Likely pathogenic (VUS class 4) |
| |
c.13538 G>A |
p.Ser4513Asn |
missense |
10 |
0 |
rs138328181 (0% ESP Cohort pop) |
0.02% (EA), 0% (AA) |
3.43 |
46 |
Contradictory |
yes |
Uncertain (VUS class 3) |
| #7 |
c.5544dupA |
p.Val1849Serfs*48 |
frame shift |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Pathogenic (class 5) |
| |
c.12603C>A |
p.Tyr4201* |
nonsense |
10 |
0 |
- |
- |
ND |
NA |
NA |
yes |
Pathogenic (class 5) |
| #8 |
c.7277 G>C |
p.Arg2426Pro |
missense |
10 |
0 |
- |
- |
5.69 |
103 |
Deleterious |
yes |
Likely pathogenic (VUS class 4) |
| |
c.8920_8923dup |
p.Tyr2975Phefs*29 |
frame shift |
10 |
0 |
- |
- |
NA |
NA |
NA |
yes |
Pathogenic (class 5) |
| #9 |
c.4954C>T |
p.Gln1652* |
nonsense |
10 |
0 |
- |
- |
ND |
NA |
NA |
yes |
Pathogenic (class 5) |
| |
c.5125C>T |
p.Gln1709* |
nonsense |
10 |
0 |
- |
- |
ND |
NA |
NA |
no6 |
Pathogenic (class 5) |
|
Patients with 2 SACS variants of uncertain clinical significance class 3 (VUS3) |
|
|
|
|
|
|
|
|
|
|
|
|
| #10 |
c.1373C>T |
p.Thr458Ile |
missense |
8 |
14/3500 |
rs61729954 (0.3% ESP Cohort pop; 3.1% AGI ASP pop) |
0.33% (EA); 0.05% (AA) |
6.26 |
89 |
Deleterious |
yes |
Uncertain (VUS class 3) |
| |
c.1373C>T |
p.Thr458Ile |
|
|
|
|
|
|
|
|
|
|
| #11 |
c.1373C>T (see patient #10) |
p.Thr458Ile |
missense |
8 |
14/3500 |
rs61729954 (0.3% ESP Cohort pop; 3.1% AGI ASP pop) |
0.33% (EA); 0.05% (AA) |
6.26 |
89 |
Deleterious |
yes |
Uncertain (VUS class 3) |
| c.2983 G>T | p.Val995Phe | missense | 10 | 11/3500 | rs142967124 (0.1% ESP Cohort pop) | 0.24% (EA); 0.02% (AA) | -0.2 | 50 | Contradictory | yes | Uncertain (VUS class 3) |
EA= European alleles; AA= African American alleles; NA = not applicable; ND = not determined; rs = reference single nucleotide polymorphism (SNP) identifier; VUS = variant of uncertain clinical significance. 1 Nomenclature of sequence variants follows the recommendations by the Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen/) and refers to the cDNA sequence NM_014363.4. 2 PhyloP was used as a conservation score as originally implemented by Pollard and colleagues. It allows the rating of nucleotides from “not conserved” (-14.1) to “highly conserved” (6.4) [13]. 3 The Grantham distance was used to evaluate physico-chemical differences of amino acids (0 = no physico-chemical differences; 215 = large differences) [14]. 4 In-silico predictions were performed using PolyPhen-2 [9], SIFT [10] and MutationTaster [11]5 Classification of novel sequence variants is according to [7]. 6 Reported in [6].
Fourteen of the novel 17 variants were not observed in controls and in the EVS (Table 1) and - in case of missense mutations -were evolutionary highly conserved (for conservation score PhyloP, see Table 1; for alignment data, see Additional file 2, each only for missense mutations). Two SACS variants - p.Thr458Ile and p.Val995Phe - were also observed in heterozygous state in 3.500 control chromosomes (p.Thr458Ile: 14/3.500=0.4%; p.Val995Phe: 11/3.500=0.3%), in dbSNP (p.Thr458Ile: 0.3% ESP Cohort pop; rs61729954; p.Val995Phe: 0.1% ESP Cohort pop; rs142967124) and in the exome variant server (p.Thr458Ile: 0.33% European alleles; p.Val995Phe: 0.24% European alleles) (see Table 1). The p.Thr458Ile variant was predicted to be deleterious by three different in silico software tools (PolyPhen-2, SIFT, MutationTaster). It leads to a change of an evolutionary highly conserved amino acid (PhyloP Score: 6.26) (Table 1 and Additional file 2). However, the physico-chemical differences of the involved amino acids are only moderate. This finding together with their occurrence in the general population leads to the classification as VUS class 3 (uncertain pathogenicity). The p.Val995Phe variant is predicted to be deleterious only by SIFT, and affects an evolutionary less conserved amino acid (PhyloP Score: -0.2) (Table 1 and Additional file 2), thus also leading to the classification as VUS class 3. Due to the frequent observation of these two variants in controls and their uncertain significance, the two patients harbouring either one of these variants (patient #10: p.[Thr458Ile];[Thr458Ile]) or the combination of them (patient #11: p.[Thr458Ile]; [Val995Phe]) were analyzed separately from the other 9 patients with two SACS variants of ≥ class 4.
One SACS variant – c.13538 G>A (p.Ser4513Asn) - was not observed in controls, but was found in the EVS (0.02%) and was only moderately conserved. This variant was found in a patient with two additional SACS variants (patient #6), namely c.11624 G>A (p.Arg3875His) and c.1647_1658del (p. Leu549_Leu552del). Only these two variants, but not the first variant, were considered to be likely pathogenic (VUS class 4), since they were absent in controls and the EVS and were highly evolutionary conserved. Unfortunately, DNA of the parents was not available for testing allele segregation.
In one index patient (patient #2), an autosomal-dominant ataxia had initially been assumed, based on the positive family history for a three-generation late-onset (>30 years), slowly progressive, pure cerebellar autosomal-dominant ataxia in her paternal relatives (in a aunt, cousin, and grandfather; or pedigree see Additional file 3). However, the father of the index patient did not show any ataxia (examined at age 80 years by M.S.) and the index patient herself showed a strikingly different early-onset ataxia. To rule out a possible digenic ataxia, also the following SCAs were excluded in the index patient: SCA 8,10, 11, 14, 15, 27 and DRPLA. The affected paternal aunt and cousin did not show any of the two (clearly pathogenic) SACS variants that were observed in the index patient. This illustrates that two different hereditary ataxias should be considered in pedigrees where the ataxia features differ strongly between pedigree members, e.g. early onset vs. late onset ataxia or multisystemic vs. pure cerebellar degeneration.
Phenotypic spectrum of index patients with two ARSACS variants of at least probable pathogenicity
78% (7/9) of the 9 index patients with at least two variants of VUS class 4 (likely pathogenic [7]) or pathogenic mutations showed the typical ARSACS phenotype of typical early-onset triad of cerebellar ataxia, lower limb pyramidal tract signs (spasticity and extensor plantar response in 6/7; only spasticity in 1/7 [patient #9]) and peripheral neuropathy, which was of axonal-demyelinating sensorimotor type in all (7/7) subjects (Table 2). 1/9 patients (patient #4) showed no spasticity, extensor plantar response or cerebellar ataxia, but marked axonal-demyelinating sensorimotor neuropathy (leading to mild subclinical afferent ataxia). The same was true for his likewise affected brother. In turn, in another patient (patient #8), cerebellar ataxia was present, but electrophysiological evidence of peripheral neuropathy absent. Neither did this patient show spasticity or an extensor plantar response. Thus, neither spasticity (absent in 2/9=22%) nor extensor plantar response (absent in 3/9=33%) nor cerebellar ataxia (absent in 1/9=11%) are obligate features in ARSACS.
Table 2.
Clinical characteristics of patients with SACS variants
| Family No. | SACS variant | Ethnical background | Current age (y)/sex | Age at onset (y) | First clinical sign | Current SARA | Cerebellar ataxia | Areflexia muscle tendon reflexes | Pyramidal features | Autonomic signs |
|---|---|---|---|---|---|---|---|---|---|---|
|
patients with 2 likely pathogenic SACS variants |
|
|
|
|
|
|
|
|
|
|
|
#1 index |
p.[Leu3102Phefs*8];[Leu3102Phefs*8] |
Turkey |
31/f |
1 |
delayed motor development |
12 |
Dysarthria, nystagmus, dysmetria UE |
ATR |
spasticity LE>UE; Bab +/+ |
urge incontinence urine |
| #1 sister |
see above |
30/f |
11 |
gait disturbance |
10 |
Nystagmus, dysmetria UE+LE |
ATR |
spasticity LE>UE; Bab +/+ |
urge incontinence urine |
|
|
#2 |
p.[Arg728*];[Phe4352Leufs*11] |
German |
38/f |
6 |
gait disturbance |
22 |
nystagmus, dysmetria UE+LE |
ATR |
spasticity, Bab +/+ |
urge incontinence urine |
|
#3 index |
p.[Gly1257*]; [Lys2862*] |
Greek |
40/m |
18 |
gait disturbance |
22 |
nystagmus, dysarthria, dysmetria UE+LE |
- |
spasticity, Bab +/+ |
urge incontinence urine |
| #3 sister |
see above |
36/f |
15 |
gait disturbance |
n.d. |
n.k. |
n.k. |
n.k. |
urge incontinence urine |
|
|
#4 index |
p.[Ile3848del]; [Ile3848del] |
Turkey |
13/m |
3 |
generalized muscle weakness, pes planus |
2 |
- |
- |
none; Bab -/- |
- |
| #4 brother |
see above |
|
6/m |
|
pes planus |
1 |
- |
- |
none; Bab -/- |
- |
|
#5; index |
p.[Leu796Tyrfs*13];[L3995dup] |
German |
27/f |
2 |
gait disturbance |
25 |
nystagmus, dysmetria UE+LE |
ATR |
spasticity, Bab +/+ |
urge incontinence urine & faeces |
| #5, sister |
see above |
25/f |
2 |
gait disturbance |
n.a. |
Dysarthria, dysmetria LE > UE |
ATR |
none, Bab -/- |
urge incontinence urine |
|
| #6 |
p.[Arg3875His]; [Leu549_Leu552del]; [Ser4513Asn] |
Macedonia |
30/m |
1 |
gait disturbance |
9 |
dysmetria UE+LE - |
ATR |
spasticity, Bab+/+ |
- |
| #7 |
p.[Val1849Serfs*48]; [Tyr4201*] |
German |
50/f |
28 |
gait disturbance |
n.a. |
nystagmus, dysarthria, dysmetria UE+ UE |
ATR |
spasticity; Bab +/+ |
urge incontinence |
| #8 |
p.[Arg2426Pro]; [Tyr2975Phefs*29] |
German |
6/m |
2 |
gait disturbance |
9 |
nystagmus, dysarthria, dysmetria UE+LE |
- |
none; Bab -/- |
incontinence |
| #9 |
p.[Gln1652*]; [Gln1709*] |
German |
41/m |
2 |
gait disturbance |
n.a. |
nystagmus, dysarthria, dysmetria UE+LE |
ATR |
spasticity; Bab -/- |
urge incontinence, erectile dysfunction |
|
patients with 2 SACS variants of unknown significance |
|
|
|
|
|
|
|
|
|
|
| #10 |
p.[Thr458Ile]; [Thr458Ile] |
Turkey |
44/m |
30 |
gait disturbance |
8 |
nystagmus, slight dysmetria UE + LE, no dysarthria |
- |
none; Bab -/- |
urge incontinence |
| #11 | p.[Thr458Ile]; [Val995Phe] | German | 43/m | 20 | gait ataxia | 21 | cerebellar oculomotor disturbance, dysarthria, dysphagia, intention tremor, dysmetria UE+LE | - | spasticity; Bab+/+ | - |
n.a.= not available; n.k., not known; y= years; m=male, f=female; ATR, Achilles Tendon Reflex; Bab=babinski sign right/left; SARA= Scale for the Assessment and Rating of Ataxia (according to [15]).
Urinary urge incontinence was present in 7/9 patients (77%), combined with faecal urge incontinence in 1 patient; erectile dysfunction was present in 1/3 male patients aged ≥16 years (33%) (Table 2). Standard funduscopy (available from 6/9 patients) revealed increased demarcation of retinal nerve fibres close to the optic disc in 1/6 (17%) patients (patient #5). No patient reported hearing loss (0%). MEPs (available from 8/9 patients) were abnormal to the legs in all patients (not evoked in 7/8, prolonged central motor conduction time in 1/8) and to the arms in 75% (prolonged central motor conduction time in all 6/8 patients), indicating that pyramidal damage does not only affect lower but also upper extremities in ARSACS (Table 3). This was true also for patient #4, in whom no clinical signs of pyramidal damage were observed, thus indicating that electrophysiology may reveal subclinical pyramidal damage in ARSACS.
Table 3.
Electrophysiology and MRI findings
| family No. | SACS variants | Sensory nerve conduction sural or radial nerve | Motor nerve conduction tibial or ulnar nerve | Motor evoked potentials | Cerebellar atrophy | Arachnoid cyst posterior fossa | T2 hypo-intensities pons | T2 hyper-intensities lateral pons/MCP | Thickening of MCP | Bilateral parietal atrophy | Thin corpus callosum (MRI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
patients with 2 likely pathogenic SACS variants |
|
|
|
|
|
|
|
|
|
|
|
| #1 |
p.[Leu3102Phefs*8];[Leu3102Phefs*8] |
Sur: no SNAP Rad: no SNAP |
Tib:reduced CMAP+MNCV Uln: reduced MNCV |
prolonged UE+LE |
+, vermal |
+ |
+ |
+ |
+ |
+ |
+ |
| #2 |
p.[Arg728*];[Phe4352Leufs*11] |
Sur: no SNAP Rad: no SNAP |
Tib: reduced MNCV Uln: reduced MNCV |
prologned UE, not evoked LE |
+, pancerebellar |
+ |
+ |
+ |
+ |
+ |
+ |
| #3 |
p.[Gly1257*];[Lys2862*] |
Sur: no SNAP Rad: no SNAP |
Tib: no MSAP Uln: reduced MNCV |
prolonged UE, not evoked LE |
+, pancerebellar |
+ |
+ |
+ |
+ |
+ |
+ |
| #4 |
p.[Ile3848del];[Ile3848del] |
Sur: no SNAP Rad: reduced CMAP+SNCV |
Tib: reduced MNCV Uln: reduced MNCV |
prolonged UE, not evoked LE |
-, only asymmetry of cerebellar hemispheres |
+ |
+ |
+ |
+ |
- |
+ |
| #5 |
p.[Leu796Tyrfs*13];[L3995dup] |
Sur: no SNAP Rad: reduced CMAP+SNCV |
Tib: reduced CMAP+MNCV Uln: reduced MNCV |
prolonged UE, not evoked LE |
+, vermal |
+ |
+ |
+ |
+ |
- |
+ |
| #6 |
p.[Arg3875His]; [Leu549_Leu552del]; [Ser4513Asn] |
Sur: reduced CMAP+SNCV Rad: reduced CMAP+SNCV |
Tib: reduced CMAP+MNCV Uln: reduced MNCV |
prolonged UE, not evoked LE |
+, vermal |
+ |
+ |
+ |
+ |
+ |
- |
| #7 |
p.[Val1849Serfs*48]; [Tyr4201*] |
Sur: reduced CMAP+SNCV |
Tib: reduced CMAP+MNCV |
not evoked LE |
+, pancerebellar |
n.a. |
n.a. |
n.a. |
n.a. |
n.a. |
n.a. |
| #8 |
p.[Arg2426Pro]; [Tyr2975Phefs*29] |
Sur: normal |
Tib: normal |
not done |
+, vermal |
- |
+ |
+ |
+ |
- |
- |
| #9 |
p.[Gln1652*]; [Gln1709*] |
Sur: no SNAP Rad: reduced SNAP+SNCV |
Tib: reduced CMAP+MNCV Uln: reduced MNCV |
not evoked LE |
+, pancerebellar |
- |
+ |
+ |
+ |
+ |
- |
|
patients with 2 SACS variants of unknown significance |
|
|
|
|
|
|
|
|
|
|
|
| #10 |
p.[Thr458Ile]; [Thr458Ile] |
Sur: normal |
Tib: normal |
normal UE+LE |
slight vermal atrophy, herniation of tonsils |
+ |
- |
- |
- |
- |
+ |
| #11 | p.[Thr458Ile]; [Val995Phe] | Sur: red SNAP Rad: red SNAP | Tib: red CMAP | prolonged UE+LE | +, pancerebellar | - | - | - | - | + | + |
Sur=sural; rad= radial; Tib= tibial, Uln=ulnar; SNAP=sensory nerve action potential; SNCV=sensory nerve conduction velocity; CMAP=compound muscle action potential; MNCV=motor nerve conduction velocity; UE=upper extremity, LE= lower extremity; MCP=middle cerebellar peduncles; n.a.= not available.
MRI (available from 8/9 patients) demonstrated cerebellar atrophy with predominance of the superior cerebellar vermis in 88% (7/8) (Table 3 and Figure 2). Interestingly, the single patient who was lacking cerebellar atrophy, did also not show any clinical signs of cerebellar dysfunction (but only mild afferent ataxia) (patient #4). His MRI did reveal, however, developmental asymmetry of the cerebellar hemispheres (Figure 3). All patients (8/8, 100%) showed linear pontine T2-hypointensities, and, in addition, also bilateral FLAIR-T2-hyperintensities of the lateral pons when merging into the middle cerebellar peduncles (MCP) as well as thickening of the MCP (Figure 2). 75% (6/8) of the patients exhibited an arachnoidal cyst in the posterior fossa, 63% (5/8) bilateral parietal cerebral atrophy (Figure 2), and 63% (5/8) a short-stretched thinning of the posterior mid-body of the corpus callosum (Figure 2). While interrater reliability for rating of cerebellar atrophy (Cohens’s kappa: 1) and parietal atrophy (Cohen’s kappa: 0.8) was very good, it was only moderate for thinning of the posterior mid-body corpus callosum (Cohen’s kappa: 0.6). The lower interrater reliability with respect to the corpus callosum, however, does not lead to a possible over-reporting of corpus callosum abnormalities, but rather to a possible underestimation as in case of disagreement the more conservative estimate was adopted for final judgement.
Figure 2.

Characteristic MRI findings in ARSACS. Axial FLAIR images of upper pons (A) and middle pons (B) show hypointense strips in the central pons representing the cortico-spinal tract (white arrows): Moreover, on the level of the mid-pons (B), they reveal diffuse slight hyperintensity of the lateral pons when merging into the middle cerebellar peduncles (stars) and thickened middle cerebellar peduncles (white arrow head) in patient #1. Also on coronal T2 images (C) the bilateral hyperintensity of the lateral pons can be seen (stars). Bilateral post-central and parietal atrophy is shown for the same patient in axial FLAIR (D, E) and sagittal T2 (F) images (white arrows). Thinning of the posterior mid-body of the corpus callosum on sagittal T2 images is shown for patient #5 (G) and patient #2 (H) (white arrows).
Figure 3.
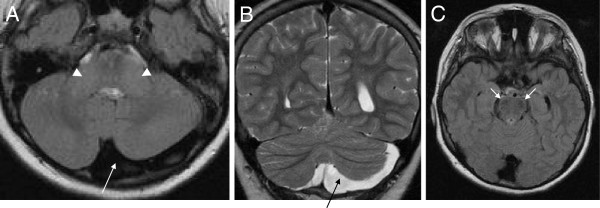
Developmental asymmetry of cerebellar hemispheres. Axial T1 (A) and coronal T2 (B) MRI images of patient #4 demonstrating developmental asymmetry of the cerebellar hemispheres (long arrows) without cerebellar atrophy. Apart from this finding, this patient shows the common infratentorial signs of ARSACS: thickening of the middle cerebellar peduncles (arrow head, A) and linear hypointensities of the mid-pons (best seen on axial FLAIR, arrows, C).
Phenotype of patients with two SACS variants of uncertain clinical significance class 3 (p.Thr458Ile and p. Val995Phe)
Compared to the aforementioned ARSACS patients, the patient with the homozygous mutation p.Thr458Ile had a relatively late onset of ataxia at age 30 years, with absence of clinical and electrophysiological signs of pyramidal damage and neuropathy (no spasticity, no extensor plantar response, normal muscle tendon reflexes, normal vibration sense, normal nerve conduction studies, normal MEPs). Funduscopy revealed no increased demarcation of retinal nerve fibres. Like in other ARSACS patients, MRI revealed minor atrophy of the superior cerebellar vermis, an arachnoid cyst, and a thinning of the posterior mid-body of the corpus callosum (Figure 4). However, in contrast to all aforementioned ARSACS patients, no linear T2-hypointensities in the pons or T2-hyperintensities of the lateral pons and thickening of the MCP were observed. Interestingly, MRI revealed herniation of the cerebellar tonsils, resembling Chiari malformation type 1 (Figure 4). The patient being compound heterozygous for the p.Thr458Ile and p.Val995Phe variants showed an early-onset triad of cerebellar ataxia, pyramidal tract signs (bilateral spasticity and extensor plantar response) and peripheral sensorimotor neuropathy. However, nerve conduction studies revealed pure axonal damage without evidence for a demyelinating component, in contrast to all other ARSACS patients with neuropathy. Moreover, atrophy was observed not only of the cerebellar vermis and hemispheres (like in the other ARSACS patients), but also of the pons. Although thinning of the posterior mid-body of the corpus callosum was present, neither linear T2-hypointensities in the pons nor T2-hyperintensities of the lateral pons and thickening of the MCP could be detected.
Figure 4.
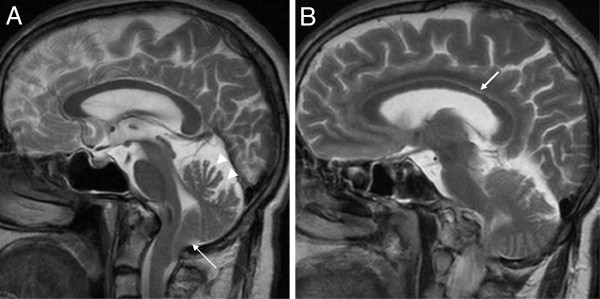
Developmental posterior fossa malformation resembling Chiari type I in the Thr458Ile+ p.Thr458Ile patient. (A) Midsagittal T2 MRI demonstrates herniation of cerebellar tonsils, resembling Chiari malformation type 1 (arrow). Also note the atrophy of the superior cerebellar vermis (arrow heads). (B) Para-midsagittal shows slight short-stretched thinning of the posterior mid-body of the corpus callosum (arrow).
Discussion
In this study we identified 9 novel index patients with two pathogenic or likely pathogenic SACS variants. These patients were part of a consecutive series of n=172 patients with early-onset ataxia. Thus, ARSACS accounts for at least 5% (9/172) of early-onset ataxias - which makes it the second most common ataxia after Friedreich’s ataxia (29%; 50/172) in our series. SACS mutations were responsible for at least 11% (9/83) of patients with unexplained ataxia. This finding is comparable to recent studies observing relative frequencies of 12% (in 232 cerebellar ataxia patients [1]) and 13% (in 85 patients with at least 2 of the 3 cardinal features of ARSACS [5]).
The majority of our ARSACS patients (78%) presented with the classical early-onset triad of cerebellar ataxia, lower limb pyramidal tract signs and axonal-demyelinating sensorimotor peripheral neuropathy, thus corroborating other recent ARSACS case series [5,6,16]. However, one index patient and his likewise affected brother did not show cerebellar ataxia or clinical pyramidal tract signs (no spasticity, no extensor plantar response), and were thus initially presumed to suffer from Charcot-Marie-Tooth (CMT) Disease. This finding corroborates a recent finding of two ARSACS siblings presenting with a CMT-like picture [17]. Importantly, however, in contrast to these previously reported siblings, our index patient and his brother did not even show mild spasticity or cerebellar ataxia. This suggests that SACS should be considered in both complicated and uncomplicated CMT.
On the other side of the phenotypic spectrum – and in sharp contrast to these CMT-like subjects – neuropathy might also be absent in ARSACS as indicated by patient #8 who did not show clinical or electrophysiological evidence of peripheral neuropathy. So far, only one ARSACS individual has been reported without clinical and electrophysiologic signs of peripheral neuropathy, suffering from a very late onset phenotype (ataxia starting at age 40 years) [5]. Our finding demonstrates that even in early-onset cases, neuropathy is not an obligate feature of ARSACS. It might, however, develop at later stages of the disease.
Interestingly, patient #8 did also show neither spasticity nor extensor plantar response. So far, only two ARSACS individuals without lower-limb spasticity were described [4], but both displayed bilateral extensor plantar response, thus suggesting that pyramidal involvement might have been likely masked by the severe neuropathy in these two individuals [1]. Since neuropathy was absent in our index patient, the lack of spasticity cannot simply be explained by a masking neuropathy. Thus, taken together, in contrast to the name of this “spastic ataxia of Charlevoix-Saguenay”, neither spasticity (absent in 2/9=22%) nor extensor plantar response (absent in 3/9=33%) nor cerebellar ataxia (absent in 1/9=11%) are obligate features of this disease. Although pyramidal tract involvement is common in ARSACS patients, as illustrated by the fact that all of our patients had abnormal MEPs, it might be only subclinical in some of them.
Increased demarcation of retinal nerve fibres is also not an obligatory feature of ARSACS. It was exhibited only by 1 (17%) of our patients. This finding is line with other recent studies that observed this feature only 1 out of 8 [5] or in none out of 12 ARSACS patients [16], respectively. Taken together, these findings illustrate that increased demarcation of retinal nerve fibres might be a specific, but not obligatory finding in ARSACS [5], especially in non-Quebec ARSACS individuals [1]. While the high frequency of urge incontinence (77%) might be due to either autonomic dysfunction or pyramidal damage [18] (or a combination of both), erectile dysfunction observed in one out of three male patients aged ≥16 years (33%) is clearly an indicator of autonomic dysfunction. So far, erectile dysfunction has only been reported in one ARSACS patient [19], but might be underdiagnosed. Thus, patients with ARSACS should be actively investigated for autonomic disturbances.
Our imaging findings extend the range of diagnostic features that help to diagnose ARSACS in patients with unexplained ataxia, spasticity or CMT. All patients with two pathogenic SACS variants showed not only the well-established linear pontine T2-hypointensities [2,3], but also bilateral FLAIR-T2-hyperintensities of the lateral pons as well as a thickening of the MCP. The structural correlate of this finding, which corroborates and extends recent findings from other groups [3,16,20], are probably abnormally large transverse pontocerebellar fibres [16]. The frequent finding of an arachnoidal cyst in the posterior fossa (75%) and of developmental cerebellar hemispheric atrophy in one patient underlines the hypothesis that disease processes in ARSACS are not only of neurodegenerative, but also of neurodevelopmental origin [16,18]. These findings might help to differentiate ARSACS from many other recessive neuropathic ataxias, since they are not found e.g. in Friedreich’s ataxia or POLG-associated ataxia [21].
It was recently suggested that structural brain damage in ARSACS is not limited to infratentorial regions [16]. Here we extend this notion, demonstrating for the first time that it also involves the cerebral cortex: 63% of our patients showed bilateral parietal cerebral atrophy. 63% of our patients also showed a short-stretched thinning of the posterior mid-body of the corpus callosum. Although this frequency is lower than in a previous study reporting a thin corpus callosum (TCC) in all patients [16], this finding substantiates the notion that disease processes in ARSACS also frequently involve key associative fibre bundles [16]. Thus, ARSACS should be part of the differential diagnosis in spastic ataxias with TCC, like e.g. SPG11, SPG15 [22] or GBA2[23].
We identified 17 novel SACS variants. Two of them – p.Thr458Ile and p.Val995Phe - were also observed in controls with a frequency of 0.4% or 0.3%, respectively. This indicates that they will likely be identified by many labs in the future, thus warranting a timely discussion whether they should be reported as pathogenic or not. The finding of a recessive mutation in a heterozygous state in controls does not per se refute a possible pathogenic role, in particular if the variant is common (as e.g. demonstrated by the expanded allele of FXN [Friedreich’s ataxia gene], which is observed with a heterozygous carrier rate of 1/60 to 1/90 in controls [24], or by the two frequent, clearly pathogenic c.1399 G>A [p.Ala467Thr] and c.2243 G>C [p.Trp748Ser] POLG mutations, which are observed with a heterozygous carrier rate of up to 1% in several European populations [25]). In particular, the p.Thr458Ile SACS variant might be pathogenic, given the in silico predictions, the high conservation index, and the MRI findings of a TCC as well as of a neurodevelopmental posterior fossa disorder consisting of an arachnoid cyst and herniation of tonsils in the homozygous carrier. Yet, for the time being, we here prefer not to claim any pathogenic significance, in particular given the large phenotypic differences compared to the other subjects with pathogenic SACS mutations. These findings illustrate a problem that is likely to occur frequently in the future: findings of variants with unknown significance may be frequent in SACS sequencing, given the fact that is has now entered clinical routine in many ataxia clinics. Thus, seemingly “atypical ARSACS phenotypes” should be scrutinized with particular caution and underlying SACS variants should ideally be analyzed functionally, e.g. with their impact on mitochondrial fission dynamics [26].
Conclusions
In sum, we here demonstrate that each feature of the classical ARSACS triad (cerebellar ataxia, spasticity, peripheral neuropathy, Charlevoix-Saguenay origin) might in fact be missing in patients with SACS mutations, so the name might be misleading. Atypical presentations may be found much more frequently in the future when ARSACS genetics becomes a standard tool of ataxia work-up by new sequencing techniques and ataxia panel diagnostics. Nevertheless, characteristic MRI features – which also extend to supratentorial regions - will help to establish the diagnosis in most cases. Moreover, the electrophysiological finding of a demyelinating component of the sensorimotor peripheral neuropathy will help to distinguish ARSACS from other common recessive ataxias like Friedreich’s ataxia, POLG or AOA2, where neuropathy is almost exclusively of axonal type [21,27].
Abbreviations
ARSACS: Autosomal recessive spastic ataxia Charlevoix-Saguenay; CMT: Charcot-Marie Tooth Disease; EVS: Exome variant server; FLAIR: Fluid attenuated inversion recovery; MCP: Middle cerebellar peduncles; MEP: Motor evoked potentials; MLPA: Multiplex Ligation-dependent Probe Amplification; MRI: Magnetic resonance imaging; NCS: Nerve conduction studies; POLG: Polymerase gamma; TCC: Thin corpus callosum; VUS: Variant of uncertain clinical significance
Competing interests
Dr. Synofzik received a research grant by the Volkswagen Foundation, a travel grant by the Movement Disorders Society and AtaxiaUK/Ataxia Ireland, and consulting fees from Actelion Pharmaceuticals Ltd.
Dr. Soehn received a research grant by the International Parkinson Fonds (Deutschland) gGmbH (IPD).
Dr. Gburek-Augustat, Dr. Schicks, Dr. Karle, Dr. Schüle, Dr. Schöning, Dr. Biskup, Dr Rudnik-Schöneborn, Dr. Hoffmann, Dr. MacLeod, Dr. Schwarz, Dr. Krüger, and Dr. Kreuz report no disclosures.
Dr. Haack was supported by the NBIA disorders association.
Dr. Senderek received a grant by the Gebert Rüf Foundation.
Dr. Bender received a travel grant by Bayer Vital and was supported by research grants of the German Research Council (DFG) BE4609/1-1 and the Wilhelm-Schuler-Stiftung.
Dr. Peter Bauer received consulting honorary and speaker’s fees from Actelion Pharmaceuticals GmbH (Germany), Actelion Pharmaceuticals (Switzerland), and Centogene (Germany). He received research grants of the German Research Council (BMBF) to EUROSCAR.
Dr. Schöls received research grants of the Deutsche Forschungsgemeinschaft (SCHO754/4-1 and SCHO754/5-1), grants of the German Research Council (BMBF) to Leukonet (01GM0644) and mitoNET (01GM0864) and E-RARE grants to EUROSPA (01GM0807) and RISCA (01GM0820).
Authors’ contributions
Dr. MS and Dr AS: design and conceptualization of the study, acquisition, analysis and interpretation of data; drafting the manuscript. Dr. JG-A, Dr. JS, Dr. KK, Dr. RS, Dr. TH, Dr. MS, Dr. SB, Dr SR-S, Dr. JS, Dr. K-TH, Dr, PM, Dr. JS, Dr. BB, Dr. SK, Dr. FK, Dr. PB: acquisition and analysis of data; revising the manuscript. Dr. LS: conceptualization of the study, acquisition, analysis and interpretation of data; revising the manuscript. All authors read and approved the final manuscript.
Supplementary Material
A) SACS sequencing. B) MLPA assay. C) Criteria for classification of sequence variants.
Alignment results. Multiple sequence alignments for novel SACS missense mutations identified in this study. Results were computed using the PolyPhen-2 interface for UCSC MultiZ46Way GRCh37/hg19 and the default Clustalx color coding: conserved A, C, F, H, I, L, M, V, W, Y or P residues are coloured blue, while conserved S or T residues are in green. Conserved positively charged residues and negatively charged residues are coloured red and magenta, respectively. Exact details can be found in the ClustalX manual (Thompson et al., 1997).
Pedigree of an ARSACS patient with a second late-onset dominant cerebellar ataxia segregating in the pedigree. The family history of index patient #2 (arrow) was positive for a three-generation late-onset (>30 years), slowly progressive, purely cerebellar autosomal-dominant ataxia. This led to a time- and cost-extensive work-up of dominant ataxia genes also in the index patient herself. After these dominant genes were all negative, also recessive genes were screened, leading to the identification of two pathogenic SACS variants (p. Arg728*; p.Phe4352Leufs*11), which were not observed in her affected relatives (WT= wild type). This observation illustrates that two different hereditary ataxias should be considered in pedigrees where the ataxia features differ strongly between pedigree members, e.g. early onset (in the index patient) vs. late onset (in her relatives), or multisystemic (in the index patient) vs. purely cerebellar (in her relatives). A timely consideration of this fact might help to find a correct diagnosis in a more timely manner and save resources.
Contributor Information
Matthis Synofzik, Email: matthis.synofzik@uni-tuebingen.de.
Anne S Soehn, Email: anne.soehn@med.uni-tuebingen.de.
Janina Gburek-Augustat, Email: Janina.Gburek-Augustat@med.uni-tuebingen.de.
Julia Schicks, Email: julia.schicks@medizin.uni-tuebingen.de.
Kathrin N Karle, Email: kathrin.karle@medizin.uni-tuebingen.de.
Rebecca Schüle, Email: rebecca.schuele-freyer@uni-tuebingen.de.
Tobias B Haack, Email: tobias.haack@helmholtz-muenchen.de.
Martin Schöning, Email: Martin.Schoening@med.uni-tuebingen.de.
Saskia Biskup, Email: saskia.biskup@medizin.uni-tuebingen.de.
Sabine Rudnik-Schöneborn, Email: srudnik-schoeneborn@ukaachen.de.
Jan Senderek, Email: Jan.Senderek@med.uni-muenchen.de.
Karl-Titus Hoffmann, Email: Karl-Titus.Hoffmann@medizin.uni-leipzig.de.
Patrick MacLeod, Email: Patrick.Macleod@viha.ca.
Johannes Schwarz, Email: Johannes.Schwarz@kliniken-muehldorf.de.
Benjamin Bender, Email: benjamin.bender@med.uni-tuebingen.de.
Stefan Krüger, Email: st.krueger@medizinische-genetik-dresden.de.
Friedmar Kreuz, Email: f.kreuz@medizinische-genetik-dresden.de.
Peter Bauer, Email: peter.bauer@med.uni-tuebingen.de.
Ludger Schöls, Email: ludger.schoels@uni-tuebingen.de.
Acknowledgements
This study was supported by the European EUROSCAR project (to L.S. and P.B.), the IZKF Aachen (project N 5–4 to S. R.-S. and J. S.), the University of Tübingen (IZKF grant 1970-0-0 to R.S.) and the Deutsche Hereditäre Ataxie Gesellschaft (DHAG) (to F.K. and S.K.).
References
- Vermeer S, van de Warrenburg BP, Kamsteeg EJ. In: GeneReviews [Internet] Pagon RA, Bird TC, Dolan CR, Stephens K, editor. Seattle (WA): University of Washington; 2012. ARSACS. http://www.ncbi.nlm.nih.gov/books/NBK1255/; accessed November 22nd, 2012. [Google Scholar]
- Gerwig M, Kruger S, Kreuz FR, Kreis S, Gizewski ER, Timmann D. Characteristic MRI and funduscopic findings help diagnose ARSACS outside Quebec. Neurology. 2010;75:2133. doi: 10.1212/WNL.0b013e318200d7f8. [DOI] [PubMed] [Google Scholar]
- Gazulla J, Benavente I, Vela AC, Marin MA, Pablo LE, Tessa A, Barrena MR, Santorelli FM, Nesti C, Modrego P. New findings in the ataxia of Charlevoix-Saguenay. J Neurol. 2012;259:869–878. doi: 10.1007/s00415-011-6269-5. [DOI] [PubMed] [Google Scholar]
- Shimazaki H, Takiyama Y, Sakoe K, Ando Y, Nakano I. A phenotype without spasticity in sacsin-related ataxia. Neurology. 2005;64:2129–2131. doi: 10.1212/01.WNL.0000166031.91514.B3. [DOI] [PubMed] [Google Scholar]
- Baets J, Deconinck T, Smets K, Goossens D, Van den Bergh P, Dahan K, Schmedding E, Santens P, Rasic VM, Van Damme P. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology. 2010;75:1181–1188. doi: 10.1212/WNL.0b013e3181f4d86c. [DOI] [PubMed] [Google Scholar]
- Vermeer S, Meijer RP, Pijl BJ, Timmermans J, Cruysberg JR, Bos MM, Schelhaas HJ, van de Warrenburg BP, Knoers NV, Scheffer H, Kremer B. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics. 2008;9:207–214. doi: 10.1007/s10048-008-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–174. doi: 10.2307/2529310. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J, Kang JS, Kremer B, Mariotti C, Melegh B, Pandolfo M, Rakowicz M, Ribai P, Rola R, Schols L, Szymanski S, van de Warrenburg BP, Durr A, Klockgether T, Fancellu R. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–1720. doi: 10.1212/01.wnl.0000219042.60538.92. [DOI] [PubMed] [Google Scholar]
- Prodi E, Grisoli M, Panzeri M, Minati L, Fattori F, Erbetta A, Uziel G, D'Arrigo S, Tessa A, Ciano C, Santorelli FM, Savoiardo M, Mariotti C. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur J Neurol Offic J Eur Federation Neurol Soc. 2013;20:138–146. doi: 10.1111/j.1468-1331.2012.03815.x. [DOI] [PubMed] [Google Scholar]
- Pyle A, Griffin H, Yu-Wai-Man P, Duff J, Eglon G, Pickering-Brown S, Santibanez-Korev M, Horvath R, Chinnery PF. Prominent sensorimotor neuropathy due to SACS mutations revealed by whole-exome sequencing. Arch Neurol. 2012;69:1351–1354. doi: 10.1001/archneurol.2012.1472. [DOI] [PubMed] [Google Scholar]
- Brodal P. The Central Nervous System:Structure and Function: Structure and Function. Oxford: Oxford University Press; 2003. p. page 392. [Google Scholar]
- Miyatake S, Miyake N, Doi H, Saitsu H, Ogata K, Kawai M, Matsumoto N. A novel SACS mutation in an atypical case with autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Intern Med. 2012;51:2221–2226. doi: 10.2169/internalmedicine.51.7374. [DOI] [PubMed] [Google Scholar]
- Gazulla J, Vela AC, Marin MA, Pablo L, Santorelli FM, Benavente I, Modrego P, Tintore M, Berciano J. Is the ataxia of Charlevoix-Saguenay a developmental disease? Med Hypotheses. 2011;77:347–352. doi: 10.1016/j.mehy.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Srulijes K, Godau J, Berg D, Schols L. Characterizing POLG ataxia: clinics, electrophysiology and imaging. Cerebellum. 2012;11:1002–1011. doi: 10.1007/s12311-012-0378-2. [DOI] [PubMed] [Google Scholar]
- Schule R, Schlipf N, Synofzik M, Klebe S, Klimpe S, Hehr U, Winner B, Lindig T, Dotzer A, Riess O, Winkler J, Schols L, Bauer P. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. 2009;80:1402–1404. doi: 10.1136/jnnp.2008.167528. [DOI] [PubMed] [Google Scholar]
- Martin E. Loss of Function of Glucocerebrosidase GBA2 Is Responsible for Motor Neuron Defects in Hereditary Spastic Paraplegia. Am J Hum Genet. 2013;92:238–244. doi: 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epplen C, Epplen JT, Frank G, Miterski B, Santos EJ, Schols L. Differential stability of the (GAA)n tract in the Friedreich ataxia (STM7) gene. Hum Genet. 1997;99:834–836. doi: 10.1007/s004390050458. [DOI] [PubMed] [Google Scholar]
- Hakonen AH, Davidzon G, Salemi R, Bindoff LA, Van Goethem G, Dimauro S, Thorburn DR, Suomalainen A. Abundance of the POLG disease mutations in europe, australia, New zealand, and the united states explained by single ancient european founders. Eur J Hum Genet. 2007;15:779–783. doi: 10.1038/sj.ejhg.5201831. [DOI] [PubMed] [Google Scholar]
- Girard M, Lariviere R, Parfitt DA, Deane EC, Gaudet R, Nossova N, Blondeau F, Prenosil G, Vermeulen EG, Duchen MR. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Proc Natl Acad Sci USA. 2012;109:1661–1666. doi: 10.1073/pnas.1113166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132:2688–2698. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) SACS sequencing. B) MLPA assay. C) Criteria for classification of sequence variants.
Alignment results. Multiple sequence alignments for novel SACS missense mutations identified in this study. Results were computed using the PolyPhen-2 interface for UCSC MultiZ46Way GRCh37/hg19 and the default Clustalx color coding: conserved A, C, F, H, I, L, M, V, W, Y or P residues are coloured blue, while conserved S or T residues are in green. Conserved positively charged residues and negatively charged residues are coloured red and magenta, respectively. Exact details can be found in the ClustalX manual (Thompson et al., 1997).
Pedigree of an ARSACS patient with a second late-onset dominant cerebellar ataxia segregating in the pedigree. The family history of index patient #2 (arrow) was positive for a three-generation late-onset (>30 years), slowly progressive, purely cerebellar autosomal-dominant ataxia. This led to a time- and cost-extensive work-up of dominant ataxia genes also in the index patient herself. After these dominant genes were all negative, also recessive genes were screened, leading to the identification of two pathogenic SACS variants (p. Arg728*; p.Phe4352Leufs*11), which were not observed in her affected relatives (WT= wild type). This observation illustrates that two different hereditary ataxias should be considered in pedigrees where the ataxia features differ strongly between pedigree members, e.g. early onset (in the index patient) vs. late onset (in her relatives), or multisystemic (in the index patient) vs. purely cerebellar (in her relatives). A timely consideration of this fact might help to find a correct diagnosis in a more timely manner and save resources.