

Abstract
Background
The CWxP motif of transmembrane helix 6 (x: any residue) is highly conserved in class A GPCRs. Within this motif, W6.48 is a big star in the theory of the global “toggle switch” because of its key role in the activation mechanism of GPCRs upon ligand binding. With all footlights focused on W6.48, the reason why the preceding residue, C6.47, is largely conserved is still unknown. The present study is aimed to fill up this lack of knowledge by characterizing the role of C6.47 of the CWxP motif.
Results
A complete analysis of available crystal structures has been made alongside with molecular dynamics simulations of model peptides to explore a possible structural role for C6.47.
Conclusions
We conclude that C6.47 does not modulate the conformation of the TM6 proline kink and propose that C6.47 participates in the rearrangement of the TM6 and TM7 interface accompanying activation.
Background
G protein-coupled receptors (GPCRs) are versatile signaling molecules that regulate a vast amount of cellular processes responding to hormones and neurotransmitters [1]. They are present in almost every eukaryotic organism, including fungi and plants, and they are highly diversified in mammalian genomes, representing 2-3% of the human proteome [2]. GPCRs transduce external signals as diverse as photons, odors, pheromones, biogenic amines, neuro-peptides, proteases, glycoprotein hormones and ions (among others) into the cell interior. The response is operated through second messenger cascades controlled by both heterotrimeric guanine nucleotide-binding proteins (G-proteins) [3] or G protein-independent pathways [4]. GPCRs constitute one of the most important pharmaceutical targets, with 30% of marketed drugs acting through them [5]. Mammalian GPCRs are classified in three major families or classes (A, B and C) from which class A is the largest and most studied [6]. Although each GPCR plays a single role in mediating physiological response, many conserved sequential and structural features are encountered among them, mainly near the cytoplasmic part where receptors bind to the G protein [7,8]. Towards the extracellular side, divergence increases [9,10] because of the wide diversity of ligands that need to be bound.
The relatively large repertoire of class A GPCR crystal structures currently available [11-29] (including complexes with agonists, antagonists and inverse agonists) shed light into the understanding of GPCRs’ molecular architecture, as well as on the events responsible for signal transmission (see Table 1). Receptors are thought to be in equilibrium among different states, being inactive ones the most populated in absence of ligand. Agonist binding alters the energetic balance towards actives states triggering activation responses. This implies changes in the conformation of a few residues that are responsible for the formation and disruption of hydrophobic and hydrogen-bond interactions between specific groups of residues. Most of the highly conserved residues often belong to “micro-switches” that swap between different conformations, and constitute an extended allosteric interface between transmembrane (TM) domains [30]. These micro-switches span from the extracellular part to the intracellular G-protein binding site and regulate the equilibrium between inactive and active conformations. This may trigger, eventually, global movements connecting ligand binding with intracellular signaling. R3.50 and D3.49 (of the DRY motif; Ballesteros and Weinstein numbering scheme will be used through this text [31]), Y5.58, Y7.53 (of the NPxxY motif), and W6.48 (of the CWxP motif), are some of the most studied micro-switches [32-36].
Table 1.
The 6.47-7.45 interaction in the analyzed crystal structures
| |
Crystal structure |
TM6-TM7 hydrogen bond |
|||||
|---|---|---|---|---|---|---|---|
| Receptor name | Structure name | PDB ID | Res.(Å) | Pair | N7.45 | D… A (Å) | Cβ-D…A (°) |
| β2-adrenergic |
β2ARINV |
2RH1 [11] |
2.4 |
C6.47-N7.45 |
CO |
3.8 |
84.5 |
| β2ARAGO |
3SN6 [12] |
3.2 |
CO |
4.4 |
81.2 |
||
| β1-adrenergic (turkey) |
β1ARINV |
2VT4 [13] |
2.7 |
C6.47-N7.45 |
CO |
3.7 |
79.3 |
| β1ARAGO |
2Y02 [14] |
2.6 |
CO |
3.9 |
85.3 |
||
| Dopamine D3 |
D3RINV |
3PBL [15] |
2.9 |
C6.47-N7.45 |
CO |
3.8 |
81.6 |
| Histamine H1 |
H1RINV |
3RZE [16] |
3.1 |
C6.47-N7.45 |
CO |
4.2 |
80.9 |
| Adenosine A2A |
A2ARINV |
4EIY [17] |
1.8 |
C6.47-N7.45 |
NH2 |
3.9 |
84.5 |
| |
A2ARAGO |
2YDV [18] |
2.6 |
|
CO |
4.3 |
74.4 |
| κ opioid |
κORINV |
4DJH [19] |
2.9 |
C6.47-N7.45 |
CO |
4.1 |
81.2 |
| μ opioid (mouse) |
μORINV |
4DKL [20] |
2.8 |
C6.47-N7.45 |
CO |
3.6 |
87.5 |
| δ opioid (mouse) |
δORINV |
4EJ4 [21] |
3.4 |
C6.47-N7.45 |
NH2 |
3.8 |
89.8 |
| N/OFQ Opioid |
N/OFQINV |
4EA3 [22] |
3.0 |
C6.47-N7.45 |
NH2 |
4.2 |
85.6 |
| Sphingosine 1-phosph. 1 |
S1PR1INV |
3V2Y [23] |
2.8 |
C6.47-N7.45 |
NH2 |
3.4 |
81.1 |
| CXCR4 chemokine |
CXCR4INV |
3ODU [24] |
2.5 |
C6.47-H7.45 |
- |
- |
- |
| M2 musc. ach. |
M2RINV |
3UON [25] |
3.0 |
T6.47-N7.45 |
NH2 |
3.2 |
120.2 |
| M3 musc. ach. (rat) |
M3RINV |
4DAJ [26] |
3.4 |
T6.47-N7.45 |
NH2 |
4 |
140.7 |
| Rhodopsin (bovine) |
bRhoINV |
1GZM [27] |
2.7 |
C6.47-T7.44 |
- |
3.7 |
101.8 |
| bRhoAGO |
3PQR [28] |
2.9 |
- |
8.8 |
- |
||
| Rhodopsin (squid) | sRhoINV | 2Z73 [29] | 2.5 | Q6.43-S7.54 | - | 2.4 | 111.2 |
(Left) Crystal structures used within the manuscript (INV: inverse agonist or antagonist; AGO: agonist); Protein Data Bank Identification Code and Resolution; receptor structures are human unless stated. (Right) Characterization of the TM6-TM7 hydrogen bond: pair involved, N7.45 rotamer (CO/NH2 indicates the closer group to C6.47 as in their crystal structure), donor(D)-acceptor(A) distance and Cβ-donor(D)···acceptor(A) angle.
The CWxP motif in TM6 (6.47-6.50) is highly conserved in non-olfactory class A GPCRs, with C6.47, W6.48 and P6.50 present in 71%, 78% and 98% of sequences, respectively (see Methods). P6.50 creates a huge kink in TM6 helix (causing a helix bend of around 35°), far bigger than a usual Proline in a transmembrane helix (around 20°) [37]. Because an outward movement of the cytoplasmic side of TM6 is known to accompany receptor activation, it was initially proposed that the Pro-kinked helix could act as a mechanical hinge [38,39]. In fact, these pioneering experiments and computer simulations on rhodopsin originated one of the central paradigms in GPCR activation: the so-called “rotamer toggle switch”. This consisted in a concerted change of C6.47, W6.48 and F6.52 side-chain rotamers between inactive and active states, which were thought to originate the opening of TM6 [40]. In specific, transition of W6.48 from gauche+ to trans and of C6.47 from trans to gauche+ was proposed by Shi and collaborators [38,39]. The low resolution structure of metarhodopsin I, determined by electron crystallography [41], supported the rotamer change of W6.48 as did more recent infrared spectroscopy experiments of rhodopsin photocycle [42]. However, and quite-unexpectedly, none of the available pairs of inactive-active structure are consistent with these rotameric changes (see Table 1 and references therein), questioning the rotamer toggle switch model. Still, it is known from mutagenesis experiments that W6.48 is essential for signaling of some (but not all) receptors [40,43]. On the other hand, there is no current explanation for the large conservation of C6.47 and for the effects on activity reported by various C6.47 mutants. In particular, C6.47T in β2-adrenergic receptor [39] and C6.47R in thyrotropin hormone receptor [44], both led to constitutive Gαs activation. Similarly, C6.47 mutants in cannabinoid receptors CB1 and CB2 modified both ligand recognition and receptor activation [45,46].
The present study reports an extensive analysis of the available GPCRs crystal structures combined with results from molecular dynamics (MD) simulations of model peptides in a membrane mimic aimed at characterizing the role of C6.47. Our results support that C6.47 is a key element of a conserved micro-switch within the TM6/TM7 interface. We show that Cys side-chain possesses unique physicochemical properties and rotameric preferences (compared to Ser and Thr) that allows formation of specific interactions not possible for other small hydrogen-bonding capable residues.
Results and discussion
Residue prevalence and side chain conformation of position 6.47
70% of class-A GPCRs have Cys at position 6.47, whereas the homologous Ser and Thr account, respectively, for only 10% and 4% of the sequences (see Methods). Accordingly, most presently available crystal structures of GPCRs contain cysteine at this position except for muscarinic receptors M2R and M3R, which feature Thr, and for squid rhodopsin, which features Ser (see Table 2). This suggests that a small residue with hydrogen-bonding capabilities is required at position 6.47, but also a preference for Cys rather than Ser or Thr. Structural analysis shows that, among receptors containing C6.47, the residue is in gauche + conformation (g+) in all cases except rhodopsin (trans, t). M2RINV and M3RINV feature T6.47 in gauche- (g-) and sRhoINV has S6.47 in trans conformation. For those receptors in which both agonist- and inverse agonist-bound structures are available (β2ARINV-β2ARAGO, β1ARINV-β1ARAGO, A2ARINV-A2ARAGO, bRhoINV-bRhoAGO) there is no change in 6.47 rotamer conformation. Thus, crystal structures do not support the rotamer change for residue 6.47 accompanying receptors activation as hypothesized by Shi et al [38,39].
Table 2.
Residues present at positions 6.47 and 7.42-7.48
| Receptor |
Position |
|||||||
|---|---|---|---|---|---|---|---|---|
| 6.47 | 7.42 | 7.43 | 7.44 | 7.45 | 7.46 | 7.47 | 7.48 | |
| β2-adrenergic |
Cg+ |
G |
Y |
V |
N |
S |
G |
F |
| β1-adrenergic (turkey) |
Cg+ |
G |
Y |
V |
N |
S |
A |
F |
| Dopamine D3 |
Cg+ |
G |
Y |
V |
N |
S |
A |
L |
| Histamine H1 |
Cg+ |
G |
Y |
I |
N |
S |
T |
L |
| Adenosine A2A |
Cg+ |
S |
H |
T g+ /t |
N |
S |
C |
L |
| κ opioid |
Cg+ |
G |
Y |
T |
N |
S |
S |
L |
| μ opioid (mouse) |
Cg+ |
G |
Y |
T |
N |
S |
C |
L |
| δ opioid (mouse) |
Cg+ |
G |
Y |
A |
N |
S |
S |
L |
| N/OFQ Opioid |
Cg+ |
G |
Y |
V |
N |
S |
C |
L |
| Sphingosine 1-phosph. 1 |
Cg+ |
A |
V |
L |
N |
S |
G |
T |
| CXCR4 chemokine |
Cg+ |
A |
F |
F |
H |
C |
A |
L |
| M2 musc. ach. |
Tg- |
C |
Y |
I |
N |
S |
T |
I |
| M3 musc. ach. (rat) |
Tg- |
C |
Y |
I |
N |
S |
T |
V |
| Rhodopsin (bovine) |
Ct |
A |
K |
T g+ /t |
Sg- |
A |
V |
Y |
| Rhodopsin (squid) | Sg+ | A | K | A | St | A | I | H |
Rotamer of C, S and T (g+/-: gauche+/-, t: trans) is indicated when relevant; residues involved in TM6-TM7 hydrogen bonds are displayed in bold.
MD simulations of A/C/T/SxxP model peptides
We have previously shown how specific Ser and Thr conformers can efficiently modulate the structure of a neighboring proline-kink by forming hydrogen bonds with the backbone carbonyl at positions i-3 or i-4 (with respect to proline, i) [37,47,48]. In the present study, we performed MD simulations of model peptides containing Ser, Thr and Cys at position i-4 with respect to proline, in a hydrophobic solvent (see Method). Analogous peptides containing C/T/SWAP motifs exhibit a similar profile (see Additional file 1: Figure S1). The aim of these simulations was to establish whether or not Cys could alter the structure of a regular proline kink and quantify the extent of the alteration compared to Ser and Thr [49]. Figure 1 shows the change in Φ and Ψ angles and in local twists and bends for peptides with respect to the reference AAAP peptide, for C/T/SAAP motifs with the C/T/S rotamer fixed to g+, g- or t (see Methods). In terms of Φ and Ψ, it can be seen that all g- conformers induce a ~15° decrease in Φ angle profile (relative to the WT) at position i-3, and an increase (8-9° for Ser and Thr, 4° for Cys) in Ψ angle profile at position i-4. Trans conformers of Ser and Thr (but not of Cys) induce a smaller but parallel effect: ~6° decrease in the Φ angle at position i-2 (and also at position i-3 for Thr) and ~10° increase in Ψ angle profile at i-3 position. Regarding bend profiles, all of them exhibit a bimodal shape, where Cys g+, Cys t and Ser g+ exhibit the smallest deviations (<2°) relative to the WT. Finally, twist angle in all g- conformers exhibits a pronounced decrease in the region between i-6 and i. Collectively, it can be concluded that Cg- (blue continuous line) and all rotamers of Ser and Thr (broken and dotted lines) modify the standard proline kink profile (black line). Opposite, profiles of both Cg+ and Ct reveal no important alterations of the standard Pro-kink (continuous lines). Taking in consideration that Cg- rotamer is forbidden because of steric clash [47] (see Table 3), cysteine turns out to be the only small hydrogen-bonding capable residue that does not modify the profile of a standard Pro-kink in its allowed conformations. On the other hand, Ser and Thr would require further adaptation of the environment in order to be tolerated at position 6.47.
Figure 1.
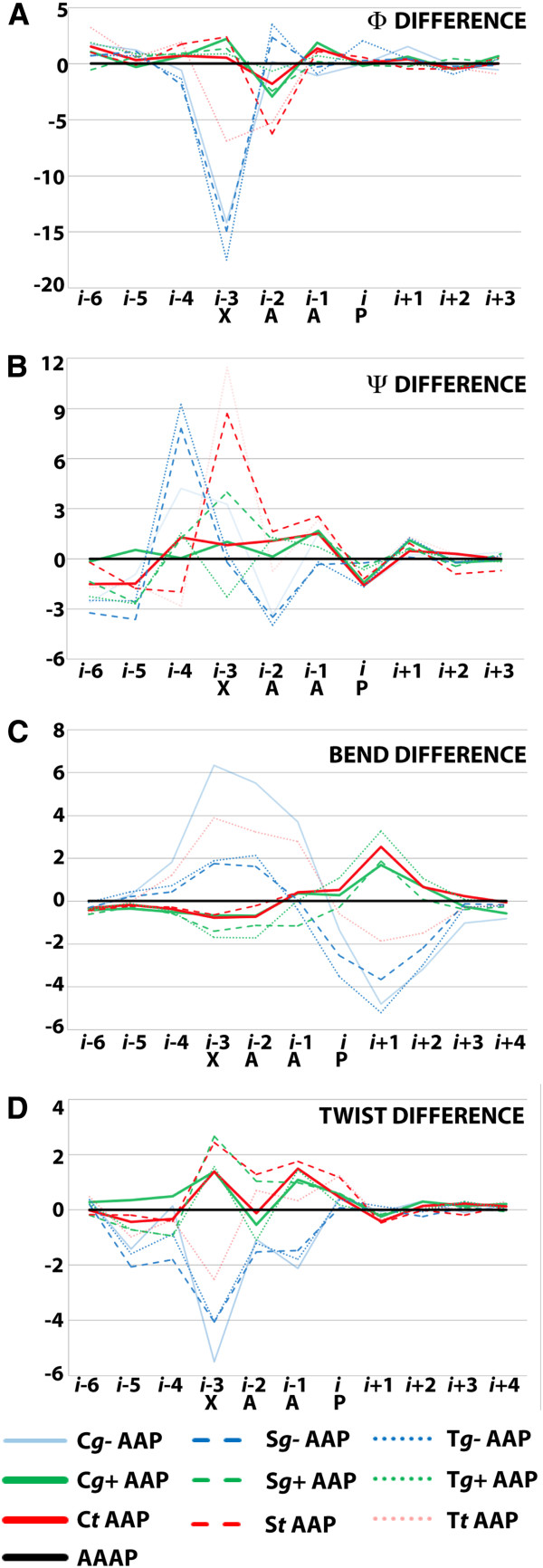
Analysis of TM6 irregularities computed from MD simulations of polyalanine peptides containing the CAAP, SAAP, TAAP motifs in the gauche-, gauche + and trans conformers. Difference between average Φ (A) and Ψ (B) dihedral angles, unit bend (C) and unit twist (D) profiles. CtAAP and Cg+AAP are the unique peptide conformations that present very similar helical geometrical parameters to AAAP. Sg+AAP peptide present certain distortion relative to AAAP peptide and Cg-AAP, Sg-AAP, StAAP, Tg+AAP, Tg-AAP, TtAAP present the most distorted helical geometrical parameters relative to AAAP peptide. Bend and twist angles assigned at position i corresponds to the value computed for (i-3, i) and (i, i + 3) and for (i-3, i), respectively.
Table 3.
Population of C/S/T rotamers in transmembrane helices from crystal structures
| Residue |
Conformation (%) |
||
|---|---|---|---|
| gauche- | gauche+ | trans | |
| Cysteine |
0 |
71 |
29 |
| Threonine |
15 |
84 |
1 |
| Serine | 20 | 52 | 28 |
Data arranged from reference [47].
TM6/TM7 interactions involving 6.47
Having discarded C6.47 as a modulator of TM6 kink, it is then likely that its high conservation is due to specific interactions played by the thiol group. Crystal structures show that the central segments of both TM6 and TM7 are in intimate contact. The maximum proximity is found between residues 6.47 and 7.42 (~3.5 Å within the closest atoms). Position 6.47 is precisely the hinge of the rigid body movement of the cytoplasmic side of TM6 associated to GPCR activation [28,50,51]. Figure 2A shows a detailed view centered at C6.47 for β2ARINV structure (PDB:2RH1) taken as representative. It can be seen that the thiol group is surrounded by three aliphatic residues (T6.43, L6.46, V7.44), one aromatic residue (F6.48), but also by hydrogen bond-capable groups (backbone carbonyl groups of residues 6.43 and 7.41 and the side chain of N7.45). This dual nature at the environment of 6.47 is common to the remaining structures and perfectly suits the physiochemical properties of Cys thiol group. Analysis of proteins of known structures show that cysteine environment are closer to those of Ile methyl groups than to Ser [52]. On the other hand, Cys is a moderately good hydrogen-bond donor as demonstrated by the fact that more than two thirds of the thiol groups in the Protein Data Bank or the Cambridge Structural Database co-occur with suitable acceptors, manly carbonyl groups [53-57]. Cysteine has also been described to participate in thiol- aromatic π-type hydrogen bonds [58].
Figure 2.

The TM6-TM7 interface in β2ARINV structure. A) Hydrophobic residues (white surface) and free carbonyl groups (red) surrounding C6.47 (grey surface). B-D) Hydrogen bond network at the TM6 and TM7 interface (indicated by dotted lines). B) In β2ARINV (left) C6.47 forms a hydrogen bond with N7.45 and eventually with the backbone of 6.43; in β2ARAGO (right) the hydrogen bond between 6.47 and N7.45 is broken and C6.47 interacts only with the backbone of 6.43. C) sRHOINV features Q6.43 bridging S6.47 and S7.45. D) In RHOINV (left) C6.47 is interacting with T7.44 g+; in RHOAGO the hydrogen bond between Cys 6.47 and T7.44 has broken. Color code is as follows: TM6 (blue), TM7 (pale red).
Based on molecular modeling and site-directed mutagenesis studies, a C6.47-N7.45 hydrogen bond interaction has recently been proposed in the gonadotropin-releasing hormone receptor [59]. Since all receptors with available structures feature N7.45 (66% conserved) except bRho/sRho (contain S:12%) and CXCR4 (contains H:8%) we assessed, by conducting systematic analysis on the available crystal structures, if this interaction was likely to exist in other GPCRs.
Available crystal structures differ in the assignment of CO and NH2 groups for N7.45 (see Table 1). This it is not surprising because even at 1.8 Å resolution (the highest obtained for a GPCR structure; see Table 1) it is not possible to correctly assign all side chain rotamers. Since it is unlikely that the orientation of the CO/NH2 groups of N7.45 is not the same in all receptors (this position is surrounded by many conserved residues) we evaluated the two possible orientations using NQ-Flipper (see Methods) [60]. The results revealed that the conformer present in β2ARINV (PDB:2RH1) is the most favorable in all structures. We, therefore, performed our analysis assuming the rotamer present in β2ARINV as the correct one. Table 1 displays S(Cys6.47) · · · O(Asn7.45) distances and Cβ-S · · · O angles measured on the crystal structures of Asn7.45-containing receptors (see Methods). In all structures containing C6.47 except β2ARAGO structure (3SN6), geometries are compatible with the existence of a hydrogen bond between C6.47 and N7.45 [53-57]. It should be noted that sulfur hydrogen bond has a strong dispersion energy component [61] and, therefore, the directionality of the hydrogen bond is less pronounced than in conventional hydrogen bonds, leading to flat potential energy surfaces that are attractive even at large distances [54,62].
Figure 2B shows a comparison between β2ARINV and β2ARAGO, displaying the breakage of the C6.47 · · · N7.45 hydrogen bond upon agonist binding. In parallel, it can be seen in the figure that carbonyl group of 6.43 in β2ARINV is oriented toward the amide group of C6.47, whereas in β2ARAGO it points towards thiol C6.47 group.
As a result, there is a decrease in the Cys6.47(S) · · · 6.43(backbone CO) distance, which suggests the stabilization of an alternative hydrogen bond in β2ARAGO. This leads us to propose that the breakage of the C6.47-N7.45 hydrogen bond is related to activation in β2AR. For A2AR the trend of that withdrawnness of N7.45 from C6.47 is clearly maintained. The fact that β1ARAGO and A2ARAGO structures reflect a smaller change as compared to β2ARAGO could be due to the fact that the structures do not account for fully active-like states, as it has previously been proposed [18,63]. Possible reasons could be the lack of TM6 opening (maybe because of absence of G-protein) or, in the case of β1ARAGO, the presence of thermostabilizing mutations (including F7.48M contacting C6.47). On the other hand, H1RINV, known for its high basal activity, exhibits a distance in between β2ARINV and β2ARAGO even in presence of antagonist [16].
Muscarinic receptors are of the few GPCRs that present a Thr in position 6.47. Despite that M2RINV and M3RINV exhibit the analogous hydrogen bond interaction between T6.47 and N7.45. The rotamer for Thr is g-, despite g+ is the most populated rotamer for Thr (see Table 2). This anomaly seems to originate from the steric clash that Thr methyl group would give with TM7 in the latter conformer. Rhodopsins belong to the small group of receptors that contain S7.45. Because serine is too short to reach the side-chain of 6.47, rhodopsins seem to have adopted alternative mechanisms to control TM6-TM7 interface. This could be related with the fact that retinal is attached to the neighboring K7.43. Figure 2C displays the structure of sRHOINV that, oddly, features S6.47 in the g+ rotamer conformation. It can be seen that the receptor contains residue Q6.43 that bridges S6.47 and S7.45. In the case of bRho (Figure 2D), bRhoINV contains an alternative TM6-TM7 hydrogen bond between C6.47 and T7.44. Comparison between bRhoINV and bRhoAGO reveals that in the former, trans C6.47 forms a hydrogen with g- T7.44, whereas in the latter, T7.44 has changed conformation to trans, breaking C6.47-T7.44 interaction (see Figure 2D). Thus, as in β2ARINV/β2ARAGO pair, there is a breakage of the C6.47-TM7 hydrogen bond associated to activation.
CXCR4INV is the only resolved receptor whose sequence contains H7.45 (instead of Asn). Although histidine is 87% conserved in chemokine receptors, CXCR4INV shows no (direct) interaction between 6.47 and H7.45.
6.47 modulates D2.50-N7.49 interaction
In β2ARINV N7.45 and N7.49 side chains form a water-mediated hydrogen bond. In β2ARAGO, release of N7.45 from C6.47 permits N7.45 to free N7.49 side-chain (of the NPxxY motif) from N7.44, enabling the interaction between N7.49 and D2.50 (Figure 3A). Stability of D2.50 is reinforced by conformation changes of residue S3.39 from g+ (pointing towards TM3’s backbone in β2ARINV) to trans (interacting with D2.50 in β2ARAGO). Unsurprisingly, S3.39 is highly conserved (71%) in class A GPCRs. Thus, C6.47-N7.45 interaction keeps N7.49 away from D2.50 and disruption of this hydrogen bond favors N7.49-D2.50 interaction.
Figure 3.

Hydrogen bond network associated to C6.47 in both inverse-agonist (left) and agonist (right) bound structures of β2AR (A) and bRho (B). Panel A: C6.47 hydrogen bonds N7.45 which retains N7.49, avoiding interaction of the latter with D2.50 (left). Breaking of C6.47-N7.45 interaction releases (right) N7.49 permitting its interaction with D2.50. Panel B: the release of both C6.47-T7.44 and Y7.48-N7.49 hydrogen bonds (left), permits N7.49 to form a hydrogen bond with D2.50. Helices are colored as follows: TM1:white, TM2:gold, TM3:red, TM6:blue and TM7:pale-red. The remaining helices have been omitted for better clarity. Relevant side-chains are shown as sticks (cyan) and hydrogen bonds are indicated by dashed lines. A crystallographic water molecule is shown as spheres.
In bRhoAGO, T7.44(t) allows Y7.48 to interact with the backbone carbonyl of 7.41. The displacement of Y7.48 away from N7.49 allows the latter to interact with D2.50 through a water molecule resolved in the crystal structure. On the other hand, interaction C6.47-T7.44 in bRhoINV disables N7.49-D2.50 interaction and retains Y7.48 close to N7.49 (Figure 3B). Unsurprisingly, Y7.48 is 80% conserved among opsins, backing the importance of this residue within this subfamily. Y7.48 could not populate the conformation found in the crystal if C6.47 would be in the g+ rotamer. This explains that C6.47 is found in the trans rotamer. On the other hand, sRho features H7.48 (relatively rare in opsins; 7%). Given the smaller side chain of His compared to Tyr, residue C6.47 can populate the g+ rotamer in the former. Still, Q6.43 would prevent N7.49 from interacting with D2.50.
Together with Y7.53, N7.49 is described to undergo a conformational change that helps the release of R3.50 from D/E3.49 (and from D/E6.30 in one third of the receptors). This permits R3.50 to adopt an extended conformation that favors the interaction with the C-terminal portion of Gα domain of G-proteins [64-66]. These conformational changes open a cavity between TMs 3, 5 and 6, allowing G-protein binding [12]. In the context of the global toggle switch model proposed by Schwartz and collaborators [30], where various non-covalent interactions form an allosteric interface that connects ligand binding with intracellular signaling, C(S,T)6.47 could be considered as an intermediate micro-switch modulating interaction between N7.45 and N7.49, thus ultimately modulating TM2-TM7 interaction via N7.49 and D2.50. In the cascade of events during GPCR activation, C6.47 micro-switch would be preceding N7.49.
The analysis of hydrogen bond networks associated to position 6.47 reveals that receptors having C(S,T)6.47 in gauche+/- conformation rarely have hydrogen-bond capable residue at position 7.44 (see Table 2). In this situation, C(S,T)6.47 forms hydrogen bond with the side chain of residue at position 7.45. On the other hand, in those structures where residue 6.47 is in the trans rotamer conformation, position 7.45 contains Ser (too short to permit a 6.47-7.45 hydrogen bond). In such cases C(S,T)6.47 forms a hydrogen bond with position 7.44 instead. Even with differences at position 6.47 in rotamer conformation (t/g+/g-) and, to a lesser extent, in residue occurrence (Cys/Ser/Thr), all crystal structures are in accordance with a hydrogen bond network that connects residue at 6.47 with N7.49 of the NPxxY motif and the highly conserved (93%) D2.50, via N7.45(or S7.44).
Conclusions
The largely conserved Cys6.47 of the CWxP motif of class-A GPCRs was initially thought to modulate the conformation of the TM6 proline-kink through a rotamer switch during the process of activation. The release of several agonist-bound (or active-like) crystal structures invalidated this possibility, as no conformational change of this residue has been observed, leaving the role and the prevalence of Cys6.47 unexplained. The present analysis of crystal structures suggests that C6.47 has an active role in rearranging the TM6/7 interface between active and inactive states. Inactive structures are characterized by an interaction between the side chains of residues C(S/T)6.47 and 7.44/7.45, and this generates a constraint in the TM6-TM7 interface that keeps N7.49 (of the NPxxY motif) away from D2.50. Because active structures do not exhibit the interaction between 7.44/7.45 and 6.47, the formation of a hydrogen bond between N7.49 and D2.50 is enabled. We propose that high conservation of Cys at this position is attributable to the requirement of a small residue with ability to form both intra- and extra- main chain hydrogen bonds without altering TM6 Pro-kink. Cys has specific rotameric preferences compared to Ser or Thr, and in addition, the thiol group forms less directional hydrogen bonds than the hydroxyl group.
Although none of the different approaches presented in this study corroborates per se our hypothesis, their combination brings to a unique possible role for Cys6.47 in class A GPCRs: Cys6.47 is the gate keeper of the hydrogen bond network involving N7.45, N.7.49 and D2.50, one of the most important micro-switches associated to GPCR activation.
Methods
Molecular dynamics simulations of model peptides
Four 25-residue long α-helical polyalanine peptides containing variants of the XAAP (being X = A, C, S and T) motif at their centers were built using PyMol [67]. Each peptide was subsequently embedded into an equilibrated cyclohexane box, to mimic membrane environment. Ten systems were generated by selecting all possible χ1 conformers (gauche+, gauche- and trans) of residue X. All simulations were performed with the GROMACS 4.0.7 simulation package [68]. The force field for the amino acids was the ported version of Amber99sb for this simulation package [69]. Cyclohexane parameters were taken from the methylene groups of Berger’s lipids [70] as employed elsewhere [71]. Systems were energy minimized and subsequently subjected to 50 ns MD simulations. Peptides were restrained to its original conformation during the first 5 ns. Additional 20 ns were discarded as equilibration and the analysis was performed on the last 25 ns. The system was kept at an isotropic pressure of 0.1 MPa using a Berendsen barostat [72]. Temperature was maintained constant at 300 K using separate V-rescale thermostats for the peptide and for cyclohexane [73]. All bonds and angles were frozen using the LINCS algorithm [74]. Lennard-Jones interactions were computed using a cutoff of 1.0 nm and the electrostatic interactions were treated using PME with the same real-space cutoff. Integration of equations of motion was performed using a time-step of 2 fs.
For each XAAP peptide, Φ and Ψ backbone dihedrals and unit bend and unit twist angles within residues i-7 to i + 4 (with position i containing X = C/S/T/A) were computed. GROMACS tools and HELANAL program [75] were used for this purpose. Unit twist angle is a useful measure of local helix distortion interpreted as follows: an ideal α-helix, with approximately 3.6 residues per turn, has a twist angle of approximately 100° (360°/3.6). Accordingly, a tighter helical segment contains <3.6 and exhibits a twist >100°, whereas a wider segment contains >3.6 residues per turn, resulting in a twist <100°.
Analysis of sequence positions
Frequencies of aminoacids at a specific position were extracted from GPCRs Motif Searcher (http://lmc.uab.cat/gmos). All data shown herein corresponds to a sequence alignment for all human non-olfactory class A GPCRs.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MO and AC carried out the molecular dynamic simulations of model peptides and did the analysis of trajectories. All authors participated in the analysis of crystal structures, development of hypothesis and writing of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Analysis of TM6 irregularities computed from MD simulations of polyalanine peptides containing the CWAP, SWAP, TWAP motifs in the gauche-, gauche+ and trans conformers. Difference between average Φ (A) and Ψ (B) dihedral angles, unit bend (C) and unit twist (D) profiles. Bend and twist angles assigned at position i correspond to the value computed for (i-3, i) and (i, i+3) and for (i-3, i), respectively.
Contributor Information
Mireia Olivella, Email: mireia.olivella@uvic.cat.
Gianluigi Caltabiano, Email: gianluigi.caltabiano@uab.cat.
Arnau Cordomí, Email: arnau.cordomi@uab.cat.
Acknowledgements
This work was supported by grants from MICINN (SAF2010-22198-C02-02), ISCIII (RD07/0067/0008) and AGAUR (2009SGR-581). AC is a receiver of a contract grant from ISCIII.
References
- Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, Leurs R. Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu Rev Pharmacol Toxicol. 2007;47:53–87. doi: 10.1146/annurev.pharmtox.47.120505.105126. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9(1):60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Sun Y, McGarrigle D, Huang XY. When a G protein-coupled receptor does not couple to a G protein. Mol Biosyst. 2007;3(12):849–854. doi: 10.1039/b706343a. [DOI] [PubMed] [Google Scholar]
- Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5(10):821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, Klomp J, Oliveira L, de Vlieg J, Vriend G. GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res. 2011;39(Database issue):D309–D319. doi: 10.1093/nar/gkq1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liapakis G, Cordomi A, Pardo L. The G-protein coupled receptor family; actors with many faces. Curr. Pharm. Des. Curr Pharm Des. 2012;18:175–185. doi: 10.2174/138161212799040529. [DOI] [PubMed] [Google Scholar]
- Caltabiano G, González A, Cordomí A, Campillo M, Pardo L. The Role of Hydrophobic Amino Acids in the Structure and Function of the Rhodopsin Family of G Protein-Coupled Receptors. Methods Enzymol. 2013;520:99–115. doi: 10.1016/B978-0-12-391861-1.00005-8. [DOI] [PubMed] [Google Scholar]
- Deupi X, Dolker N, Lopez-Rodriguez ML, Campillo M, Ballesteros JA, Pardo L. Structural models of class a G protein-coupled receptors as a tool for drug design: insights on transmembrane bundle plasticity. Curr Top Med Chem. 2007;7(10):991–998. doi: 10.2174/156802607780906799. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Cordomi A, Caltabiano G, Pardo L. Impact of helix irregularities on sequence alignment and homology modeling of G protein-coupled receptors. ChemBioChem. 2012;13(10):1393–1399. doi: 10.1002/cbic.201200189. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK. High-Resolution Crystal Structure of an Engineered Human beta2-Adrenergic G Protein Coupled Receptor. Science. 2007;5854:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469(7329):241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2011;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475(7354):65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337(6091):232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474(7352):521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485(7398):327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485(7398):400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485(7398):395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335(6070):851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482(7386):547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482(7386):552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Edwards PC, Burghammer M, Villa C, Schertler GFX. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343(5):1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471(7340):651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453(7193):363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30(5):249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated Methods for Modeling G-Protein Coupled Receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455(7212):497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454(7201):183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Lin SW, Sakmar TP. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35(34):11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- Ramon E, Cordomi A, Bosch L, Zernii EY, Senin II, Manyosa J, Philippov PP, Perez JJ, Garriga P. Critical role of electrostatic interactions of amino acids at the cytoplasmic region of helices 3 and 6 in rhodopsin conformational properties and activation. J Biol Chem. 2007;282(19):14272–14282. doi: 10.1074/jbc.M611091200. [DOI] [PubMed] [Google Scholar]
- Olivella M, Deupi X, Govaerts C, Pardo L. Influence of the environment in the conformation of alpha-helices studied by protein database search and molecular dynamics simulations. Biophys J. 2002;82(6):3207–3213. doi: 10.1016/S0006-3495(02)75663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visiers I, Ballesteros JA, Weinstein H. Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 2002;343:329–371. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277(43):40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285(6):3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 2004;23(18):3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S, Zaitseva E, Caltabiano G, Schertler GF, Sakmar TP, Deupi X, Vogel R. Tracking G-protein-coupled receptor activation using genetically encoded infrared probes. Nature. 2011;464(7293):1386–1389. doi: 10.1038/nature08948. [DOI] [PubMed] [Google Scholar]
- Pellissier LP, Sallander J, Campillo M, Gaven F, Queffeulou E, Pillot M, Dumuis A, Claeysen S, Bockaert J, Pardo L. Conformational toggle switches implicated in basal constitutive and agonist-induced activated states of 5-hydroxytryptamine-4 receptors. Mol Pharmacol. 2009;75(4):982–990. doi: 10.1124/mol.108.053686. [DOI] [PubMed] [Google Scholar]
- Biebermann H, Winkler F, Handke D, Teichmann A, Gerling B, Cameron F, Eichhorst J, Gruters A, Wiesner B, Kuhnen P. New pathogenic thyrotropin receptor mutations decipher differentiated activity switching at a conserved helix 6 motif of family A GPCR. J Clin Endocrinol Metab. 2012;97(2):E228–E232. doi: 10.1210/jc.2011-2106. [DOI] [PubMed] [Google Scholar]
- Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (-)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68(6):1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol. 2008;15(11):1207–1219. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Deupi X, Olivella M, Haaksma EEJ, Pardo L. Serine and threonine residues bend alpha-helices in the chi(1) = g(-) conformation. Biophys J. 2000;79(5):2754–2760. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X, Olivella M, Sanz A, Dolker N, Campillo M, Pardo L. Influence of the g- conformation of Ser and Thr on the structure of transmembrane helices. J Struct Biol. 2009;169(1):116–123. doi: 10.1016/j.jsb.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Deupi X, Olivella M, Govaerts C, Ballesteros JA, Campillo M, Pardo L. Ser and Thr residues modulate the conformation of pro-kinked transmembrane alpha-helices. Biophys J. 2004;86(1):105–115. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, Schertler GF. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471(7340):656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469(7329):175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano N, Ota M, Nishikawa K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999;458(1):69–71. doi: 10.1016/s0014-5793(99)01122-9. [DOI] [PubMed] [Google Scholar]
- Zhou P, Tian F, Lv F, Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins. 2009;76(1):151–163. doi: 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- Wennmohs F, Staemmler V, Schindler M. Theoretical investigation of weak hydrogen bonds to sulfur. J Chem Phys. 2003;119(6):3208–3218. [Google Scholar]
- Allen FH, Bird CM, Rowland RS, Raithby PR. Hydrogen-bond acceptor and donor properties of divalent sulfur (Y-S-Z and R-S-H) Acta Crystallogr Sect B-Struct Commun. 1997;53:696–701. [Google Scholar]
- Gregoret LM, Rader SD, Fletterick RJ, Cohen FE. Hydrogen bonds involving sulfur atoms in proteins. Proteins. 1991;9(2):99–107. doi: 10.1002/prot.340090204. [DOI] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P. Different types of interactions involving cysteine sulfhydryl group in proteins. J Biomol Struct Dyn. 1998;15(6):1059–1072. doi: 10.1080/07391102.1998.10509001. [DOI] [PubMed] [Google Scholar]
- Duan GL, Smith VH, Weaver DF. Characterization of aromatic-thiol pi-type hydrogen bonding and phenylalanine-cysteine side chain interactions through ab initio calculations and protein database analyses. Mol Phys. 2001;99(19):1689–1699. [Google Scholar]
- Lu ZL, Coetsee M, White CD, Millar RP. Structural determinants for ligand-receptor conformational selection in a peptide G protein-coupled receptor. J Biol Chem. 2007;282(24):17921–17929. doi: 10.1074/jbc.M610413200. [DOI] [PubMed] [Google Scholar]
- Weichenberger CX, Sippl MJ. NQ-Flipper: recognition and correction of erroneous asparagine and glutamine side-chain rotamers in protein structures. Nucleic Acids Res. 2007;35(Web Server issue):W403–W406. doi: 10.1093/nar/gkm263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal HS, Wategaonkar S. Sulfur, not too far behind O, N, and C: SH…pi hydrogen bond. J Phys Chem A. 2009;113(46):12774–12782. doi: 10.1021/jp907747w. [DOI] [PubMed] [Google Scholar]
- Steiner T. The hydrogen bond in the solid state. Angew Chem. 2002;41(1):49–76. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol. 2011;21(4):541–551. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Urizar E, Claeysen S, Deupi X, Govaerts C, Costagliola S, Vassart G, Pardo L. An activation switch in the rhodopsin family of G protein-coupled receptors: the thyrotropin receptor. J Biol Chem. 2005;280(17):17135–17141. doi: 10.1074/jbc.M414678200. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276(31):29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Jongejan A, Sansuk K, Hacksell U, Timmerman H, Brann MR, Weiner DM, Pardo L, Leurs R. Constitutively active mutants of the histamine H1 receptor suggest a conserved hydrophobic asparagine-cage that constrains the activation of class A G protein-coupled receptors. Mol Pharmacol. 2008;73(1):94–103. doi: 10.1124/mol.107.038547. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 15r3. Schrodinger, LLC; 2012. [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Sorin EJ, Pande VS. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys J. 2005;88(4):2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72(5):2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordomí A, Caltabiano G, Pardo L. Membrane Protein Simulations Using AMBER Force Field and Berger Lipid Parameters. J Chem Theory Comput. 2012;8(3):948–958. doi: 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684. [Google Scholar]
- Bussi G, Zykova-Timan T, Parrinello M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J Chem Phys. 2009;130(7):074101. doi: 10.1063/1.3073889. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Kollman PA. Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J Comput Chem. 1992;13(8):952–962. [Google Scholar]
- Bansal M, Kumar S, Velavan R. HELANAL: a program to characterize helix geometry in proteins. J Biomol Struct Dyn. 2000;17(5):811–819. doi: 10.1080/07391102.2000.10506570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of TM6 irregularities computed from MD simulations of polyalanine peptides containing the CWAP, SWAP, TWAP motifs in the gauche-, gauche+ and trans conformers. Difference between average Φ (A) and Ψ (B) dihedral angles, unit bend (C) and unit twist (D) profiles. Bend and twist angles assigned at position i correspond to the value computed for (i-3, i) and (i, i+3) and for (i-3, i), respectively.