

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant genetic disease with a wide spectrum of vascular malformations involving multiple organs. Nine-16% of patients with HHT harbor brain arteriovenous malformations (AVMs), which can cause intracranial hemorrhage. Our objective was to study clinical manifestations of brain AVM in patients with HHT and correlate these with the specific gene mutated. We reviewed records of 171 patients with HHT and brain AVMs. A history of intracranial hemorrhage was found in 27% (41/152) patients, with a mean (range) age of 26 +/− 18 (0–68) years. All patients with intracranial hemorrhage were neurologically asymptomatic prior to intracranial hemorrhage. Multiple brain AVMs were found in 23% (170/39) of patients on initial examination. Genetic test results were available in 109 (64%) patients. Mutations in ENG, ACVRL1, and SMAD4 were present in 75 (69%), 18 (17%) and 2 (2%), respectively. A history of intracranial hemorrhage was reported in 24% of patients with an ENG mutation and 27% of patients with an ACVRL1 mutation, with a mean (range) age of 26 +/− 16 (2–50) and 18 +/− 21 (0–48) years, respectively. No statistically significant differences in age at first brain AVM diagnosis, prevalence of intracranial hemorrhage history, age at intracranial hemorrhage, or other manifestations of brain AVMs were observed among gene groups. In conclusion, no evidence for differences in brain AVM characteristics was observed among HHT gene groups, although we cannot exclude clinically important differences. Larger studies are needed to further guide brain AVM screening decisions in patients with HHT.
Keywords: Hereditary Hemorrhagic Telangiectasia, Brain Arteriovenous Malformation, Genotype, Intracranial Hemorrhage
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is inherited in an autosomal dominant pattern and is characterized by vascular malformations. It can involve multiple organs including brain, lung, liver, skin, and mucous membranes, and causes a broad range of clinical manifestations, from epistaxis to hemoptysis and stroke. Nine-16% of patients with HHT have brain arteriovenous malformations (BAVMs)[Bayrak-Toydemir et al 2006; Letteboer et al 2006; Sabba et al 2007], which can cause intracranial hemorrhage (ICH), disability, and death.
Mutations in three genes, endoglin (ENG)[McAllister et al 1994; McDonald et al 1994; Shovlin et al 1994], activin A receptor type II-like 1 (ACVRL1)[Berg et al 1997; Vincent et al 1995], and SMAD family member 4 (SMAD4) [Gallione et al 2004], have been recognized as the cause of HHT1, HHT2, and the Juvenile Polyposis-HHT overlap syndrome (JP-HHT), respectively. Previous HHT gene-phenotype correlation studies demonstrated an association between ENG mutation and BAVMs[Bayrak-Toydemir et al 2006; Letteboer et al 2006; Sabba et al 2007], though BAVMs were also reported in small numbers of patients with ACVRL1[Bayrak-Toydemir et al 2006; Berg et al 2003; Kjeldsen et al 2005; Letteboer et al 2006] and SMAD4 mutations[Gallione et al 2010]. However, no study to date has reported or compared the clinical manifestations of BAVMs, most notably ICH, among HHT genes.
The purpose of this cross-sectional study was to describe the clinical presentations of patients with HHT and BAVMs, and to correlate them with the mutated HHT genes in a large series of retrospectively collected patients.
MATERIALS AND METHODS
Patient selection
We retrospectively identified patients diagnosed with BAVMs and HHT from the Yale HHT Center (Yale University) (1988 to 2010) and the Toronto HHT Centre (University Health Network and St. Michael’s Hospital, University of Toronto) (1984 to 2009). Both centers routinely screen patients with HHT for BAVMs after the diagnosis of HHT. In addition, we included patients with BAVMs and HHT recruited as part of the NIH-ORD-NINDS Brain Vascular Malformation Consortium (BVMC) 6203 study, enrolled from May 2010 to March 2011. These patients were recruited through the following sites; Washington University HHT Center, Georgia Health Science University HHT Center, University of California at San Francisco (UCSF) Center for Cerebrovascular Research and the HHT Foundation International. Institutional review board approval for human research was obtained by each institution.
Diagnosis of HHT
The diagnosis of HHT was based on clinical criteria [Shovlin et al 2000] or genetic test results, as per International HHT Guidelines [Faughnan et al 2011]. The clinical diagnostic criteria, also known as the Curaçao Criteria, are: (1) Spontaneous recurrent nosebleeds; (2) Mucocutaneous telangiectasia at characteristic sites (lips, oral cavity, nose or fingers); (3) Visceral involvement such as pulmonary, hepatic or central nervous system arteriovenous malformations; and (4) An affected first degree relative according to these criteria. Patients were classified as “definite HHT” when at least three criteria were met, as “possibly HHT” when two criteria were met, or “unlikely HHT” when one or no criteria were met. We included only those with “definite HHT” clinical diagnosis or positive genetic test results for HHT (mutation in any one of the ENG, ACVRL1, or SMAD4 genes).
Diagnosis of BAVM
Eligible patients had a previous diagnosis of BAVMs, confirmed by chart review, magnetic resonance imaging (MRI), cerebral angiography, surgical pathology, or autopsy.
Data Collection
The following data were collected retrospectively to the extent possible from patient records, HHT Center databases or the BVMC database: personal or familial HHT genetic test results from a clinical laboratory, including pathogenic mutations on each gene; gender; age at first BAVM diagnosis; number of BAVMs at first BAVM diagnosis, history of ICH; age at ICH; history of seizure; epistaxis; diagnosis of pulmonary AVMs; presence of HHT-related gastrointestinal (GI) bleeding and presence of symptomatic liver vascular malformations (VMs). We defined “BAVM multiplicity” as harboring two or more BAVMs, and “no mutation identified (NMI)” when the patient had undergone genetic testing for ENG and ACVRL1, but no mutation was identified (despite “definite” clinical HHT diagnosis).
Mutation nomenclature followed HGVS guidelines and was based on the following reference sequences; ENG, NG_009551.1; ACVR1, NG_009549.1; and SMAD4, NG_013013.2.
Data Analyses
Demographic and clinical characteristics of patients with BAVM and HHT were reported as means with standard deviation for continuous variables, and total number and proportion for categorical variables. Patients with BAVM were grouped by the gene that was mutated. Gene groups were evaluated using one-way ANOVA for continuous variables and Fisher’s exact tests for categorical variables. Fisher’s exact P-values for categorical variables are reported and P<0.05 was considered statistically significant. 95% confidence intervals (CI) were calculated for means (age at first BAVM diagnosis, age at ICH) and proportions (ICH history) by HHT gene group, except for SMAD4 because the sample size was too small (n=2) to obtain reliable estimates. Scatterplots (dot plots) were generated to display age at first BAVM diagnosis and ICH by HHT gene group. All statistical analyses were conducted using Intercooled Stata, version 11 (Stata Corp, College Station, TX).
RESULTS
Demographic Data
Of the 171 patients with HHT and BAVM, 103 were included from Yale HHT Center and 51 were recruited from the Toronto HHT Centre. An additional 17 were included from the BVMC 6203 study, of which eight were recruited through HHT Foundation International, five from the Washington University HHT Center, two from the Georgia HHT Center, and two from UCSF.
Of the 171 patients, 69 (40%) were male. The mean age at first diagnosis of BAVMs was 29 +/− 18 years, with a range of 0 – 73 years. Multiplicity information was available for 170 of 171 patients. Thirty-nine (23%) of these patients harbored more than one BAVM and the mean number of BAVMs per patient was 1.5 +/− 1.3 for the 170 patients. Information on ICH was available for 152 patients, and 41 (27%) of the patients had a history of ICH. The mean age at ICH was 26 +/− 18 years, with a broad range of 5 weeks – 68 years of age. Twenty-eight (17%) of 168 patients had a history of a seizure at or before the time of BAVM diagnosis. In the 171 patients, recurrent epistaxis was present in 78%, pulmonary AVM in 61%, HHT-related GI bleeding in 8%, and symptomatic liver VMs in 3% (Table I).
Table I.
Demographic and Clinical Characteristics of 171 Patients with HHT and BAVM
| Characteristics | |
|---|---|
| Gender (N=171) | |
| Male (#, %) | 69 (40%) |
| Genotype (N=109) | |
| ENG mutation (#, %) | 75 (69%) |
| ACVRL1 mutation (#, %) | 18 (17%) |
| SMAD4 mutation (#, %) | 2 (2%) |
| No mutation identified (#, %) | 14 (13%) |
| Age at first BAVM Diagnosis (mean years, SD) (N=165) | 29 ± 18 |
| History of ICH (#, %) (N=152) | 41 (27%) |
| Age at ICH (mean years, SD) (N=39) | 26 ± 18 |
| History of Seizure (#, %) (N=164) | 28 (17%) |
| Multiple BAVMs (#, %) (N=170) | 39 (23%) |
| Number of BAVMs (mean, SD) (N=170) | 1.5 ± 1.3 |
| History of other HHT manifestations | |
| Epistaxis (#, %) (N=169) | 132 (78%) |
| Pulmonary AVM (#, %) (N=166) | 101 (61%) |
| HHT-related GI bleeding (#, %) (N=166) | 13 (8%) |
| Symptomatic Liver AVM (#, %) (N=161) | 5 (3%) |
(HHT = Hemorrhagic Hereditary Telangiectasia, BAVM = Brain Arteriovenous Malformation, SD = Standard Deviation, ICH = Intracranial Hemorrhage, AVM = Arteriovenous Malformation, GI = Gastrointestinal)
Gene Mutations
Of the 171 patients, genetic testing information was available in 109 (64%). Of these, 75 (69%) had an ENG mutation, 18 (17%) had an ACVRL1 mutation, and two (2%) had a SMAD4 mutation. In 14 (13%) patients, no mutation was identified (NMI group) in either ENG or ACVRL1. Two of these 14 patients also had negative SMAD4 mutation testing (Table I). The detailed information for each mutation, which was available in 57 patients (47 with ENG mutations and ten with ACVRL1 mutations) from 45 families, is shown in the Supplementary Table.
Age at First BAVM Diagnosis and HHT Genes
The age at first BAVM diagnosis was available in 165 patients (97%), with a mean age of 29 +/− 18 years. The mean age (95% CI) at first BAVM diagnosis in patients with ENG, ACVRL1, or SMAD4 mutations, or NMI was 28 (24 to 32), 26 (17 to 35), 11 (not available), and 22 (12 to 32) years, respectively (Table II and Figure 1). There was no significant difference in age at first BAVM diagnosis among the HHT genes (P=0.352).
Table II.
Demographic and Clinical Manifestations of Patients with BAVM Stratified by HHT Mutated Gene
|
ENG N=75 |
ACVRL1 N=18 |
SMAD4 N=2 |
NMI N=14 |
P Value | |
|---|---|---|---|---|---|
| Male (#, %) | 30 (40%) | 4 (22%) | 0 (0%) | 5(36%) | P =0.417 |
| Age of first BAVM Dx (mean years, SD) | 28 ± 17 | 26 ± 17 | 11 ± 7 | 22 ± 17 | P = 0.352 |
| Hx of ICH (#, %) | 16 (24%) | 4 (27%) | 0 (0%) | 8 (57%) | P = 0.069 |
| Age at ICH (mean years, SD) | 26 ± 16 | 18 ± 21 | N/A | 17 ± 10 | P = 0.386 |
| Multiple BAVMs (#, %) | 21 (28%) | 3 (17%) | 0 (0%) | 3 (21%) | P = 0.783 |
| Number of BAVMs (mean, SD) | 1.5 ± 0.8 | 1.2 ± 0.5 | 1.0 | 1.4 ± 1.1 | P =0.607 |
| History of Seizure (#, %) | 9 (12%) | 1 (6%) | 0 (0%) | 2 (14%) | P = 0.820 |
| History of Epistaxis (#, %) | 59 (79%) | 14 (78%) | 2 (100%) | 10 (71%) | N/A |
| History of Pulmonary AVM (#, %) | 56 (75%) | 9 (50%) | 1 (100%) | 7 (50%) | N/A |
| History of HHT-related GI bleeding (#, %) | 7 (9%) | 3 (18%) | 0 (0%) | 0 (0%) | N/A |
| History of symptomatic Liver VM (#, %) | 1 (1%) | 2 (11%) | 0 (0%) | 0 (0%) | N/A |
(BAVM: Brain Arteriovenous Malformation, HHT = Hereditary Hemorrhagic Telangiectasia, NMI = no mutation identified on ENG or ACVRL1 gene, Dx = Diagnosis, SD = Standard Deviation, ICH = Intracranial Hemorrhage N/A = not applicable, AVM = Arteriovenous Malformation, GI = Gastrointestinal)
Figure 1.
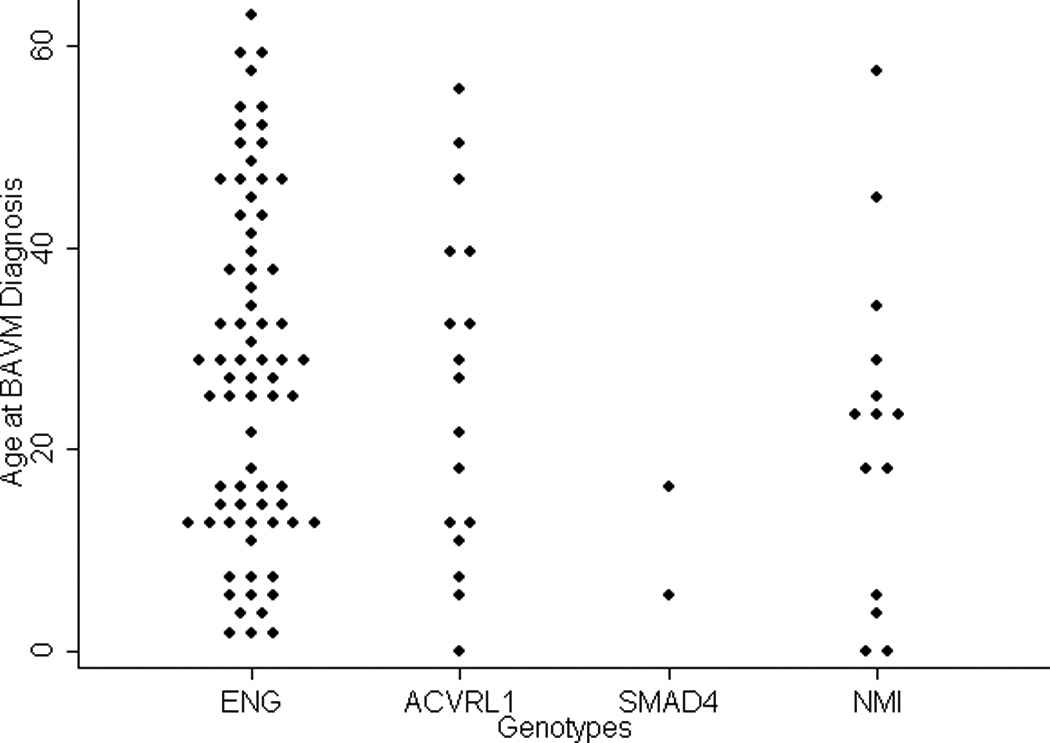
Dot plots of the age of patients at BAVM diagnosis (years) are displayed, stratified by the HHT mutated gene. BAVM: brain arteriovenous malformation. NMI: no mutation identified group
Multiplicity of BAVMs and HHT Genes
Multiplicity of BAVMs was present in 39 (23%) of 170 patients at the time of first BAVM diagnosis, with a mean number of BAVMs per patient of 1.5 +/− 1.3. Multiplicity of BAVMs and the mean number of BAVMs per patient in each gene group are indicated in Table II. There was no significant difference in the prevalence of BAVM multiplicity (P=0.783) or in the mean number of BAVMs per patient (P=0.607) among the HHT genes.
History of ICH and HHT Genes
Forty-one (27%) of the 152 patients in whom ICH history information was available had a history of ICH before or after BAVM diagnosis. A history of ICH was present in 24% (95% CI: 14% to 35%) patients with ENG mutations, 27% (8% to 55%) patients with ACVRL1 mutations, and 57% (29% to 82%) patients with NMI, but in neither of the two patients with a SMAD4 mutation (Table II). There was no significant difference in the frequency of ICH history among the HHT genes (P=0.069).
Age at ICH and HHT Genes
The mean age (95% CI) at ICH in those with an ENG mutation, an ACVRL1 mutation or NMI was 26 (17 to 34), 18 (not available) and 17 (8 to 26) years, respectively. Detailed information is shown in Table II. The age at ICH for each gene, which was not reported for patients with SMAD4 since neither had ICH, was widely distributed from infancy through adulthood (Figure 2). There was no significant difference in the age at ICH among the HHT genes (P=0.386).
Figure 2.
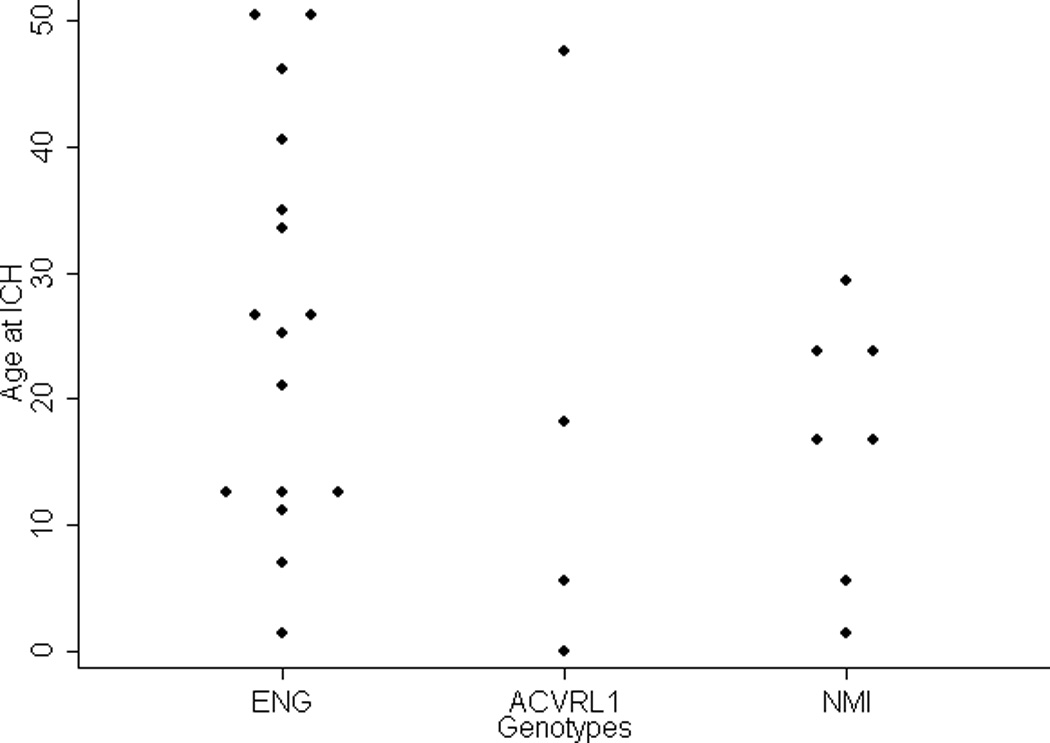
Dot plots of the age of patients at intracranial hemorrhage (years) are displayed, stratified by HHT mutated gene. NMI: no mutation identified group
Seizure, ICH, and HHT Genes
Nine patients (12%) with ENG mutation, one (6%) with ACVRL1 mutation and two (14%) with NMI had a history of seizure (Table II), with no significant difference among these groups (P=0.820).
Of total 28 patients with seizure in this study, 13 also had a history of ICH. In all patients with both a seizure and a history of ICH, their initial seizures occurred at the time of ICH. In some patients without ICH, seizures led to the BAVM diagnosis or BAVM treatment.
Other Systemic Vascular Malformations and HHT Genes
The presence of recurrent epistaxis and other HHT-related visceral AVMs with each HHT gene group is shown in Table II. Recurrent epistaxis and pulmonary AVM were frequent in our study population, whereas HHT-related GI bleeding and symptomatic liver VM were infrequent, for all HHT genes.
DISCUSSION
In this series of patients with HHT and BAVMs, we confirmed that BAVMs are present in patients with mutations in all three genes known to be associated with HHT. In addition, we show that ICH can occur in patients with ENG or ACVRL1 mutations or in the NMI group, from infancy throughout adulthood. We conclude that there is no clear evidence that decisions regarding screening for BAVM in HHT should be based on which gene is mutated or age, though larger studies are needed to confidently detect or rule out clinically significant differences among groups with mutations in the various genes.
In this study, we confirmed that not only those patients with ENG mutations are at risk for BAVM, but also those with other HHT gene mutations, as 33 (31%) of patients with BAVM had non-ENG mutations. In addition, we demonstrated 12 (43%) patients with ICH had non-ENG mutations. Prior gene-phenotype studies of HHT reported prevalence of BAVMs by gene, but none have reported clinical manifestations of BAVMs such as ICH or seizure [Bayrak-Toydemir et al 2006; Berg et al 2003; Kjeldsen et al 2005; Letteboer et al 2006; Sabba et al 2007]. We did not observe a correlation of the frequency of ICH history or age at ICH with the gene that was mutated, although the study was not designed specifically to test this hypothesis. An important observation was that the age at ICH ranged widely from infancy throughout adulthood, regardless of which gene was mutated. There was a trend in the NMI group to have a higher frequency (57%) of ICH than other HHT gene groups in this series. This may reflect unknown HHT loci with a higher risk of ICH, but another plausible explanation is that it reflects a screening bias for completion of HHT genetic test (either by clinicians, patients or both) in patients with more severe clinical presentation.
In 13 patients with history of both ICH and seizure, the initial seizure did not occur prior to ICH, but rather at the time of ICH. Based on our series, seizure may be a clue for BAVM diagnosis, but cannot be relied upon as a warning sign to predict those who will develop ICH.
Previous studies reported that 32–50% of patients with HHT and BAVM(s) have multiple BAVMs, and that the average number of BAVMs has been 2.00–2.67[Bharatha et al 2012; Matsubara et al 2000; Putman et al 1996]. The multiplicity of BAVMs in the current study is similar to previous reports, and there was no significant association with a specific HHT gene.
We have reported specific mutations when available (Supplementary Table). Interestingly, there were four ENG mutations and one ACVRL1 mutation that were detected in multiple, apparently unrelated families. It is possible that these mutations may confer greater risk for development of BAVMs among patients with HHT, though this would contradict haplo-insufficiency model as the currently accepted mechanism of HHT [Abdalla and Letarte 2006]. Alternatively, these families may be distantly related, though we did not evaluate this.
In the current study of patients with HHT and BAVMs, pulmonary AVMs, which can cause hemoptysis, brain abscess and stroke, were common regardless of the mutated gene. This observation should be considered in clinical and genetic counseling, and provides additional evidence for timely screening for pulmonary AVMs in this high-risk group.
We have limited sample size, and the clinical information is retrospective and is based on the data available from routine clinical care. For example, we did not have genetic information in 62 (36.3%) patients. Given the missing genetic data, as well as small sample size in the ACVRL1 and SMAD4 groups and incomplete information for clinical presentation, it is possible that we have missed an association of gene and phenotype. A second example is that we could not assess the risk of ICH for each HHT gene, given the small number of patients in each HHT gene groups and the cross-sectional nature of the study. However nothing limits the validity of our primary observations that BAVMs can occur with mutations in any of the genes and that ICH also can occur in the ENG, ACVRL1, or NMI group, from infancy through adulthood. Further studies of BAVMs in HHT will require larger sample sizes to adequately test for differences.
The International HHT Guideline process found only limited evidence and insufficient expert agreement to generate a recommendation for routine BAVM screening [Faughnan et al 2011]. This study may be helpful to clinicians and patients in their decision process about BAVM screening, though the current study was not designed to assess the significance of routine BAVM screening or the outcomes of management (surgical resection, endovascular embolization, radiosurgery or combination of them, or just observation). Based on our study, there is no evidence that clinicians should incorporate HHT genetic testing data or patient age into decisions about screening for BAVMs. Further studies powered to test such differences are needed to guide BAVM screening decisions for patients with HHT.
Supplementary Material
ACKNOWLEDGEMENTS
This research and fellowship for TN were granted by the Brain Vascular Malformation Consortium (BVMC). The BVMC (U54NS065705) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through the NIH Office of Rare Diseases Research (ORDR) and the National Institute of Neurological Disorders and Stroke (NINDS). M.E.F. was also supported by the Keenan Research Centre and Li Ka Shing Knowledge Institute of St. Michael’s Hospital and the Nelson Arthur Hyland Foundation.
REFERENCES
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet Part A. 2006;140:463–470. doi: 10.1002/ajmg.a.31101. [DOI] [PubMed] [Google Scholar]
- Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, Lux A, McKinnon W, Marchuk D, Guttmacher A. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40:585–590. doi: 10.1136/jmg.40.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997;61:60–67. doi: 10.1086/513903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharatha A, Faughnan ME, Kim H, Pourmohamad T, Krings T, Bayrak-Toydemir P, Pawlikowska L, McCulloch CE, Lawton MT, Dowd CF, Young WL, Terbrugge KG. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012;43:72–78. doi: 10.1161/STROKEAHA.111.629865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous ME, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJ, White RI, Young LH, Zarrabeitia R. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87. doi: 10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD, Clericuzio C, Danesino C, Drautz J, Fahl J, Fan Z, Faughnan ME, Ganguly A, Garvie J, Henderson K, Kini U, Leedom T, Ludman M, Lux A, Maisenbacher M, Mazzucco S, Olivieri C, Ploos van Amstel JK, Prigoda-Lee N, Pyeritz RE, Reardon W, Vandezande K, Waldman JD, White RI, Jr, Williams CA, Marchuk DA. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet Part A. 2010;152A:333–339. doi: 10.1002/ajmg.a.33206. [DOI] [PubMed] [Google Scholar]
- Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- Kjeldsen AD, Moller TR, Brusgaard K, Vase P, Andersen PE. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med. 2005;258:349–355. doi: 10.1111/j.1365-2796.2005.01555.x. [DOI] [PubMed] [Google Scholar]
- Letteboer TG, Mager JJ, Snijder RJ, Koeleman BP, Lindhout D, Ploos van Amstel JK, Westermann CJ. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet. 2006;43:371–377. doi: 10.1136/jmg.2005.035451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara S, Mandzia JL, ter Brugge K, Willinsky RA, Faughnan ME. Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. AJNR Am J Neuroradiol. 2000;21:1016–1020. [PMC free article] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westermann CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, Markel DS, Zolotor A, McKinnon WC, Vanderstoep JL, Jackson CE, Iannuzzi M, Collins FS, Boehnke M, Porteous ME, Guttmacher AE, Marchuk DA. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat Genet. 1994;6:197–204. doi: 10.1038/ng0294-197. [DOI] [PubMed] [Google Scholar]
- Putman CM, Chaloupka JC, Fulbright RK, Awad IA, White RI, Jr, Fayad PB. Exceptional multiplicity of cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) AJNR Am J Neuroradiol. 1996;17:1733–1742. [PMC free article] [PubMed] [Google Scholar]
- Sabba C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M, Dicuonzo F, Guant G. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007;5:1149–1157. doi: 10.1111/j.1538-7836.2007.02531.x. [DOI] [PubMed] [Google Scholar]
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, Seidman CE, Seidman JG. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet. 1994;6:205–209. doi: 10.1038/ng0294-205. [DOI] [PubMed] [Google Scholar]
- Vincent P, Plauchu H, Hazan J, Faure S, Weissenbach J, Godet J. A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet. 1995;4:945–949. doi: 10.1093/hmg/4.5.945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.