

Abstract
Background
Hyaluronic acid (HA) fillers are an established intervention for correcting facial volume deficiency. Few studies have evaluated treatment outcomes for longer than 6 months. The purpose of this study was to determine the durability of an HA filler in the correction of midface volume deficiency over 24 months, as independently evaluated by physician investigators and subjects.
Methods
Subjects received treatment with Juvéderm™ Voluma™ to the malar area, based on the investigators’ determination of baseline severity and aesthetic goals. The treatment was administered in one or two sessions over an initial 4-week period. Supplementary treatment was permissible at week 78, based on protocol-defined criteria. A clinically meaningful response was predefined as at least a one-point improvement on the MidFace Volume Deficit Scale (MFVDS) and on the Global Aesthetic Improvement Scale (GAIS).
Results
Of the 103 subjects enrolled, 84% had moderate or significant volume deficiency at baseline. At the first post-treatment evaluation (week 8), 96% were documented to be MFVDS responders, with 98% and 100% graded as GAIS responders when assessed by the subjects and investigators, respectively. At week 78, 81.7% of subjects were still MFVDS responders, with 73.2% and 78.1% being GAIS responders, respectively. Seventy-two subjects completed the 24-month study, of whom 45 did not receive supplementary Voluma™ at week 78. Forty-three of the 45 (95.6%) subjects were MFVDS responders, with 82.2% and 91.1% being GAIS responders, respectively. At end of the study, 66/72 subjects were either satisfied or very satisfied with Voluma™, with 70/72 indicating that they would recommend the product to others. Adverse events were transient and infrequent, with injection site bruising and swelling being the most commonly reported.
Conclusion
Voluma™ is safe and effective in the correction of mild to severe facial volume deficiency, achieving long-term clinically meaningful results. There was a high degree of satisfaction with the treatment outcome over the 24 months of the study.
Keywords: hyaluronic acid, dermal fillers, volume deficit, volume deficiency, mid-face, malar
Introduction
Facial appearance is defined by properties of the skin, muscle, bony anatomy, and amount and distribution of subcutaneous adipose tissue. Loss of bone mass and soft tissue volume, redistribution of fat, as well as decreased skin elasticity and thickness contribute to the formation of wrinkles and folds characterizing the signs of aging. These events are typically noticeable by 30 years of age in most individuals.1–3 With respect of the midface, flattening and furrowing of the central area of the midcheek is observed, with displacement medially exaggerating the depth of the nasolabial folds.4–7 In addition, the lower eyelid lengthens and the upper border of the malar fat pad becomes visible, leading to tear trough formation.4–6,8 Therefore, appropriate replacement of lost volume and provision of structure should correct these signs of aging, creating a more youthful appearance.
Hyaluronic acid (HA) is a naturally occurring glycosaminoglycan and a key component of the extracellular matrix in adult tissue. Approximately 50% of the total HA in the body is located in the dermis.9 Due to its high affinity for water, HA plays an integral role in the regulation and maintenance of hydration within tissues.10,11 HA-based dermal fillers have been developed through the application of proprietary-based manufacturing technologies which achieve resistance to biological degradation and to shear stress in vivo, leading to improved treatment durability while enabling administration at an acceptable extrusion force. The documented safety and efficacy of these products have established HA-based fillers as a standard treatment for correction of facial lines and folds and, more recently, to restore or create structure and volume.7,11 One product, Juvéderm™ Voluma™, is prepared with a combination of low molecular weight (<1 mDa) and high molecular weight (>1 mDa) hyaluronic acid. This results in a more mobile fiber network, facilitating a high degree of covalent binding to the cross-linking agent: 1,4-butanediol, as observed with 1H-nuclear magnetic resonance spectroscopy analysis (Data on file, Allergan Inc, 2012). This more efficient cross-linking capacity influences both the rheological properties (eg, G′ values) and the swelling ratio of the filler, ie, the amount of water a filler is capable of retaining. Voluma™ has a high G′ and a low swelling ratio, facilitating high lift capacity, which is desirable for deeper injections, with little change in the volume of the implant following its deposition (Data on file, Allergan Inc, 2012). In biocompatibility testing, during which the filler was in contact with tissue for more than 30 days, Voluma™ was determined to be noncytotoxic, nonirritant, nontoxic, nonsensitizing, nongenotoxic, and nonpyrogenic. Further, there is no evidence that 1,4-butanediol is carcinogenic (Data on file, Allergan Inc, 2012).
To document how these physicochemical properties might contribute to clinical performance over time, the primary objective of the current study was to evaluate the durability of Voluma™ in the correction of midface volume deficiency over 24 months, using validated assessment scales and by subject-reported outcomes.
Materials and methods
Subjects
Eligible study subjects were aged between 30 and 60 years, with bilateral mild (2), moderate (3), significant (4), or severe (5) midface volume deficit, as defined by the validated midface volume deficit scale (MFVDS; Data on file, Allergan, 2012). The midface region was defined as the medial malar, lateral malar/cheek, and the buccal/submalar areas. Exclusion criteria included any pre-existing condition which might affect the evaluation of treatment outcome including: visible scars, wounds or lesions, active inflammation, infection, or connective tissue disease in the face. Subjects were also excluded for any of the following: filler treatment in the midface or in the nasolabial folds in the preceding 12 months; fat injections, surgical procedures in the face or neck in the preceding 3 months, or prior treatment with permanent or semipermanent facial implants in the treatment area. Pregnancy, nursing mothers, a history of HA hypersensitivity, bleeding disorders, or use of aspirin, anticoagulants or antiplatelet therapy within 10 days of the screening/baseline visit, were also exclusions. Females of childbearing potential were required to have a negative urine pregnancy test and to use adequate contraception while participating in the study.
The study was approved by a central institutional review board and conducted at six Australian private aesthetic clinics, in full accordance with Good Clinical Practice regulations and guidelines, including the International Conference on Harmonization. All subjects were required to provide written informed consent and sign a photography release form prior to any study-related procedures. This study is listed on the Clinical Trials.gov registry (NCT01029535).
Treatment
Commercially available Voluma™ (Allergan Inc, Irvine, CA, USA) was used for this study. Before administration of the study product, the subjects removed makeup and the treatment area was cleaned with antiseptic solution. The product was administered with either a 23 g needle or a 18 g; 70 mm cannula. Topical or injectable anesthesia was permitted to the treatment area, as was the use of ice before and after treatment. The mode of administration (needle or cannula), site of administration (submalar, lateral, and/or medial), principal administration technique (bolus, fanning, and/or crosshatching), injection technique (retrograde and/or antegrade), and depth of injection (supraperiosteal or subcutaneous) were at the discretion of the physician investigator. At baseline (week 0), the investigators were instructed to correct the midface volume deficiency in each subject to a post-treatment rating of minimal (1) or mild (2), based on the MFVDS, with consideration of the subject’s pretreatment deficiency and the maximum volume of product permissible for administration. At week 0, a maximum volume of 2 mL per side was allowed. Additional study product at week 4 could be administered (up to 2 mL per malar) if the following protocol-defined criteria were fulfilled: presence of asymmetry in the malar region and/or only a 1 grade improvement in the subject’s pretreatment MFVDS score was achieved following the initial (week 0) treatment session. The first post-treatment evaluation for efficacy was conducted at week 8. Further treatment at the midface was not permitted until week 78 (18 months). At this visit, the study product could only be administered if the subject desired retreatment and the MFVDS score had worsened by 1 grade or more from the score documented at the week 8 visit (when the peak therapeutic effects of the study treatment were anticipated). Subjects not fulfilling this retreatment criteria at week 78 were reassessed using the same criteria at week 104 (the final visit) and were treated only if the retreatment criteria were met. Subjects treated at the week 78 visit could not be retreated again at week 104. Assessment for retreatment at the week 78 or 104 visits occurred after evaluation of treatment outcome by the investigators and the subjects at these visits, based on the outcome parameters detailed below. The administration of a filler product for the treatment of tear troughs was permissible only after completion of the week 78 evaluation in selected subjects, and the discretion of the investigator. No other aesthetic treatments were allowed to the midface during the study.
Outcome parameters
The severity of the midface volume deficiency was graded by the investigators at baseline (week 0) and at the scheduled on-study clinic visits (weeks 8, 52, 78, and 104), based on the MFVDS. This validated scale ranges from none (0) to severe (5) with a photonumeric guide comprising reference photographs labeled with differentiating descriptions for each severity grades (Data on file, Allergan Inc, 2012). All investigators were instructed on how to use this scale prior to study initiation. In addition, at each of these on-study visits, both investigators and subjects were asked to independently evaluate the aesthetic improvement in the subject’s midface from his/her pretreatment state, using the Global Aesthetic Improvement Scale (GAIS). This involved referencing the two-dimensional digital photographs of the frontal and lateral views of the subject taken prior to administration of the study treatment at week 0, with the subjects also viewing themselves in a mirror. The GAIS is a five-point Likert scale, ranging from “much improved” to “much worse”. The subjects also indicated their degree of satisfaction with the study treatment on a four-point scale and, further, whether they would recommend the treatment to others. At each clinic visit, the subjects always completed their self-evaluation independently and prior to the investigator’s evaluation. Assessment of treatment outcome using these scales at weeks 78 and 104 clinic visits was always conducted prior to evaluation for retreatment, as stated previously. A 1 grade or greater improvement in the MFVDS or GAIS from baseline was considered to be clinically meaningful (“responder”).
Evaluation of the safety and tolerability of the study product was based on spontaneous reporting of adverse events by the subjects, as well as through nonleading questioning of the study subjects’ general health at each study visit, or over the telephone on days 3 and 30 after retreatment at week 78 or at week 104 in those subjects who received retreatment. The study investigators also assessed the treatment area for injection site reactions at the scheduled clinic visits.
Statistical analysis
All eligible subjects receiving treatment at baseline (week 0) were included in the safety analyses. A responder analysis was performed at weeks 8, 52, 78, and 104, based on the proportion of subjects achieving a 1 grade or better improvement from baseline for both sides of the face, as evaluated using the MFVDS (investigator assessment) or the GAIS (independent investigator and subject self-assessments). The efficacy outcome data are presented for those subjects who received no further study treatment after week 0 or 4. As a consequence, subjects who received retreatment after outcome evaluation at week 78 were excluded from the week 104 analysis. Results are expressed as observed cases of subjects achieving each event with 95% confidence intervals (CI) at each time point.
Results
Subjects
A total of 103 subjects were enrolled and received the study treatment. All subjects met the entry criterion of an MFVDS score between 2 and 5. The majority of subjects had pretreatment grades of 3 (44%) or 4 (40%), consistent with a moderate or significant volume deficiency, while 9% and 7% of subjects presented with grades of 2 (mild) and 5 (severe), respectively, as documented by the physician investigators. Examples of subjects with a pretreatment MFVDS score of 3, 4, or 5 are presented in Figure 1. The mean age of the subjects was 47 (range 29–60) years, with the majority being female (81%). Most of the subjects (96%) had a Fitzpatrick score of 2, 3, or 4. A total of 82 (80%) and 72 (71%) of the subjects completed the week 78 and final (week 104) visits, respectively. The reasons for premature subject withdrawal are listed in Table 1.
Figure 1.
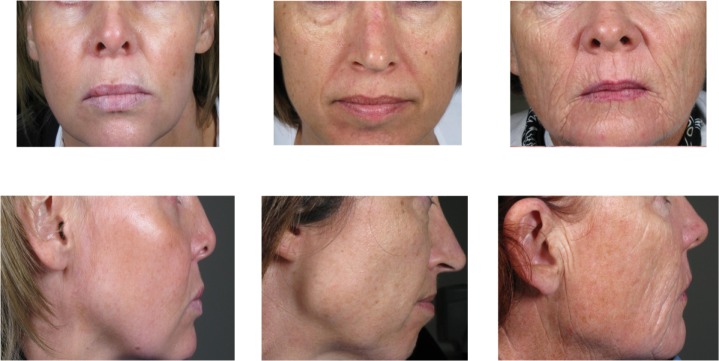
Study subjects with a pre-treatment MFVDS grade of 3, 4, and 5 respectively, as evaluated by the physician investigator.
Abbreviation: MFVDS, Midface Volume deficient Scale.
Table 1.
Reasons for withdrawal from the study
| Reason | n |
|---|---|
| Lost to follow-up | 24 |
| Protocol violation | 2 |
| Withdrew consent | 2 |
| Adverse event | 1 |
| Death | 1 |
Dosing and method of administration
The mean volume injected at the week 0 to both the right and left malar regions was 1.7 mL per side. At week 4, 68% of subjects met the prespecified criteria for supplementary treatment of the right malar and 63% of the left malar and were retreated (mean volume administered was 1.3 mL per side). The mean volume administered over this initial 4-week period of the study for the total study population was 5.1 mL. Of the 82 subjects who completed the week 78 visit evaluation, 33 (40%) fulfilled criteria for retreatment and were retreated (mean volume, 1.3 mL per side). At the completion of the study (week 104), 26 (36.1%) of the 72 subjects attending this visit were retreated following confirmation that they fulfilled the protocol-defined criteria. Again, evaluation of treatment outcome, based on the MFVDS and GAIS instruments, always occurred prior to assessment for retreatment at these visits.
Efficacy
Subjects with ≥1 grade improvement on MFVDS (physician evaluation)
The proportion of subjects who achieved a 1 grade or better improvement in their baseline MFVDS score (MFVDS responders) for each of the clinic visits is presented in Figure 3. Based on this outcome measure, 96% of subjects were responders at week 8. This treatment effect was maintained in 88.2% and 81.5% of the subjects at weeks 52 and 78, respectively, without supplementary treatment after week 0 or 4 (Figure 3A). For evaluation of outcome at week 104, only a subset of 45 of the 72 subjects who completed the study were included in this analysis. These subjects did not receive supplementary treatment with the study product at the week 78 visit and up to the time of the last outcome evaluation in this study. In this cohort, 43/45 (95.6%) of subjects were still MFVDS responders at week 104, as evaluated by the investigators (Figure 3B).
Figure 3.
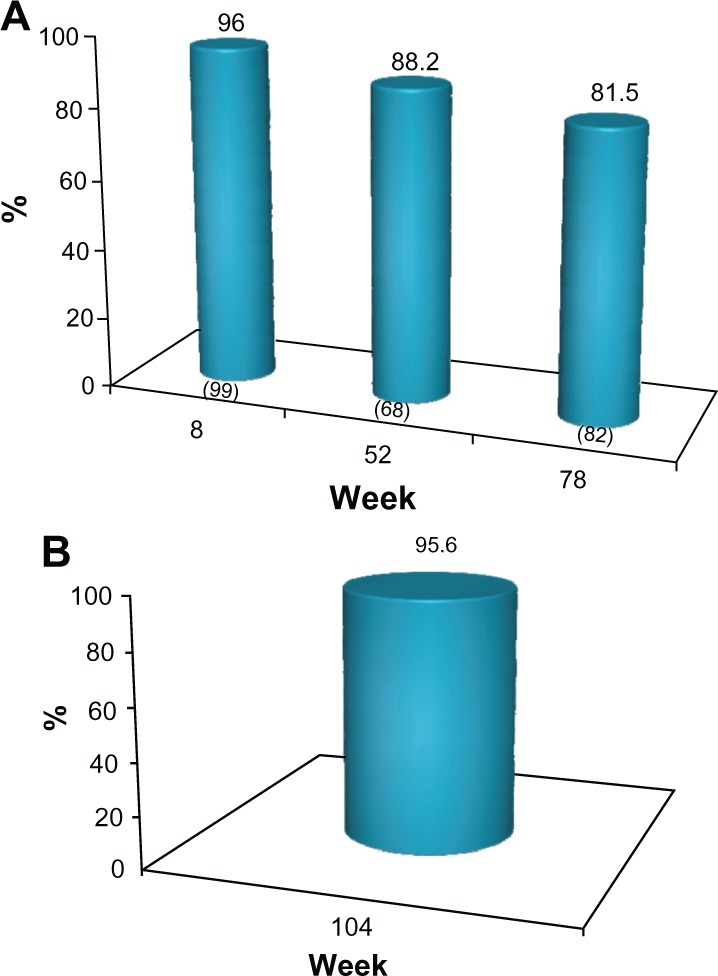
(A) Proportion of subject with ≥1 point improvement from baseline in MFVDS score, (“MFVDS responder”) as evaluated by the physician investigators (all subjects). No subject received further study treatment after week 0 or 4. Numbers in parentheses are the sample size at each visit. (B) Proportion of responders from the sub-group of 45 subjects who completed the study and who received no further treatment with the study product after the week 0 or 4.
Abbreviation: MFVDS, Midface Volume Deficit Scale.
The MFVDS responder rate with the 95% CI at each of the scheduled clinic visits is also shown in Table 2. To illustrate this improvement, the subject in Figure 2 maintained a 1 grade improvement in MFVDS, relative to her baseline score over the duration of the study following treatment at weeks 0 and 4 only.
Table 2.
Proportion of subjects with maintenance of a clinically meaningful correction of their midface volume deficiency during the study, based on physician MFVDS and physician and subject GAIS scoresa,b
| % responders (95% CI) |
||||
|---|---|---|---|---|
| Outcome parameter (n)c |
Week 8 99 |
Week 52 68 |
Week 78 82 |
Week 104 45d |
| MFVDS (physician investigator evaluation) |
96.0 (90.0, 98.9) |
88.2 (78.1, 94.8) |
81.7 (71.6, 89.4) |
95.6 (84.9, 99.5) |
| GAIS (physician investigator evaluation) |
100 (96.3, 100) |
88.2 (78.1, 94.8) |
78.1 (67.5, 86.4) |
91.1 (78.8, 97.5) |
| GAIS (subject evaluation) |
98.0 (92.9, 99.8) |
79.4 (67.9, 88.3) |
73.2 (62.2, 82.4) |
82.2 (68.0, 92.0) |
Notes:aImprovement of ≥1 grade from baseline score for each scale
data presented for cohort of subjects who received no further treatment with the study product after the week 0 or 4 clinic visit
number of subjects evaluated at each clinic visit
In comparison, the responder rate for all 72 patients attending this visit, including those retreated at week 78 (n = 27) was: MFVDS: 91.7%, GAIS (physician investigator evaluation): 90.3%, GAIS (subject’s evaluation): 86.1%.
Abbreviations: CI, confidence interval; GAIS, global aesthetic improvement scale; MFVDS, Midface Volume Deficit Scale.
Figure 2.

Study subject with a pre-treatment MFVDS grade of 4 who maintained a ≥1 grade improvement from baseline/pre-treatment state for the duration of the 104 week study after Voluma™ treatment at weeks 0 and 4 only, as evaluated using the MFVDS (physician investigator assessment) and the GAIS (independent physician investigator and subject evaluations).
Abbreviations: GAIS, Global Aesthetic Improvement Scale; MFVDS, Midface Volume Deficit Scale.
Subjects with ≥1 grade improvement on GAIS (independent physician and subject evaluation)
The proportion of subjects who achieved a 1 grade or better improvement in their pre-treatment appearance (GAIS responders) at each clinic visit is presented in Figures 4 and 5 for investigator assessment and subject self-evaluation, respectively. At the week 8 visit, the investigators rated 100% of subjects as GAIS responders. This was consistent with subject self-evaluation, for which a 98% response rate was reported. At week 52, the investigators determined that 88.2% of subjects responded, while the subject GAIS responder rate was 79.4%. At week 78, this improvement continued to be maintained in 78.1% and 73.2% of the subjects, respectively (Figures 4A and 5A). In the cohort of 45 subjects who completed the study and who did not receive supplementary study treatment after the week 0 or 4 visits, 91.1% and 82.2% were documented as GAIS responders at the week 104 visit, respectively (Figures 4B and 5B).
Figure 4.
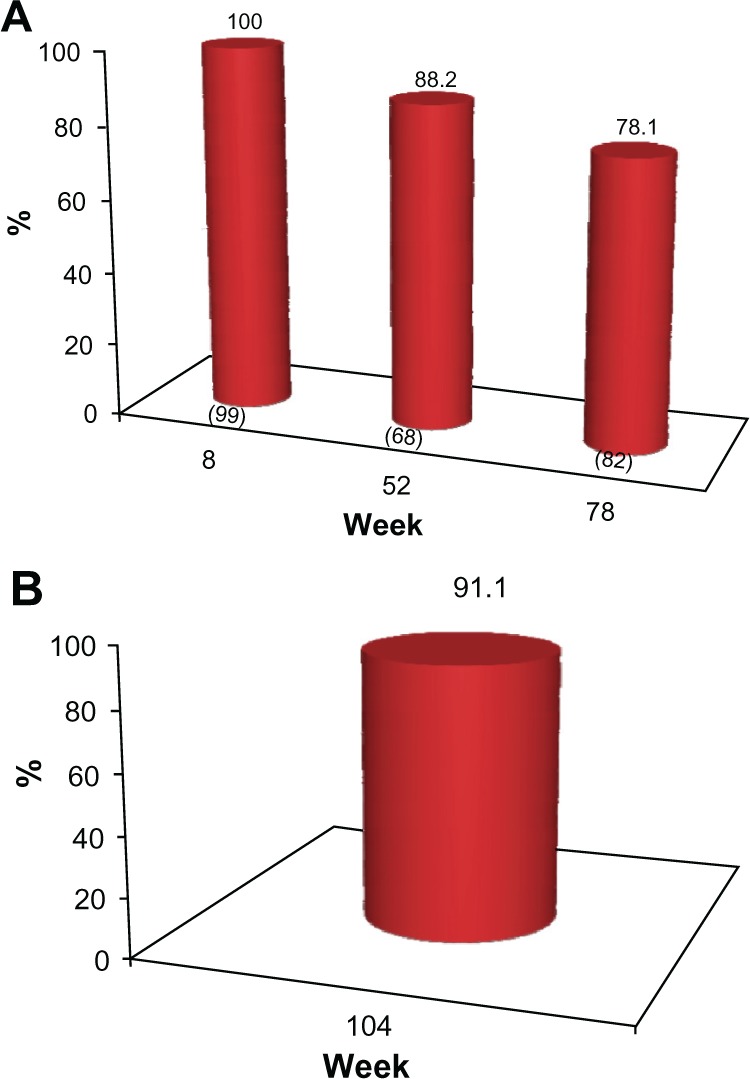
(A) Proportion of subjects with ≥1 point improvement in their pre-treatment appearance based on GAIS (“GAIS responder”), as evaluated by the physician investigators. No subject received further study treatment after week 0 or 4. Numbers in parentheses are the sample size at each visit. (B) Proportion of subjects from the subgroup of 45 who completed the study and who received no further treatment after week 0 or 4 with ≥1 point improvement in their pre-treatment appearance, based on the GAIS (“GAIS responder”) at week 104, as evaluated by the physician investigators.
Abbreviation: GAIS, Global Aesthetic Improvement Scale.
Figure 5.
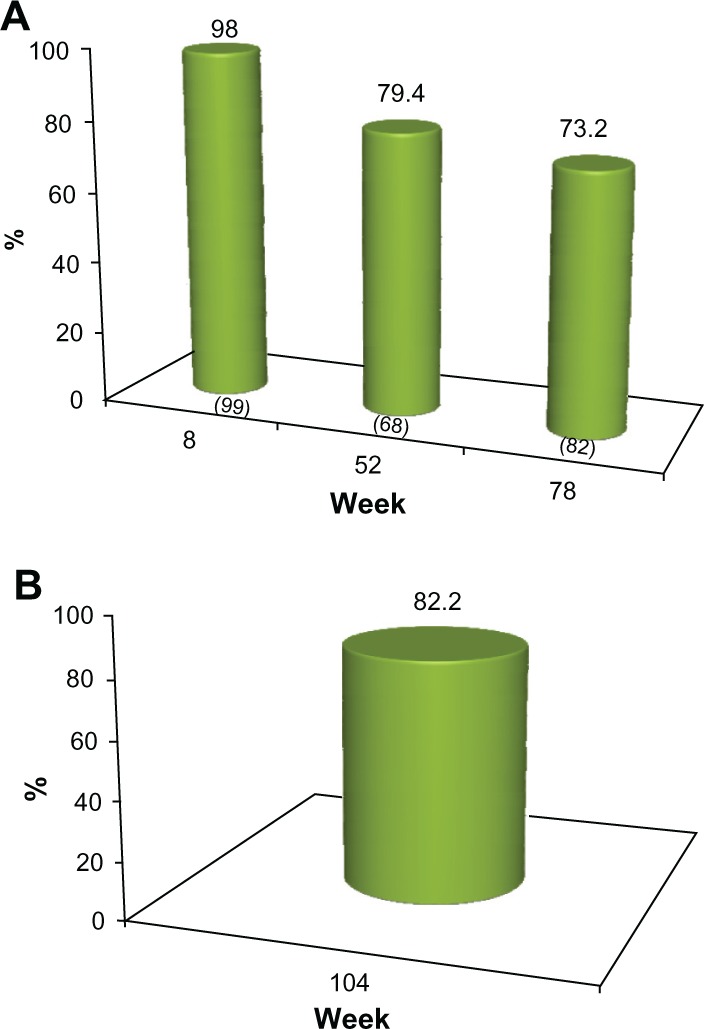
(A) Proportion of subjects with ≥1 point improvement in their pre-treatment appearance based on GAIS (“GAIS responder”), as evaluated by the subjects. No subject received further study treatment after week 0 or 4. Numbers in parentheses are the sample size at each visit. (B) Proportion of subjects from the sub-group of 45 who completed the study and who received no further treatment after week 0 or with a ≥1 point improvement in their pre-treatment appearance, based on the GAIS (“GAIS responder”) at week 104, as self-evaluated by the subjects.
Abbreviation: GAIS, Global Aesthetic Improvement Scale.
The responder rate for the GAIS at each clinic visit is also presented in Table 2 for both investigator assessment and subject self-evaluation. The subject in Figure 2 also maintained a 1 grade improvement in GAIS score over the duration of the study, relative to her pre-treatment appearance, as independently assessed by the investigator and by the subject. Again, this subject did not receive further treatment with the study product after week 4.
Subject treatment evaluation
Of the 72 subjects who completed the 24-month study, 66 (91.6%) were either satisfied or very satisfied with the study treatment, based on the five-point Likert scale employed in this study, while 70 (97.2%) indicated that they would recommend the treatment to others. This high level of satisfaction with the study treatment was documented at the first post-treatment evaluation at week 8 and this was consistently reported by the subjects at each clinic visit thereafter (Figures 6 and 7).
Figure 6.

Treatment Satisfaction of the Study Subjects (all observed subjects).
Figure 7.
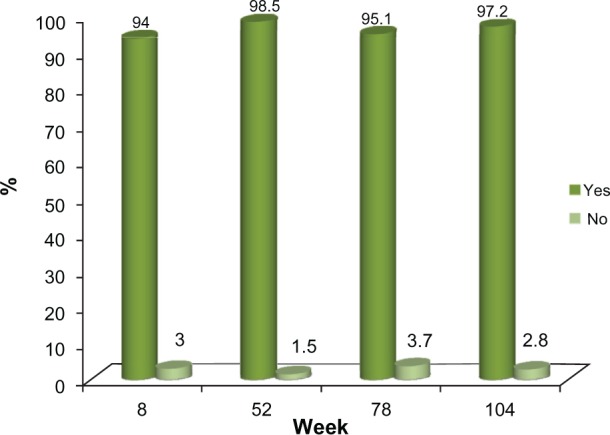
Study Treatment Recommendation.
Note: Study Subject’s response to the following question: “Would you recommend Voluma™to others”? (all observed subjects).
Safety and tolerability
The study treatment was well tolerated, with the majority of treatment-related adverse events being transient, of mild to moderate severity, and localized to the injection sites; principally bruising and swelling. Treatment-related injection site reactions are presented in Table 3 for two observational periods to document the two intervals following administration of the study product: weeks 0–8 and weeks 78–108, including the 4 week post-treatment follow-up period for subjects receiving retreatment at week 104. Any adverse events occurring after week 8 were documented during this second observational period.
Table 3.
Incidence of injection site reactions following the two observational periods (weeks 0–8 and weeks 78–108)
| Event | n (%)a | n (%)b |
|---|---|---|
|
| ||
| Weeks 0–8 | Weeks 78–108 | |
| Bruisingc | 42 (40.8) | 2 (1.9) |
| Swelling | 15 (14.6) | 5 (4.9) |
| Pain/tenderness | 8 (7.8) | 1 (1.0) |
| Erythema | 2 (1.9) | 0 (0) |
| Eyelid edema | 1 (1.0) | 0 (0) |
| Vasovagal syncope | 1 (1.0) | 0 (0) |
Notes:aFrequency of adverse events reported during the period: week 0–8 in 103 subjects receiving the study treatment at week 0
frequency of adverse events reported during the period: week 78–108 following re-retreatment with the study product in eligible subjects at either week 78 or week 104. Subjects receiving the study treatment at week 104 were followed for a total of 4 weeks post-treatment for safety and tolerability. New adverse events reported between weeks 8 and 78 were documented in the second observational period
includes lump/hematoma.
There was a single case of swelling in the left tear trough area which became more severe and generalized over time. This event occurred approximately 17 weeks after the week 4 treatment with the study product and 2 months after bilateral administration of Juvéderm Ultra™ to the tear troughs, and lead to bilateral hardening of the Voluma™ implant. Oral prednisolone 5 mg/day was administered over 5 days and hyalase (100–150U) was injected three times over a 3-week period. The swelling completely resolved approximately one month after the third hyalase session. One subject was withdrawn from the study after the week 52 visit following a positive pregnancy test. The pregnancy went to term with no complications. One subject died approximately 3 months after the week 78 visit. The investigator did not consider that this event was related to the trial or to the study treatment.
Discussion
This study enrolled 103 Australian subjects who were predominantly women in their mid-40s with mild to severe mid-face volume deficiency, most likely related to a combination of age-related and environmental factors. The objective of the study was to assess the safety and efficacy of Voluma™ in achieving a clinically meaningful correction in the mid-face in this study cohort and, furthermore, to assess whether this initial correction would persist over the duration of the 24-month observational period of the study. To this end, after one or two treatment sessions over the initial 4-week period of the study, further treatment to the midface with the study product (or any other aesthetic treatment) was not permitted prior to the week 78 visit and only in those subjects who fulfilled the protocol-defined criteria for retreatment at this visit. So that the durability of study product could be appropriately evaluated, only those patients who did not receive Voluma™ after week 0 or 4 were included in the efficacy analysis. Because all the 82 subjects attending the week 78 visit were evaluated for treatment efficacy prior to assessment for retreatment, all 82 were included in the analysis at week 78. At week 104, 45 of the 72 subjects who attended this visit had not received retreatment after week 4, and thus this cohort of 45 was included in the efficacy analysis at week 104.
Two validated assessment instruments, the physician’s MFVDS. and the GAIS, were used in this study. The GAIS was completed independently by the subjects and the physician investigators on separate case report forms, with the subject self-evaluation always occurring prior to meeting with the physician investigator. A 1 grade or better improvement from baseline was considered to be a clinically meaningful outcome (ie, “treatment responder”) for both of these assessment scales.
When analyzed, Voluma™ treatment administered over the initial 4-week period of the study (mean volume: 5.1 mL) led to a clinically meaningful correction in the midface for almost all subjects at the first post-treatment assessment (week 8). Thereafter, this outcome was achieved in approximately 80% of the subjects at week 52 and over 70% of the subjects at week 78, based on these validated assessment scales (Figures 3A, 4A, and 5A). At the last study visit (week 104), over 80% of 45 subjects completing this visit without retreatment after week 0 or 4 maintained a clinically meaningful volume correction, based on these two assessment scales (Figures 3B, 4B, and 5B). Consistent with these observations, at the end of each of the scheduled study visits, over 90% of subjects indicated that they were satisfied or very satisfied with the study treatment and would recommend the product to others.
The enduring treatment effects of Voluma™ to 18–24 months following one or two treatment sessions over the first 4 weeks of the study is likely to be related to the unique physicochemical and structural properties of Voluma™ that impart lift effects as well as a resistance to biological degradation and shear stress.11,12
The number of subjects who withdrew prior to the end of the study, principally due to loss of follow-up (Table 1) requires comment. Based on the high satisfaction rate documented by the subjects at each of the scheduled study visits (Figures 6 and 7), it seems possible that subjects lost to follow-up were still satisfied with their treatment results and thus were not interested in being retreated. The adverse events recorded suggest the treatment was well tolerated, so safety-related issues for this premature withdrawal seem unlikely. Also, no subject asked for treatment with hyaluronidase to remove the study product. In this trial, subjects were offered the opportunity for retreatment at week 78 or 104 at no cost to them, if they qualified, and thus there was an overall incentive for subjects to remain in the trial for its duration.
The study treatment was well tolerated, with most adverse events being injection site reactions of mild to moderate severity. Notably, the incidence of adverse events in those subjects who received retreatment at the week 78 or 104 clinic visits was substantially lower than that documented during the first 8-week period of the study, when up to two treatments could be administered (Table 3). None of investigators had previously injected the study product, other than during a brief training session conducted at the prestudy investigators meeting. Therefore, an improved injection technique on the part of the investigators gained during the study could have contributed to this improved tolerability. Therefore, more extensive experience with injecting Voluma™ prior to the study might have reduced the overall incidence of injection site reactions. Lower relative injection volumes (mean volume per side, 1.7 versus 1.3 versus 0.9 mL for weeks 0, 78, and 104, respectively) could also be a possible explanation for the reduced incidence of adverse events after between weeks 78 and 108.
The single case of edema around the left lower eyelid, which preceded a generalized swelling and the hardening of the Voluma™ implant requires further discussion. This event occurred approximately 17 weeks after the study product treatment at week 4 and approximately 6 weeks after the non-study related administration of Juvéderm Ultra™ to the tear troughs. Such an event is rare with HA fillers and the etiology is unclear and may relate to multiple factors, including the amount of filler administered, location of the implant, the process and degree of degradation of the implant, and indeed a subject’s predisposition to such an event (ie, anatomy, prior surgery, and/or filler treatments in the area). The relative importance of each of these potential factors is unknown.8
A potential limitation of this study is the utilization of Likert scales to evaluate treatment outcome. These scales are, by their very nature, subjective instruments. Nevertheless, they are a practical tool for continuous tracking of treatment outcomes and have been validated and used as the primary efficacy parameter in similar studies published in the peer reviewed literature.13,14 Measuring volumetric changes with three-dimensional camera systems15 might address the deficiencies of these subjective rating scales. However, the cost of these systems and the associated data analysis is likely to be prohibitive in most postregistration studies conducted at multiple sites. Another potential issue in this study was the failure to monitor the subject’s body weight during the study to evaluate the impact of any significant weight fluctuation on facial adiposity and appearance.
Conclusion
In summary, this 24-month prospective, observational study is the longest and largest of its type ever conducted to assess the durability of an HA-based filler in the correction of facial volume deficiency. In this study, in which a majority of subjects had a moderate to significant volume deficiency of the midface at baseline, treatment with the study product, administered in one or two sessions over the initial 4-week period of the study, was sufficient to achieve a clinically relevant correction in over 95% of subjects when independently evaluated by investigators and subjects at the first post-treatment visit (week 8), using two validated assessment scales, ie, the MFDVS and GAIS. At week 78, this clinically relevant improvement was maintained in over 70% of the 82 subjects completing this visit, based on the same assessment scales, while the outcome was enduring in over 88% of the 45 subjects who completed this 24-month study, with no further study treatment after either week 0, week 4, or week 78. There was a high degree of consistency in the documentation of a clinically meaningful correction during the study, as measured independently by the investigators and subjects using the GAIS instrument. Relevant to this, the subjects indicated a high level of satisfaction with the treatment outcome when evaluated at each scheduled clinic visit, including at the end of the study. Voluma™ is a safe and effective alternative to invasive surgical procedures for the correction of midface volume deficiency.
Acknowledgment
This work was published on behalf of the Juvéderm Voluma™ Long-Term MidFace Volume Deficit Study Group (Protocol VOL-AP-01). Permission has been obtained from the patient for the reproduction of the images in Figure 2.
Footnotes
Disclosure
The study was sponsored by Allergan Australia, NSW, Australia. The data management and statistical analysis for this study was conducted independently by John Wlodarczyk Consulting Services, NSW, Australia. PC, IC, GG, SL, PM, and TS were paid investigators and were provided with the study product by Allergan for administration in this study. GG has also been a paid investigator for studies conducted on behalf of Kythera, Galderma, and Peplin. GG has also been a paid consultant to Allergan, Galderma, Valeant, Peplin, and Q-Med. JR and MH are paid employees of Allergan.
References
- 1.Ascher B, Coleman S, Alster T, et al. Full scope of effect of facial lipoatrophy: a framework of disease understanding. Dermatol Surg. 2006;32:1058–1069. doi: 10.1111/j.1524-4725.2006.32230.x. [DOI] [PubMed] [Google Scholar]
- 2.Sadick NS, Smoller B. A study examining the safety and efficacy of a fractional laser in the treatment of photodamage on the hands. J Cosmet Laser Ther. 2009;11:29–33. doi: 10.1080/14764170802612992. [DOI] [PubMed] [Google Scholar]
- 3.Donofrio LM. Fat distribution: a morphologic study of the ageing face. Dermatol Surg. 2000;26:1107–1112. [PubMed] [Google Scholar]
- 4.Monheit GD, Thomas JA, Murphy DK, et al. Photographic documentation from a double-blind, randomized, multicenter study comparing new hyaluronic acid-based fillers vs. crosslinked bovine collagen; Poster 202 presented at the Annual Scientific Meeting of the American Academy of Dermatology; July 26–30, 2006; San Diego, CA. [Google Scholar]
- 5.Coleman SR, Grover R. The anatomy of the aging face: volume loss and changes in 3-dimensional topography. Aesthet Surg J. 2006;26(1S):S4–S9. doi: 10.1016/j.asj.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 6.Mendelson BC, Jacobsone SR. Surgical anatomy of the midcheek: facial layers, spaces, and the midcheek segments. Clin Plast Surg. 2008;35:395–404. doi: 10.1016/j.cps.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Raspaldo H. Volumizing effect of a new hyaluronic acid sub-dermal facial filler: a retrospective analysis based on 102 cases. J Cosmet Laser Ther. 2008;10:134–142. doi: 10.1080/14764170802154607. [DOI] [PubMed] [Google Scholar]
- 8.Funt DK. Avoiding malar edema during midface/cheek augmentation with dermal fillers. J Clin Aesthet Dermatol. 2011;4:32–36. [PMC free article] [PubMed] [Google Scholar]
- 9.Kablik J, Monheit GD, Yu L, Chang G, Gershkovich J. Comparative physical properties of hyaluronic acid dermal fillers. Dermatol Surg. 2009;35 (Suppl 1):302–312. doi: 10.1111/j.1524-4725.2008.01046.x. [DOI] [PubMed] [Google Scholar]
- 10.Comper WD, Laurent TC. Physiological function of connective tissue polysaccharides. Physiol Rev. 1978;58:255–315. doi: 10.1152/physrev.1978.58.1.255. [DOI] [PubMed] [Google Scholar]
- 11.Tezel A, Fredrickson GH. The science of hyaluronic acid dermal fillers. J Cosmet Laser Ther. 2008;10:35–42. doi: 10.1080/14764170701774901. [DOI] [PubMed] [Google Scholar]
- 12.Sall I, Ferard G. Comparison of the sensitivity of 11 crosslinked hyaluronic acid gels to bovine testis hyaluronidase. Polym Degrad Stab. 2007;92:915–919. [Google Scholar]
- 13.Goodman GJ, Bekhor P, Rich M, Rosen RH, Halstead MB, Rogers JD. A comparison of the efficacy, safety, and longevity of two different hyaluronic acid dermal fillers in the treatment of severe nasolabial folds: a multicenter, prospective, randomized, controlled, single-blind, within-subject study. Clin Cosmet Investig Dermatol. 2011;4:197–205. doi: 10.2147/CCID.S26055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ascher B, Bayerl C, Brun P, et al. efficacy and safety of a new hyaluronic acid dermal filler in the treatment of severe nasolabial lines – 6-month interim results of a randomized, evaluator-blinded, intra-individual comparison study. J Cosmet Dermatol. 2011;10:94–98. doi: 10.1111/j.1473-2165.2011.00550.x. [DOI] [PubMed] [Google Scholar]
- 15.Meier JD, Glasgold RA, Glasgold MJ. 3D photography in the objective analysis of volume augmentation including fat augmentation and dermal fillers. Facial Plast Surg Clin N Am. 2011;19:725–735. doi: 10.1016/j.fsc.2011.07.012. [DOI] [PubMed] [Google Scholar]