

Abstract
Castration resistant prostate cancer remains a major clinical burden and novel therapeutic options are urgently required to improve survival. Tasquinimod is an orally administered quinoline-3-carboxamide with potent antiangiogenic and antitumorigenic action that has shown promise in the treatment of advanced prostate cancers. This review explores both preclinical and clinical findings to date. In summary, tasquinimod has been shown to demonstrate a potent in vitro and in vivo anticancer action and completed early phase clinical trials have demonstrated good drug tolerance and prolonged progression-free survival. Although Phase III clinical trials are on-going, the findings to date highlight the promise of this drug in the treatment of advanced prostate cancer.
Keywords: prostate cancer, castration resistant prostate cancer, antiangiogenesis, S100A9, HDAC4, ABR-215050, quinoline-3-carboximide
Introduction
Prostate cancer is the most common male malignancy in Europe and the United States.1 Although organ-confined prostate cancer is often curable, 20%–30% of men present with metastatic cancer for which radical therapy is not possible.2 In cases of advanced prostate cancer, the mainstay of treatment is with androgen deprivation therapy. However, its effects are transient and disease progression to castration resistant prostate cancer (CRPC) inevitably occurs. Despite improved survival in CRPC using taxanes (docetaxel and cabazitaxel) and abiraterone, patients remain incurable and in the United States more than 32,000 men die annually of metastatic disease.3–7 Molecular characterizations of CRPC have revealed that androgen receptor pathways remain active despite androgen deprivation therapy and therefore considerable translational research is focused on the targeted inhibition of the androgen receptor.8 Alternative strategies for treating prostate cancer include targeting the tumor microenvironment.9 In order for cancer cells to survive, they require a continuous supply of both oxygen and nutrients. The diffusion limit of oxygen in tissue is between 100 and 200 μm and thus, for a tumor to grow beyond this size it requires neovascularization.10 The use of drugs targeting the molecular pathways involved in angiogenesis offers a novel approach to prostate tumor treatment. This review explores a role for such an antiangiogenic agent, tasquinimod, in the treatment of advanced prostate cancer.
Mechanisms of tumor angiogenesis
Vascular tissue homeostasis is maintained by a balance between proangiogenic and antiangiogenic factors resulting in quiescent endothelial cells. During normal angiogenesis, downregulation of the angiogenic suppressor thrombospondin-1 (TSP-1) is coupled with upregulation of angiogenic stimulators, hypoxia-induced factor-1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF).10 This same process is “hijacked” by cancer cells and upregulation of these factors leads to a switch from a quiescent to neovascularizing state referred to as the “angiogenic switch.”11,12 This switch permits exponential tumor growth and is characterized by angiogenic sprouting and new vessel formation, which specifically feeds hypoxic and necrotic areas of the tumor.12 Furthermore, this is a process that cancer cells employ to access the circulation to metastasize and subsequently undergo neovascularization at new sites.13 As a result, tumor vasculature plays a role in disease progression and offers an attractive target for therapy.
Development of tasquinimod
Tasquinimod is a second generation quinolone-3-carboxamide, developed from the first generation agent linomide (4-hydroxyl-N, 1-dimethy-2-pxp-N-phenyl-1, 2-dihydroquinoline-3-carboxamide; roquinimex). Linomide is an orally administered antiangiogenic drug, which demonstrated potent prostate cancer growth inhibition in vivo.14–18 However, clinical trials of linomide were stopped due to unacceptable toxicity.19,20 Animal studies suggested that linomide induces a dose-dependent proinflammatory reaction that may have accounted for the toxicity seen in clinical trials.21 Subsequently, a number of second generation quinolone-3-carboxamide based agents were generated and identified a lead compound as tasquinimod (4-hydroxy-5-methoxy-N,1-dimethyl-2-oxo-N-[(4-trifluoromethyl) phenyl]-1,2-dihydroquinoline-3-caboxamide, TASQ, ABR-215050) (molecular weight: 406.35 g/mol, Chemical Abstracts Service registry number #254964-60-8) (Figure 1).22 In vitro, tasquinimod demonstrated inhibition of endothelial capillary tube formation and endothelial outgrowth from aortic rings.22 In addition, tasquinimod inhibited blood vessel development in chicken egg chorioallantoic membranes and in vivo endothelial network formation within Matrigel.22 Tasquinimod targeted only proliferating endothelial cells and, therefore, specifically affected the formation of new vasculature and did not cause the regression of preformed endothelial tubules.23 This effect was enhanced when cells were placed in a stressful microenvironment (low glucose, high CO2, low O2, acidic media, similar to those found in the tumors’ microenvironment).24 Tasquinimod was shown to have 30–60 fold greater antitumor efficacy compared to linomide.22 The specific inhibition of demethylation of the quioline-3-carboboxamide N-methyl moiety by the addition of a trifluoromethyl group (Figure 1) resulted in a loss in the unwanted proinflammatory activity seen with linomide.22 Additionally, unlike some other linomide derivatives, tasquinimod remained water soluble, allowing it to be administered orally.22 Although targets for tasquinimod have now been identified (discussed in the next section), these have not been attributed to any specific moiety of the compound. However, it is known that the angiogenic effects seen with tasquinimod are a result of targeting multiple components of tumor neoangiogenesis.
Figure 1.
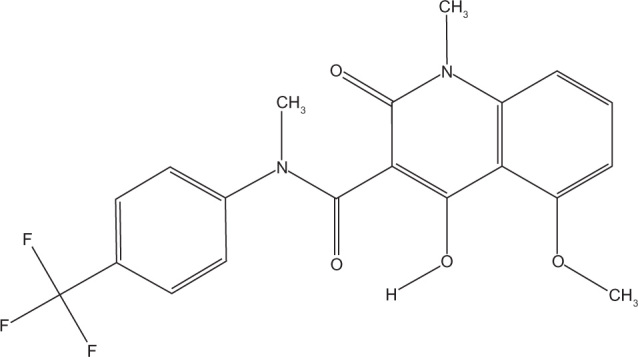
Structure of tasquinimod (TasQ, ABR-215050).
Mechanisms of tasquinimod action
The anti-tumor action of tasquinimod is from the suppression of the angiogenic switch.25 Drug treatment with tasquinimod in both in vivo and in vitro studies leads to a significant upregulation in the expression of TSP-1 and down-regulation of HIF-1α and VEGF.25,26 Further investigations have shown that tasquinimod has an indirect effect on regulating these proangiogenic factors through two distinct mechanisms that are outlined below.
The first mechanism of tasquinimod activity is through high-affinity binding and sequestering of the calcium binding protein S100A9.27 In the presence of zinc, S100A9 is a ligand for the proinflammatory receptor Toll-like receptor 4 (TLR4).27–29 In turn, activated TLR4 stimulates the invasion of tumor tissue by myeloid-derived suppressor cells (MDSCs). In a hypoxic environment, these inflammatory cells upregulate HIF-1α that promotes MDSCs differentiation into tumor associated macrophages (TAMs). It is the TAMs that then secrete proangiogenic factors such as VEGF, independent of the tumor cells.27,30–32 Of relevance to prostate cancer, the expression of S100A9 was confirmed within CD68+VE macrophages in clinical samples and loss of TLR4 (TLR4 −/−) or S100A9 (S100A9 −/−) in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model of prostate cancer delayed tumor growth.27
The second mechanism of tasquinimod action is through targeting the histone acetylation of genes that regulate expression of proangiogenic factors.24 Hypoxia is shown to induce a global modification in the epigenetic landscape, which is associated with gene-specific histone modifications induced at promoter regions of hypoxia-responsive genes. Treatment of prostate cancer cells with tasquinimod inhibited this response at proangiogenic genes. Although, tasquinimod did not affect expression levels or enzymatic activity of histone deacetylase (HDAC) enzymes it was shown to bind to HDAC4 in the zinc-binding regulatory domain to lock the protein in a conformation preventing HDAC4/N-CoR/HDAC3 complex formation.24 Blocking of this interaction prevents HDAC3 deacetylation of proteins, through HDAC4 binding, and inhibition of HDAC4 client transcription factors such as HIF-1α.33,34 Therefore, tasquinimod suppresses HIF-dependent transcription during hypoxia, in both prostate cancer and endothelial cells.24 Knockdown of HDAC4 with specific small interfering RNA demonstrated the same response as tasquinimod treatment in both in vitro and in vivo experiments.24
The antiangiogenic activities of tasquinimod are therefore due to the dual inhibition of S100A9/TLR4 in MDSCs and the inhibition of HDAC4/N-CoR/HDACs deacetylation of HIF1-α in both endothelial and tumor cells, inhibiting hypoxia induced angiogenesis (Figure 2).24,27
Figure 2.
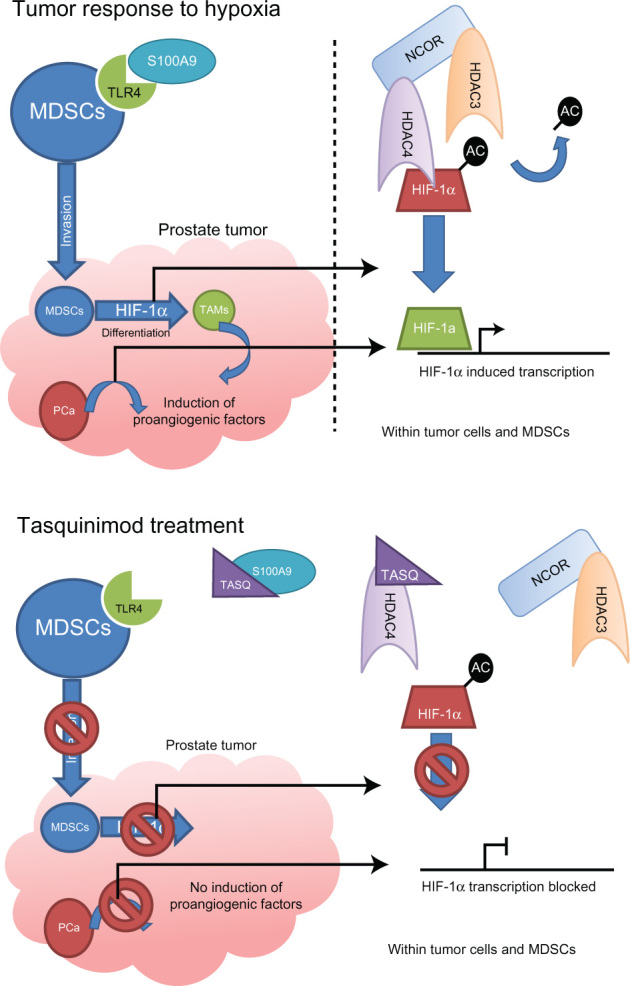
Mechanisms of action of tasquinimod.
Notes: Upper panel shows the normal response to hypoxia in a tumor, with invasion of MDSCs and differentiation to TAMs through HIF-1α along with tumor cell hypoxic response, with the underlying molecular mechanism depicted on the right hand side. The lower panel shows how tasquinimod (TASQ) can efficiently disrupt both MDSCs invasion and hypoxic response in cells within the tumor through disruption of TLR4/S100A9 and HDAC4/N-CoR/HDAC3 interactions.
Abbreviations: HDAC, histone deacetylase; HIF-1α, hypoxia-induced factor-1 alpha; MDSCs, myeloid-derived suppressor cells; TAMs, tumor associated macrophages; TLR4, Toll-like receptor 4.
Tasquinimod in preclinical models of prostate cancer
Androgen deprivation therapy alone has been demonstrated to decrease production of VEGF and repress expression of TSP-1, blocking the angiogenic switch resulting in inhibition of vascular growth. However, this response is temporary and angiogenesis is restored in progression to CRPC.15,35,36 In order to investigate if this angiogenic inhibition with androgen deprivation therapy could be maintained, the addition of tasquinimod was investigated in human prostate cancer xenografts (CWR-22Rv1, CWR-22R-H, LAPC-4, LNCaP, PC-3, and DU-145). Tasquinimod treatment, when combined with androgen deprivation therapy, demonstrated a synergistic action leading to a significant reduction in tumor growth.37 This effect was shown to be an antiangiogenic response as observed by a significant decrease in the tumor blood vessel density.37 Treatment sequence with serial hormone and then tasquinimod, or vice versa, was also compared and showed that enhanced antitumor activity remained with tasquinimod either before or after androgen deprivation therapy.37 Additionally, tasquinimod treatment was combined with docetaxel treatment and androgen deprivation therapy to explore its role in combination therapy for metastatic prostate cancer. This demonstrated enhanced tumor growth inhibition and survival above androgen deprivation therapy plus docetaxel alone.37
Animal studies investigating the effects of tasquinimod on metastasis, using both orthotopic and intratibial xenograft models, have been explored.26 With intratibial injections to mimic bone metastases, treatment with tasquinimod during tumor inoculation reduced tumor take rate, even when treatment was delayed for 1 week after inoculation.26 This reduction of tumor establishment was not limited to primary engraftment sites, with tasquinimod treatment also significantly inhibiting the establishment of lung metastases and the frequency of lymph node metastases within orthotropic implant models.26
These preclinical data demonstrated the potential utility of tasquinimod when coupled within conventional chemotherapy and hormonal based approaches to treat advanced prostate cancer. In addition, there is also evidence that tasquinimod potentiates treatment with radiotherapy, and may be applicable to radical radiotherapy of nonmetastatic disease or radiotherapy to metastases for local symptom control. Of relevance to antiangiogenic therapies, radiotherapy not only targets tumor cells, but also damages endothelial cells already existing in the cancer mass.38 Radiotherapy-induced endothelial damage induces a transient hypoxia, causing a compensatory stimulation of angiogenesis through upregulated expression of VEGF and HIF-1α.39 This reflex mechanism reduces endothelial cell death and increases tumor endothelial cell proliferation, eventually resulting in improved tumor oxygenation following radiotherapy.40–43 Targeting this rebound neoangiogenesis associated with radiotherapy offers an additional approach to tumor treatment, which was explored with tasquinimod. In vivo xenograft studies of human prostate cancer (CWR22R-H) demonstrated that tasquinimod treatment significantly improved the outcomes from radiation treatment by reducing tumor volumes.23 On further investigation, 51Cr-labeled red blood cells demonstrated a 40% reduction in tumor vascular volume that correlated with a reduction in oxygenation in a tumor specific manner.23 In addition, the optimal timing schedule for initiating tasquinimod was investigated, revealing that treatment after completion of the fractionated radiation was more efficacious than treatment before radiation doses.23
Clinical trials of tasquinimod
The promising preclinical data on tasquinimod in prostate cancer has led to clinical trials. An open-label Phase I clinical trial in 32 patients with CRPC and no previous treatment with chemotherapy evaluated the safety and tolerability of tasquinimod, with secondary measures of pharmacokinetics and efficacy.44 The initial maximum tolerated dose was calculated at 0.5 mg of tasquinimod daily; but with stepwise intrapatient dose escalation, a dose of 1.0 mg per day was well tolerated. The dose limiting toxicity with the drug was sinus tachycardia and asymptomatic hyperamylasemia.44 On a fixed dose of 0.5 mg of tasquinimod, inflammation measured by transient laboratory abnormalities was found to be the most common adverse event associated with tasquinimod treatment (41% of patients). At 1.0 mg daily tasquinimod, common adverse events consisted of nausea (57%), fatigue (43%), anemia (29%), myalgia (25%), hypoesthesia (25%), and pain in extremities (25%). The pharmacokinetic study revealed that in the 0.5 mg fixed dose group, the maximum plasma concentration (mean 0.26 μmol/L), was attained at 2.6 hours and the elimination half-life was 40 ± 16 hours (mean ± standard deviation). Two patients on the Phase I trial saw a greater than 50% drop in serum prostate-specific antigen (PSA) levels that lasted for 7 and 13 weeks, respectively. Interestingly fewer patients in the trial demonstrated additional bone lesions than expected (three out of 15 patients), and the median time of disease progression (seen as an increase in PSA of greater than 25%) was seen to be delayed (19 weeks).44
A Phase II randomized double-blind placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic CRPC has been completed.45 In this multicenter trial, 206 men were assigned to either oral once-daily 1.0 mg tasquinimod (after a titration phase of 0.25 mg/day escalating to 1.0 mg/day over 4 weeks) or placebo in a 2:1 ratio. The primary end point in the 201 evaluable patients was disease progression at 6 months – characterized by Response Evaluation Criteria in Solid Tumors (RECIST) end-points, Prostate Cancer Working Group (PCWG2) recommended end-points,46 pathological fracture, spinal cord compression, or pain criteria.45 In this trial, 69% of patients treated with tasquinimod versus 37% in placebo were progression-free at 6 months (P ≤ 0.001; Cochran-Mantel-Haenszel; hazard ratio = 0.49; 95% confidence interval: 0.36–0.67). A median progression-free survival of 7.6 months in tasquinimod treated patients and 3.3 months for placebo treated patients was observed P = 0.0042; hazard risk = 0.57; 95% confidence interval: 0.39–0.85. Furthermore, this study evaluated progression-free survival according to PCWG2 CRPC subtypes, defined by site of metastatic lesion that correlates with clinical outcomes:46 improvement in progression-free survival was demonstrated in patients with visceral metastases (6 months versus 3 months, P = 0.045; hazard ratio = 0.41; 95% confidence interval: 0.16–1.02), bone metastases with or without nodal metastases (8.8 months versus 3.4 months, P = 0.019; hazard ratio = 0.56; 95% confidence interval: 0.34–0.92) and bone metastases only (12.1 versus 5.4 months P = 0.016; hazard ratio = 0.45; 95% confidence interval: 0.23–0.88). PCWG2 CRPC subtype of lymph node metastases demonstrated a nonsignificant increased progression-free survival of 6.1 months compared with 3.1 months (P = 0.54; hazard ratio = 0.73; 95% confidence interval: 0.27–2.00). Using RECIST assessment of disease response, tasquinimod treatment resulted in a partial response in 3%, stable disease in 58%, and progressive disease in 39%; compared with placebo where there was a partial response in 0%, stable disease in 35%, and progressive disease in 65% (8.4 months versus 3.8 months, P = 0.0029; hazard ratio = 0.51; 95% confidence interval: 0.32–0.80). No statistically significant differences were present in the time to PSA progression between the groups. However, the positive findings based on the use of the PCWG2 end-points,46 which remove the focus from PSA-based biochemical assessment, confirmed the feasibility of conducting trials to measure clinical benefit when there is no significant change in this traditional biomarker. Toxicity seen in the Phase II trial mirrored those seen in the Phase I study, with slightly higher frequencies of adverse events (Table 1) that were thought to be related to the greater number of older men in the study; however, these adverse events were transient and manageable.45 Treatment associated transient increase in white cell counts, amylase, lipase, CRP, and fibrinogen were self-limiting and generally returned to normal within 6 months.
Table 1.
Frequency of adverse events in tasquinimod versus placebo treated patients
| Adverse event | Tasquinimod | Placebo |
|---|---|---|
| Grade 1–4 | 89% | 94% |
| Grade 3–4 | 40% | 12% |
Notes: Common adverse events ranked by frequency: fatigue, nausea, constipation, back pain, decreased appetite, pain in extremities, flatulence, arthralgia, anemia, diarrhea, insomnia, weight loss, abdominal pain, vomiting, blood amylase increase, blood lipase increase, myalgia, peripheral edema, musculoskeletal pain, deep vein thrombosis, and myocardial infarction.
Potential clinical applications of tasquinimod in advanced prostate cancer
Currently patients presenting with advanced prostate cancer begin treatment with luteinizing hormone-releasing hormone analog/antagonist (LHRHa) therapy, moving onto maximum androgen blockade with the use of an antiandrogen alongside LHRHa (Figure 3). Once patients stop responding to this therapy, antiandrogen withdrawal has been shown to transiently help tumor regression.47 Progression after this stage is characterized CRPC, and it is usual for fit patients to begin first-line chemotherapy with docetaxel, which has been shown to increase survival by an average of 2–3 months.3 Following sustained tumor growth at this stage of the disease, patients either continue chemotherapy with cabazitaxel or start therapy targeting testosterone production with treatment with the cytochrome P17 inhibitor abiraterone.4,5 Preclinically, tasquinimod has been demonstrated to work synergistically with many of these therapeutic options and a number of clinical trials are currently in progress to assess tasquinimod alongside chemotherapy (docetaxel and cabazitaxel).44,45
Figure 3.
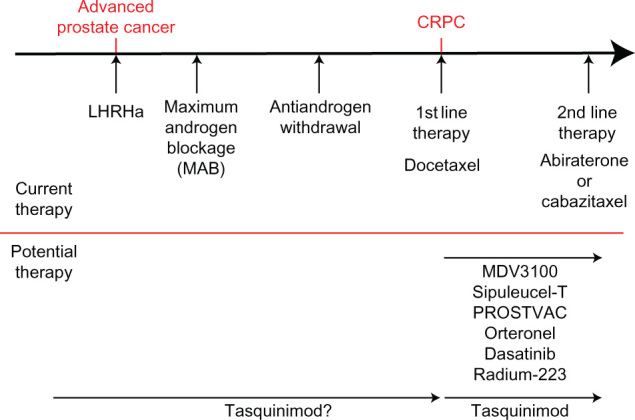
Prostate cancer therapeutic options.
Notes: Upper panel shows the progression of advanced prostate cancer, with the current mainstream treatment options used at each stage. Lower section shows some of the promising therapies coming through and how they could fit into this treatment regime.
Abbreviations: CRPC, castration resistant prostate cancer; LHRHa, luteinizing hormone-releasing hormone analog/antagonist.
In addition to tasquinimod, a large number of new treatments are showing great therapeutic promise, including next generation androgen receptor antagonist MDV3100,48 steroidogenesis targeting orteronel,49–51 tyrosine kinase inhibitors dasatinib,52 vaccine based therapies sipuleucel-T and PROSTVAC® (Bavarian-Nordic, Kvistgard, Denmark),53,54 and internal radiotherapy through Radium-223 (Figure 3).55 As with tasquinimod, these therapies are currently being investigated for the treatment of CRPC; however, tasquinimod has also shown promising preclinical utility in a hormone naïve setting.22,37 Furthermore, in contrast to some emerging therapies tasquinimod appears to be relatively well tolerated by patients, although formal head-to-head comparisons are not yet available.44,45 With this in mind, tasquinimod may have potential to be utilized to slow disease progression from an earlier point in the treatment pathway, in conjunction with first line hormone therapy (Figure 3).
Previous experiences with angiogenic inhibition therapies, such as VEGF inhibitors, have been compromised by the development of disease resistance and subsequent relapse (reviewed extensively by Bergers and Hanahan 2008).56 In brief, resistance manifests through a number of mechanisms including: (1) utilization of alternative proangiogenic pathways to drive relapse; for example, targeted VEGF therapy can be overcome through fibroblast growth factor signaling;57 (2) tumor recruitment of proangiogenic nonmalignant cells such as MDSC and endothelial progenitors;58,59 (3) vasculature maintenance and regrowth by the support of pericytes;58–61 (4) the hypoxic promotion of invasion by cancer cells, allowing survival elsewhere in the body and subsequently overcoming the angiogenic therapy.62,63 In these instances the angiogenic inhibition being utilized has targeted a single specific pathway to prevent neoangiogenesis, such as the targeting of VEGF with bevacizumab, and as such have been shown to have minimal efficacy in prostate cancer treatment.64 However, tasquinimod has the advantage that it targets both tumor cell and host cell induced neoangiogenesis through two distinct mechanisms and not a single target.24,27
As a single agent, tasquinimod demonstrated strong antitumor activity in Phase II clinical trials; however, it must be noted that 39% of patients still did not respond to therapy.45 These clinical findings are likely to reflect the fact that prostate cancer is a highly heterogeneous disease, and similar patterns of nonresponders are also seen in other therapies in advanced prostate cancer (nonresponse rate in docetaxel is 35%–38%,3 cabazitaxel is 48%,4 and abiraterone is 32%).5 As we are currently unable to predict those patients who would be responsive or unresponsive to the therapies utilized for CRPC, it is likely that combination therapy will be the best strategy to target as many tumor cells as possible. In time, with developments in personalized medicine, it may become possible to tailor individual therapies according to the molecular characteristics of the tumor within a patient. Until this time, combination therapies targeting as many as possible tumor pathways appear to offer an attractive strategy, as long as side effects are acceptable. Within CRPC, not only is androgen receptor signaling maintained but an upregulation in the expression of steroidogenesis associated genes is seen such as SRD5A1, SRD5A3, and AKR1C3.51 The recent development of compounds such as the bifunctional AKR1C3 inhibitor/androgen receptor antagonist could play a strong role in such combination therapies, and compliment the dual mechanisms of action seen with tasquinimod.50 Combinations of such drugs alongside tasquinimod could allow intensive therapy targeting multiple pathways in both the androgen receptor signaling axis and angiogenesis.
Conclusion
In order for tumors to grow they must support themselves with their own blood supply. Neovascularization is achieved through a complex interplay between tumor cells and normal cells stimulated by hypoxia.9,11 Due to its potent reduction of the hypoxic response in cancer cells, endothelial cells, MDSCs, and TAMs, tasquinimod inhibits tumor angiogenesis whilst sparing already formed vasculature. In vivo and in vitro data highlight a potent anticancer effect as a monotherapy in addition to greatly improving the response to combination therapies with androgen deprivation therapy, docetaxel, or radiotherapy. Phase I and II studies have demonstrated tasquinimod to be well-tolerated and lead to significant improvements in progression-free survival from metastasis, by a median of 4.3 months, in patients with minimally symptomatic CRPC. The data reviewed highlights tasquinimod as an extremely promising and much needed therapeutic tool for use in CRPC. A definitive assessment of tasquinimod in clinical CRPC treatment is still required and the results of an ongoing major Phase III clinical trial are awaited.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain J, Melia J, Moss S, Brown J. The diagnosis, management, treatment and costs of prostate cancer in England and Wales. Health Technol Assess. 1997;1(3):i–vi. 1–53. [PubMed] [Google Scholar]
- 3.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 4.de Bono JS, Oudard S, Ozguroglu M, et al. TROPIC Investigators Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 5.Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–1495. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46(4):765–781. doi: 10.1016/j.ejca.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 8.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 9.Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008;1(4):158–164. doi: 10.1593/tlo.08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain RK, Au P, Tam J, Duda DG, Fukumura D. Engineering vascularized tissue. Nat Biotechnol. 2005;23(7):821–823. doi: 10.1038/nbt0705-821. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 12.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 13.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143(2):401–409. [PMC free article] [PubMed] [Google Scholar]
- 14.Hartley-Asp B, Vukanovic J, Joseph IB, Strandgården K, Polacek J, Isaacs JT. Anti-angiogenic treatment with linomide as adjuvant to surgical castration in experimental prostate cancer. J Urol. 1997;158(3 Pt 1):902–907. doi: 10.1097/00005392-199709000-00069. [DOI] [PubMed] [Google Scholar]
- 15.Joseph IB, Isaacs JT. Potentiation of the antiangiogenic ability of linomide by androgen ablation involves down-regulation of vascular endothelial growth factor in human androgen-responsive prostatic cancers. Cancer Res. 1997;57(6):1054–1057. [PubMed] [Google Scholar]
- 16.Vukanovic J, Hartley-Asp B, Isaacs JT. Inhibition of tumor angiogenesis and the therapeutic ability of linomide against rat prostatic cancers. Prostate. 1995;26(5):235–246. doi: 10.1002/pros.2990260503. [DOI] [PubMed] [Google Scholar]
- 17.Vukanovic J, Isaacs JT. Linomide inhibits angiogenesis, growth, metastasis, and macrophage infiltration within rat prostatic cancers. Cancer Res. 1995;55(7):1499–1504. [PubMed] [Google Scholar]
- 18.Vukanovic J, Passaniti A, Hirata T, Traystman RJ, Hartley-Asp B, Isaacs JT. Antiangiogenic effects of the quinoline-3-carboxamide linomide. Cancer Res. 1993;53(8):1833–1837. [PubMed] [Google Scholar]
- 19.Wolinsky JS, Narayana PA, Noseworthy JH, et al. Linomide in relapsing and secondary progressive MS: part II: MRI results. MRI Analysis Center of the University of Texas-Houston, Health Science Center, and the North American Linomide Investigators. Neurology. 2000;54(9):1734–1741. doi: 10.1212/wnl.54.9.1734. [DOI] [PubMed] [Google Scholar]
- 20.Noseworthy JH, Wolinsky JS, Lublin FD, et al. Linomide in relapsing and secondary progressive MS: part I: trial design and clinical results. North American Linomide Investigators. Neurology. 2000;54(9):1726–1733. doi: 10.1212/wnl.54.9.1726. [DOI] [PubMed] [Google Scholar]
- 21.Jonsson S, Andersson G, Fex T, et al. Synthesis and biological evaluation of new 1,2-dihydro-4-hydroxy-2-oxo-3-quinolinecarboxamides for treatment of autoimmune disorders: structure-activity relationship. J Med Chem. 2004;47(8):2075–2088. doi: 10.1021/jm031044w. [DOI] [PubMed] [Google Scholar]
- 22.Isaacs JT, Pili R, Qian DZ, et al. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate. 2006;66(16):1768–1778. doi: 10.1002/pros.20509. [DOI] [PubMed] [Google Scholar]
- 23.Dalrymple SL, Becker RE, Zhou H, DeWeese TL, Isaacs JT. Tasquinimod prevents the angiogenic rebound induced by fractionated radiation resulting in an enhanced therapeutic response of prostate cancer xenografts. Prostate. 2012;72(6):638–648. doi: 10.1002/pros.21467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isaacs JT, Antony L, Dalrymple S, et al. Tasquinimod is an allosteric modulator of HDAC4 survival signaling within the compromised cancer microenvironment Cancer ResEpub November132012 [DOI] [PMC free article] [PubMed]
- 25.Olsson A, Björk A, Vallon-Christersson J, Isaacs JT, Leanderson T. Tasquinimod (ABR-215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer. 2010;9:107. doi: 10.1186/1476-4598-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jennbacken K, Welén K, Olsson A, et al. Inhibition of metastasis in a castration resistant prostate cancer model by the quinoline-3- carboxamide tasquinimod (ABR-215050) Prostate. 2012;72(8):913–924. doi: 10.1002/pros.21495. [DOI] [PubMed] [Google Scholar]
- 27.Kallberg E, Vogl T, Liberg D, et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS One. 2012;7(3):e34207. doi: 10.1371/journal.pone.0034207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Björk P, Björk A, Vogl T, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7(4):e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 30.Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Squadrito ML, De Palma M. Macrophage regulation of tumor angiogenesis: implications for cancer therapy. Mol Aspects Med. 2011;32(2):123–145. doi: 10.1016/j.mam.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. 2009;85(6):996–1004. doi: 10.1189/jlb.0708446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bottomley MJ, Lo Surdo P, Di Giovine P, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283(39):26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahm A, Paolini C, Pallaoro M, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA. 2007;104(44):17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joseph IB, Nelson JB, Denmeade SR, Isaacs JT. Androgens regulate vascular endothelial growth factor content in normal and malignant prostatic tissue. Clin Cancer Res. 1997;3(12 Pt 1):2507–2511. [PubMed] [Google Scholar]
- 36.Colombel M, Filleur S, Fournier P, et al. Androgens repress the expression of the angiogenesis inhibitor thrombospondin-1 in normal and neoplastic prostate. Cancer Res. 2005;65(1):300–308. [PubMed] [Google Scholar]
- 37.Dalrymple SL, Becker RE, Isaacs JT. The quinoline-3-carboxamide anti-angiogenic agent, tasquinimod, enhances the anti-prostate cancer efficacy of androgen ablation and taxotere without effecting serum PSA directly in human xenografts. Prostate. 2007;67(7):790–797. doi: 10.1002/pros.20573. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300(5622):1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 39.Vergis R, Corbishley CM, Norman AR, et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 2008;9(4):342–351. doi: 10.1016/S1470-2045(08)70076-7. [DOI] [PubMed] [Google Scholar]
- 40.Sonveaux P, Brouet A, Havaux X, et al. Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: implications for tumor radiotherapy. Cancer Res. 2003;63(5):1012–1019. [PubMed] [Google Scholar]
- 41.Stuben G, Thews O, Pöttgen C, Stuschke M, Sack H. Tumour oxygenation during fractionated radiotherapy – comparison with size-matched controls. Acta Oncol. 1999;38(2):209–213. doi: 10.1080/028418699431636. [DOI] [PubMed] [Google Scholar]
- 42.Crokart N, Jordan BF, Baudelet C, et al. Early reoxygenation in tumors after irradiation: determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys. 2005;63(3):901–910. doi: 10.1016/j.ijrobp.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 43.Hou H, Lariviere JP, Demidenko E, Gladstone D, Swartz H, Khan N. Repeated tumor pO(2) measurements by multi-site EPR oximetry as a prognostic marker for enhanced therapeutic efficacy of fractionated radiotherapy. Radiother Oncol. 2009;91(1):126–131. doi: 10.1016/j.radonc.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bratt O, Häggman M, Ahlgren G, Nordle O, Bjöork A, Damber JE. Open-label, clinical phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br J Cancer. 2009;101(8):1233–1240. doi: 10.1038/sj.bjc.6605322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pili R, Häggman M, Stadler WM, et al. Phase II randomized, doubleblind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29(30):4022–4028. doi: 10.1200/JCO.2011.35.6295. [DOI] [PubMed] [Google Scholar]
- 46.Scher HI, Halabi S, Tannock I, et al. Prostate Cancer Clinical Trials Working Group Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupont A, Gomez JL, Cusan L, Koutsilieris M, Labrie F. Response to flutamide withdrawal in advanced prostate cancer in progression under combination therapy. J Urol. 1993;150(3):908–913. doi: 10.1016/s0022-5347(17)35646-x. [DOI] [PubMed] [Google Scholar]
- 48.Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375(9724):1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaoka M, Hara T, Hitaka T, et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J Steroid Biochem Mol Biol. 2012;129(3–5):115–128. doi: 10.1016/j.jsbmb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl) amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22(10):3492–3497. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitsiades N, Sung CC, Schultz N, et al. Distinct Patterns of Dysregulated Expression of Enzymes Involved in Androgen Synthesis and Metabolism in Metastatic Prostate Cancer Tumors. Cancer Res. 2012;72(23):6142–6152. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu EY, Wilding G, Posadas E, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15(23):7421–7428. doi: 10.1158/1078-0432.CCR-09-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheikh NA, Petrylak D, Kantoff PW, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother. 2013;62(1):137–147. doi: 10.1007/s00262-012-1317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker CC, Pascoe S, Chodacki A, et al. A randomized, double-blind, dose-finding, multicenter, phase 2 study of radium chloride (ra 223) in patients with bone metastases and castration-resistant prostate cancer. Eur Urol. 2013;63(2):189–197. doi: 10.1016/j.eururo.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 59.Du R, Lu KV, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jain RK, Booth MF. What brings pericytes to tumor vessels? J Clin Invest. 2003;112(8):1134–1136. doi: 10.1172/JCI20087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mancuso MR, Davis R, Norberg SM, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116(10):2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bikfalvi A, Moenner M, Javerzat S, North S, Hagedorn M. Inhibition of angiogenesis and the angiogenesis/invasion shift. Biochem Soc Trans. 2011;39(6):1560–1564. doi: 10.1042/BST20110710. [DOI] [PubMed] [Google Scholar]
- 63.Rubenstein JL, Kim J, Ozawa T, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2(4):306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ogita S, Tejwani S, Heilbrun L, et al. Pilot Phase II Trial of Bevacizumab Monotherapy in Nonmetastatic Castrate-Resistant Prostate Cancer. ISRN oncology. 2012;2012:242850. doi: 10.5402/2012/242850. [DOI] [PMC free article] [PubMed] [Google Scholar]