

Abstract
The mammalian target of rapamycin (mTOR) is a protein kinase that regulates protein translation, cell growth, and apoptosis. Rapamycin (RPM), a specific inhibitor of mTOR, exhibits potent and broad in vitro and in vivo antitumor activity against leukemia, breast cancer, and melanoma. Recent studies showing that RPM sensitizes cancers to chemotherapy and radiation therapy have attracted considerable attention. This study aimed to examine the radiosensitizing effect of RPM in vitro, as well as its mechanism of action. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and colony formation assay showed that 10 nmol/L to 15 nmol/L of RPM had a radiosensitizing effects on pancreatic carcinoma cells in vitro. Furthermore, a low dose of RPM induced autophagy and reduced the number of S-phase cells. When radiation treatment was combined with RPM, the PC-2 cell cycle arrested in the G2/M phase of the cell cycle. Complementary DNA (cDNA) microarray and reverse transcription polymerase chain reaction (RT-PCR) revealed that the expression of DDB1, RAD51, and XRCC5 were downregulated, whereas the expression of PCNA and ABCC4 were upregulated in PC-2 cells. The results demonstrated that RPM effectively enhanced the radiosensitivity of pancreatic carcinoma cells.
Keywords: radiation, pancreatic carcinoma, mTOR, rapamycin
Introduction
Pancreatic cancer is the most lethal of the solid tumors and the fourth leading cause of cancer-related death in North America.1,2 The incidence of pancreatic cancer has been gradually increasing, even though the incidence of other common cancers has declined.3 About 80% to 85% of patients with pancreatic cancer present with locally advanced or metastatic disease that precludes curative resection and have a poor prognosis.1,3 Despite developments in detection and treatment, the 5-year survival rate of pancreatic cancer is only about 4%.3
Radiotherapy allows tumor control through the cytotoxic action of ionizing radiation. Radiotherapy is used frequently in the management of multiple tumor types, including locally advanced pancreatic cancer. Although modern technologies permit precise radiation delivery to the tumor mass, while minimizing the exposure of the surrounding healthy tissues, the efficacy of radiotherapy remains limited by the intrinsic or acquired radioresistance of tumors. Enhancing tumor radiosensitivity could reduce the amount of radiation required for definitive treatment and improve clinical outcome. The recent interest in targeting the components of signaling pathways prompts an urgent search for agents that enhance the sensitivity of tumor cells to irradiation.
The signaling pathways that activate mammalian target of rapamycin (mTOR) are altered in many human cancers, and these alterations are associated with prognosis and treatment response.4 Inhibiting mTOR has been found to enhance the cytotoxicity of DNA-damaging agents in several cancer cells.5–7 Rapamycin (RPM) is a lipophilic macrolide antibiotic initially developed as a fungicide and immunosuppressant.8 Previous studies have reported that RPM has antiproliferative effects on some tumors.9–15 RPM also acts as a specific mTOR inhibitor. In several, but not all cancer cell lines, RPMs (RPM and its derivatives) increase the irradiation cytotoxicity in vitro through multiple mechanisms, including the autophagy pathway.16–19 RPMs may also increase the effects of radiotherapy in tumor xenograft models.20 This study aimed to examine the radiosensitizing effect of RPM on pancreatic cancer in vitro and its molecular mechanism.
Material and methods
Reagents
The reagents used in our research were: tetal bovine serum (Gibco® FBS; Life Technologies, Carlsbad, CA, USA); Roswell Park Memorial Institute (RPMI) medium (Gibco RPMI 1640; Life Technologies); 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St Louis, MO, USA); reverse transcription polymerase chain reaction (RT-PCR) kit (catalog #sc-8319 Ampliqon A/S, Odense, Denmark); TRIzol® Reagent (Life Technologies); mTOR monoclonal antibody (Santa Cruz Biotechnology Inc, Dallas, TX, USA); Rapamycin (RPM) (Sigma-Aldrich); monodansylcadaverine (MDC) (Sigma-Aldrich).
Cell culture and irradiation
Human pancreatic cancer cell lines, PC-2 and PANC-1 were obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were cultured in RPMI 1640 maximal medium containing 10% inactivated (56°C, 30 minutes) fetal bovine serum, 1 × 105 U/L penicillin, and 100 mg/L streptomycin, in a humidified atmosphere with 5% CO2 incubator at 37°C.
The PC-2 and PANC-1 cells were then treated with 2, 4, 6, 8, and 10 Gy of X-ray irradiation, using a Varian Clinac® 2100EX linear accelerator (Varian Medical Systems Inc, Palo Alto, CA, USA), at a dose rate of 400 cGy/min. The distance between the cells and the radiation source was maintained at 1 meter. After irradiation, the medium was immediately replaced with fresh medium. The dishes were then returned to the incubator for further culture, and the cells were harvested at 0, 24, 48, 72, 96, and 120 hours.
MTT assay
The viability of pancreatic cancer cells was assessed using an MTT dye reduction assay (Sigma-Aldrich), which was conducted as described in a previous work.21 After irradiation, the cells were seeded in a 96-well plate at a density of 1 × 104 cells/well, cultured for 12 hours, then treated with different concentration (10, 20, 30, 40, and 50 μmol/L) RPM for 0–96 hours. At the end of the treatment, MTT, 50 μg/10 μL, was added and the cells were incubated for another 4 hours. Dimethyl sulfoxide (DMSO), 200 μL, was added to each well after removal of the supernatant. After shaking the plate for 10 minutes, cell viability was assessed by measuring the absorbance at 490 nm, using an enzyme-labeling instrument (EX-800 type, Bio-Tek, Winooski, VT, USA); all measurements were performed four times. A cell growth curve was completed using time as the abscissa and absorbance value (A value) (mean ± standard deviation [SD]) as the ordinate.
Colony formation assay
A colony formation assay was applied for measurement of the clonogenic cell survival, as described in a previous work.22 After the treatment with radiation, the cells were plated into 60 mm petri dishes, with standard culture media. After 2 weeks, the cells were fixed with 4% formaldehyde, stained with crystal violet, and colonies containing more than 50 cells were counted and normalized to their corresponding nonirradiated control. The surviving fraction for a given treatment was calculated as the plating efficiency of the irradiated samples relative to that of the sham irradiated ones. Each point on survival curves represents the mean surviving fraction from at least three independent experiments. The multitarget click model in GraphPad Prism 5.0 (GraphPad Software Inc, San Diego, CA, USA) was used to fit the cell survival curves.
The surviving fractions (SF) were calculated as follows: SF = number of clones formed after irradiation of the cells/the number of cells inoculated × cell planting rate (the 0 Gy group was calculated as the planting rate).
The radiosensitive enhancement ratio = SF of the control group/SF2 of the drug-treated group.
MDC staining of autophagic vacuoles
MDC staining of autophagic vacuoles was performed for the autophagy analysis, as described in a previous work.23 The PC-2 cells were divided into a control group, a 10 nmol/L RPM group, and a 15 nmol/L RPM group. The cells were incubated for 48 hours on coverslips. Autophagic vacuoles were labeled with 0.05 mmol/L MDC in phosphate-buffered saline (PBS) at 37°C for 10 minutes. Then, the cells were washed three times with PBS. The autophagic vacuoles in the PC-2 cells were observed under a fluorescence microscope (Olympus BX60; Olympus Corp, Tokyo, Japan). The fluorescence intensity of MDC was measured at an excitation wavelength of 380 nm, emission wavelength of 530 nm.
Cell cycle analysis
For the cell cycle, exponentially growing PC-2 cells were cultivated and synchronized for 24 hours in serum-free medium, and then, the medium was exchanged with a complete medium before irradiation.24 At 24 hours postirradiation, the cells were harvested and fixed in 70% ice-cold ethanol, at −20°C, overnight. Before analysis, the cells were washed with PBS, and DNA content was labeled with 50 nmol/L propidium iodide (PI) in the presence of 1 mg/mL ribonuclease (RNase) (Sigma-Aldrich) for 30 minutes at room temperature, in the dark. The presence of apoptotic cells was detected by determination of the “sub-G1” population. DNA distributions were analyzed using a FACScan™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), with CellQuest™ Pro software (BD Biosciences), for the proportions of cells in G1/G0, S, and G2/M phases of the cell cycle. This assay was performed in triplicate.
Microarray and data analysis
PC-2 cells, including both treated and nontreated were collected after 48-hour culture. Total RNA was extracted using the TRIzol reagent, following the manufacturer’s instructions. The RNA concentration was determined using the NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Inc, Waltham, MA, USA), and RNA integrity was examined by an Agilent 2100 Bioanalyzer System (Agilent Technologies Inc, Santa Clara, CA, USA). Only samples with an RNA integrity number above 8.0 were used for further analysis.
The samples were subjected to mRNA expression profiling on 4 × 44 K Agilent Whole Human Genome Microarray slides (catalog G4112F; Agilent Technologies Inc), using a single-color array method.25 Total RNA (200 ng) was labeled and amplified according to the manufacturer’s instructions, using a Low Input Quick Amp Labeling Kit (Agilent Technologies Inc). Labeled RNA was purified and hybridized to the microarray slides, according to the manufacturer’s protocol. After washing the microarray slides, array scanning and feature extraction was performed with the default scenario, with the Agilent High-Resolution Microarray Scanner and Feature Extraction 9.5.3 software (Agilent Technologies Inc). Total gene signal normalization at the 75th percentile of raw signal values and baseline transformation at the median of all samples was performed using the GeneSpring® software 10.1 (Agilent Technologies Inc), following the instructions of the manufacturer. Before the statistical analysis of microarray data, flag (100% present in at least one group) and fold change (fold change greater than two between RPM-treated and control cell cultures) filters were applied by the GeneSpring software.
Three independent experiments were performed, and expression signals were converted into numerical data using the ImaGene™ software 5.5 (BioDiscovery Inc, Hawthorne, CA, USA), and the raw data was saved as a Microsoft Excel file. Data were subsequently exported to GeneSpring 7.0 (Agilent Technologies Inc) for background subtraction, based on negative controls and per spot and per chip intensity dependent normalization (nonlinear or lowess normalization). These corrected, normalized signals could then be used to estimate the relative abundance of genes with the ratio of Cy-3 and Cy-5 signal intensities. Changes in gene expression were considered significant if the detection P-value was less than 0.05 in at least two of three arrays and the fold change was at least two.
RT-PCR validation
Five genes with the most significant differences in expression and highest fold change revealed by whole-genome microarray experiments were subjected to further validation by RT-PCR. As previously described elsewhere,26 cells collected at specified times were used to extract total RNA, using the TRIzol reagent and following the manufacturer’s instructions. Briefly, 1 μg RNA synthesized complementary (c)DNA through reverse transcriptase, under the following conditions: 5 minutes at 70°C, extended for 60 minutes at 42°C, enzyme inactivated at 95°C for 3 minutes and terminated reaction at 4°C The renaturation temperature was 55°C (cycling 20–25 times). The amplification condition was as follows: The sample was pre-denatured for 3 minutes at 95°C, then denatured for 30 seconds at 95°C, renatured for 30 seconds at 55°C, and finally, extended for 30 seconds at 72°C. The primer sequences were designed with Primer Premier 5.0 software (Premier Biosoft, Palo Alto, CA, USA). The primer sequences and annealing temperature of the genes are shown in Table 1. The PCR product was detected with agarose gel electrophoresis, and an ethidium bromide imaging system was used to perform density index analysis. The expression intensity of the destination gene mRNA was denoted with the ratio of the photodensity of the RT-PCR products of the destination gene and β-actin.
Table 1.
Primer sequences and annealing temperature of the genes in RT-PCR
| Gene | Gene sequences | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|
| DDB1 | Forward primer 5′-ATCATCCGGAATGGAATTGGAA-3′ Forward primer 5′-TCAGACCGCAGTGGCCATAA-3′ |
83 | 60 |
| RAD51 | Forward primer 5′-TGGGAACTGCAACTCATCTGG-3′ Forward primer 5′-GCGCTCCTCTCTCCAGCAG-3′ |
157 | 61 |
| XRCC5 | Forward primer 5′-GCCATATCAGTGAACCTTTAGAGAC-3′ Forward primer 5′-GGAACTGGAACTCAAGGCAAG-3′ |
268 | 59 |
| PCNA | Forward primer 5′-CTGTAGCGGCGTTGT-3′ Reverse primer 5′-ACTTTCTCCTGGTTTGG-3′ |
362 | 52 |
| ABCC4 | Forward primer 5′-GCTCACTGGATTGTCTTCATTTTC-3′ Reverse primer 5′-CTCGGTTACATTTCCTCCTCCAT-3′ |
147 | 59 |
| β-actin | Forward primer 5′-CCTGAGGCTCTTTTCCAGCC-3′ Reverse primer 5′-TAGAGGTCTTTACGGATGTCAACGT-3′ |
110 | 60 |
Notes: Five genes, including DDB1, RAD51, XRCC5, PCNA, and ABCC4 were confirmed to their expression, in microarray analysis using RT- PCR, and were significantly expressed after RPM treatment in PC-2 cells. The primer pairs and the predicted sizes of the amplified PCR products and the annealing temperatures of PCR were listed. β-actin is the housekeeping gene for RT- PCR analysis. The primer sequences were designed with Primer Premier 5.0 software (Premier Biosoft, Palo Alto, CA, USA).
Abbreviations: PCR, polymerase chain reaction; RPM, rapamycin; RT, reverse transcription.
Statistical analysis
All data were expressed by mean ± standard error of the mean (SEM). The statistical analysis was performed using SPSS 13.0 for Windows software (IBM, Armonk, NY, USA). Oneway analysis of variance (ANOVA) and Student’s t-test were used to analyze the statistical differences between groups under different conditions. The statistical analysis of the microarray data was performed using the GeneSpring 10.1 (Agilent Technologies Inc) software. A P-value < 0.05 was considered statistically significant.
Results and discussion
Antitumor effect of RPM on pancreatic cancer cells
RPM has been shown to have in vitro or in vivo growth inhibitory effects on a number of cancers, including gallbladder cancer, Kaposi’s sarcoma, laryngeal cancer, and prostate cancer.9–15 In vitro RPM combined with inhibition of the Notch pathway has shown greater efficacy than RPM alone in the treatment of patients with pancreatic cancer.27 RPM also exhibits dose-dependent antiproliferative effects on pancreatic carcinoma cell lines in vitro, both alone and in combination with FTY720.28 RPM is well tolerated in clinical use. However, the efficacy of inhibiting mTOR in tumor tissues has not been correlated with its antitumor effects.29
After treatment with different doses of RPM for 48 hours, the MTT results showed that RPM inhibited the proliferation of the pancreatic cancer cells in a dose-dependent manner. Figure 1 shows that RPM concentrations ≤ 20 nmol/L resulted in cell viabilities exceeding 80.0% in both PC-2 cells and PANC-1 cells. The median inhibitory concentration (IC50) of RPM was 44.3 nmol/L for PC-2 cells and 47.1 nmol/L for PANC-1 cells. Hence, we chose a low-dose (5∼15 nmol/L) of RPM for the further radiosensitization study.
Figure 1.
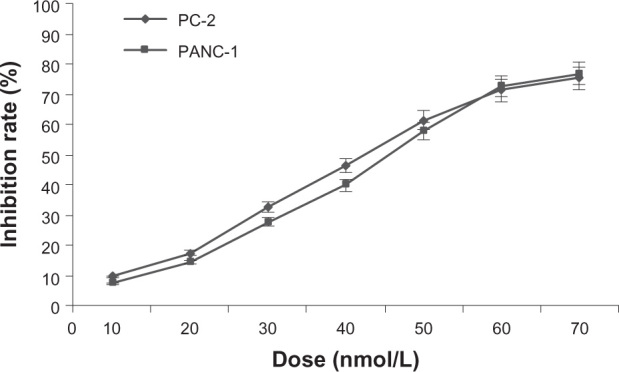
Growth inhibiting effects of RPM on pancreatic cancer cell lines PC-2 and PANC-1.
Notes: Pancreatic cancer cells were treated with RPM (0, 10, 20, 30, 40, 50, 60, and 70 nmol/L) for 48 hours. Cell viability was determined by the MTT method. This assay was performed in triplicate. A dose-dependent inhibition of cell growth could be observed (P < 0.05, ANOVA analysis).
Abbreviations: ANOVA, analysis of variance; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; RPM, rapamycin.
Effect of radiation combined with RPM on proliferation of pancreatic cancer cells
Previous reports demonstrated that the RPMs sensitize certain cancer cells that were resistant to chemotherapeutic agents and radiotherapy.16–19,30 These facts suggest that mTOR is an important target for anticancer therapeutics development.31
After x-ray irradiation at 4 Gy, cell viability was determined using the MTT method. As shown in Figure 2, there was no significant difference between the 5 nmol/L RPM treatment group and the control group (P > 0.05). In the 10 nmol/L and 15 nmol/L RPM treatment groups, cell survival was significantly inhibited compared with the control group (P < 0.05). The difference was not statistically significant (P > 0.05) between the 10 nmol/L and the 15 nmol/L RPM treatment groups.
Figure 2.
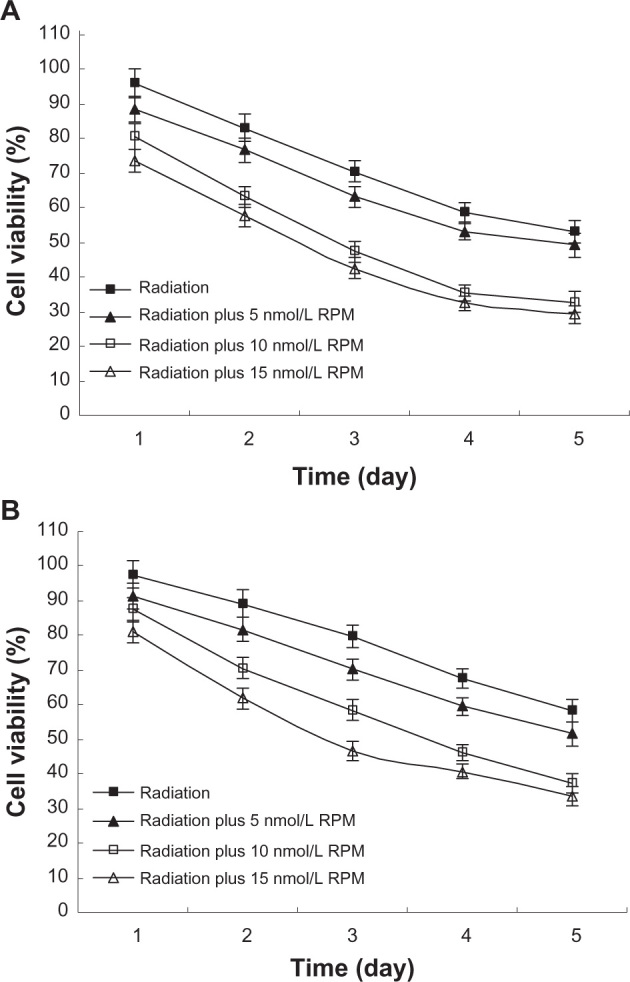
Effect of radiation plus RPM on cell viability of pancreatic cancer cells with MTT assay, in (A) PC-2 cells and (B) PANC-1 cells.
Notes: After 4 Gy X-ray irradiation, cell viability was determined by the MTT method. This assay was performed in triplicate. in the 10 nmol/L and the 15 nmol/L RPM treatment groups, cell survival was significantly inhibited compared with the control group. (P < 0.05, ANOVA analysis.)
Abbreviation: ANOVA, analysis of variance; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; RPM, rapamycin.
Effect of radiation combined with RPM on radiosensitivity of PC-2 cells
Saito et al32 provided noninvasive evidence of RPM-induced vascular renormalization and the resultant transient increase in tumor oxygenation. The improved oxygenation from RPM treatment provides a temporal window for anticancer therapies to enhance radiotherapy response. Mauceri et al33 tested the effects of combined treatment with RAD001, a different rapalog, and fractionated radiation, using a xenograft model of human non–small cell lung cancer cells (A549 cells). The results suggest that RAD001 increases the antitumor activity of radiation. Furthermore, combination therapy with RPM before irradiation normalized the tumor vasculature, thereby improving tumor oxygenation, and increasing the sensitivity of alveolar rhabdomyosarcoma xenografts to adjuvant irradiation.34
In this study, cells were irradiated after treatment with different RPM concentrations for 6 hours. The radiosensitivity of pancreatic cancer cells was determined using a colony formation assay. The multitarget click model in GraphPad Prism 5.0 was used to fit the cell survival curves. The radiosensitization was not significant in the 5 nmol/L RPM treatment group compared with the control group (P > 0.05). The 10 nmol/L and 15 nmol/L RPM treatment groups exhibited significantly increased radiosensitivity in both the PC-2 cells and PANC-1 cells (Figure 3). The difference between the 10 nmol/L and 15 nmol/L RPM treatment groups was not statistically significant (P > 0.05). The results show that RPM has significant radiosensitizing effects at 10 nmol/L to 15 nmol/L, with 10 nmol/L providing the best radiosensitization.
Figure 3.

Survival fraction of pancreatic cancer cells treated by different dose of irradiation (A) PC-2 cells; (B) PANC-1 cells.
Notes: Pancreatic cancer cells were treated with different concentrations for 6 hours before radiation. The radiosensitivity of pancreatic cancer cells was determined by a colony formation assay. The multitarget click model in GraphPad Prism 5.0 (GraphPad Software Inc, San Diego, CA, USA) was used to fit the cell survival curves. This assay was performed in triplicate. The radiosensitizing effect was observed in the 10 nmol/L and 15 nmol/L RPM treatment groups.
Abbreviation: RPM, rapamycin.
Effects of RPM on autophagy by MDC-labeled method
Autophagy often contributes to the demise of tumor cells. This mechanism may provide a method for radiosensitizing cancer cell types that are refractory to apoptosis induction. However, the data suggest that aside from promoting cell death, radiotherapy combined with autophagy inducers also favors the emergence of a subpopulation of senescent tumor cells that are unable to proliferate but that are still metabolically active.35–38 Using multidrug-resistant v-Haras-transformed NIH3T3 cells, Eum and Lee demonstrated that RPM-induced cell death might result from two different mechanisms.39 At high RPM concentrations (≥100 nM), cell death occurs via an autophagy-dependent pathway, whereas at lower concentrations (≤10 nM), cell death occurs after a G1-phase cell cycle arrest.
We used a fluorescence microscope with monodansyl-cadaverine (MDC) staining to determine whether a low dose of RPM induces autophagy in PC-2 cells. MDC is a specific marker for autophagic vacuoles.40 The MDC-labeled autophagic vacuoles appeared as distinct dot-like structures distributed in the cytoplasm and in the perinuclear area, under a fluorescence microscope. The fluorescence density and MDC-labeled particles of the PC-2 cells were higher in the 10 nmol/L and 15 nmol/L RPM treatment groups than in the control group (Figure 4). This result indicates that low doses of RPM induced the formation of MDC-labeled vacuoles. The results indicate that autophagy was activated when the PC-2 cells underwent RPM-induced death.
Figure 4.
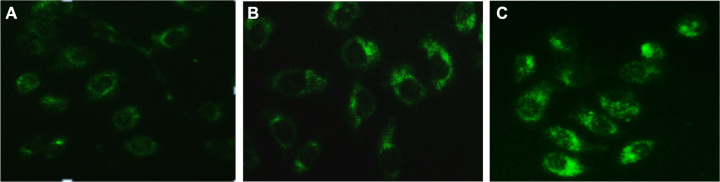
MDC-labeled autophagic vacuoles in PC-2 cells by fluorescence microscope after RPM treatment. Autophagic vacuoles were labeled with 0.05 mmol/L MDC in PBS, at 37°C for 10 minutes. (A) 0 nmol/L RPM group; (B) 10 nmol/L RPM group; and (C) 15 nmol/L RPM group (×200).
Abbreviations: MDC, monodansylcadaverine; PBS, phosphate-buffered saline; RPM, rapamycin.
Effects of radiation combined with RPM on the cell cycle distribution, under flow cytometry (FCM)
Radiation has been shown to induce transient cell cycle delays in the G1, S, and G2 phases, culminating in a G2/M interphase arrest that often takes place within 4 to 12 hours after irradiation.41 This occurrence allows the cells to repair DNA strand breaks before continuing cell division or alternatively, to initiate programmed cell death if the cellular damage is too severe for repair.42 Double-stranded DNA breaks may induce checkpoint responses in any phase of the cell cycle. There is evidence to suggest that the cells in different phases of the cell cycle have different radiosensitivities. The cells in the S phase have strong radioresistance. However, the cells in the G2/M phase are more sensitive to radiation, so the G2/M phase is a sensitive period for the irradiation and chemotherapy of tumor cells.43
In lung cancer cell lines, RPM was not found to affect the G2/M phase arrest induced by irradiation, at 12 hours, but caused a significant accumulation of cells in the G1 phase, with a corresponding decrease in S phase cells at 24 hours, after irradiation. Albert et al16 also reported that everolimus pretreatment in breast cancer MDA-MB-231 cells did not alter the cell cycle distribution at 8 hours after irradiation but increased the proportion of G2/M cells by twofold at 24 hours. Another pattern of response was observed in lung cancer A459 cells and breast cancer SKBR3 cells, wherein RPM treatment was found to have abrogated the G2/M delay at 8 hours after irradiation.17,44 Interestingly, in another study, the RPM treatment of prostate cancer cells, either concurrently with or before irradiation, resulted in an accumulation of relatively radioresistant G1 cells and a reduction in relatively more radiosensitive G2/M cells.45
In the present study, the effects of RPM and RPM with irradiation, on the cell cycle distribution were analyzed using FCM. Figure 5 shows a significant increase in the proportion of G0/G1 phase cells and a reduction in the proportion of S-phase cells, and the variations of G2/M phase cells between the four groups showed no significant difference (P > 0.05), after treatment with different RPM concentrations. The G2/M phase cells significantly increased after 4 Gy of irradiation, whereas the percentage of the S-phase cells decreased. In addition to the increase in the percentage of G2/M phase cells, the G0/G1 and the S-phase fractions were reduced when RPM was combined with 4 Gy X-ray irradiation. The percentages of cells in the S phase in the 10 nmol/L and 15 nmol/L RPM treatment groups were significantly lower than those in the control group and in the radiotherapy alone group. Tese results suggest that irradiation combined with RPM induced the cell cycle arrest of the PC-2 cells in the G2/M phase.
Figure 5.

Effects of radiation plus RPM on the cell cycle of PC-2 cells, under FCM.
Notes: The cell cycle distributions in PC-2 cells were determined using PI staining and FCM analysis, after RPM treatment or radiation plus RPM for 24 hours. (1) blank control group; (2) 5 nmol/L RPM group; (3) 10 nmol/L RPM group; (4) 15 nmol/L RPM group; (5) radiation alone group; (6) radiation plus 5 nmol/L RPM group; (7) radiation plus 10 nmol/L RPM group; and (8) radiation plus 15 nmol/L RPM group. The results presented were representative of three independent experiments. *P < 0.05, **P < 0.01 versus the control group; #P < 0.05, ##P < 0.01 versus the radiation alone group.
Abbreviations: FCM, flow cytometry; PI, propidium iodide; RPM, rapamycin.
The sub-G1 population exhibited apoptosis-associated chromatin degradation. The ratios of cell apoptosis in the control group and the RPM treatment groups (5, 10, and 15 nmol/L) were 2.56%, 3.95%, 6.41%, and 9.25%, respectively. Significant differences were observed between the RPM treatment groups and the control group (P < 0.05). The apoptosis rate increased by varying degrees when the PC-2 cells were treated with different RPM concentrations and irradiation dosages. The apoptosis rate was more than three times that of the control group when RPM was combined with 4 Gy of irradiation, significantly higher than that of irradiation alone and the RPM treatment groups (P < 0.01).
Gene expression by microarray in RPM-treated PC-2 cells
cDNA microarray is a powerful tool for simultaneously and quickly identifying changes in the expression of large numbers of genes.46 Several drug targets are components of complex signaling pathways, and the activation of signaling pathways leads to changes in multiple mRNA expression. Thus, microarrays can provide a detailed quantitative assessment of the consequences of this activation and allows efficient identification of the mechanisms of drug action.47,48
In this study, we performed cDNA microarray to characterize the radiosensitization mechanism of RPM. We used an Agilent Whole Human Genome Microarray chip to assess the expression profiles in RPM-treated cells. As shown in Figure 6, after treated with 10 nmol/L RPM for 48 hours, 527 functionally related genes were identified to be differentially expressed in the treated cells, including 340 downregulated genes and 187 upregulated genes. Table 2 shows 44 differentially expressed DNA transcription-and repair-related genes, including 19 upregulated genes and 25 downregulated genes. The genes that exhibited significant differential expression include DDB1, RAD51, XRCC5, PCNA, and ABCC4.
Figure 6.
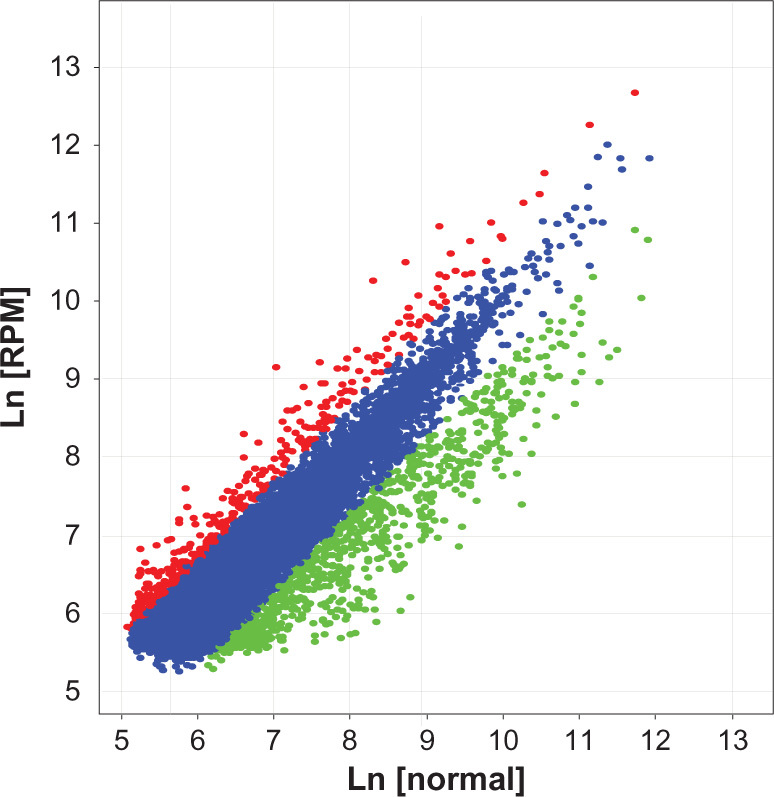
Scatter plot of the gene differential expression in PC-2 cells induced by RPM.
Notes: The Cy3 (RPM treated group) and Cy5 (untreated control) channel intensities from the two-color DNA microarray experiments were shown in the scatter plot. The variables appear in a linear relationship, and the linear correlation between them is 0.935. The red plots represented upregulated genes, and the green plots represented downregulated genes.
Abbreviation: RPM, rapamycin.
Table 2.
The DNA damage repair and transcription genes differentially expressed in PC-2 cells induced by RPM
| Genbank ID | Gene description | Ratio (Cy5/Cy3) |
|---|---|---|
| NM_016931 | Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) | 0.16 |
| NM-001923 | Damage-specific DNA binding protein (DDB1) | 0.20 |
| NM_018682 | Myeloid/lymphoid or mixed-lineage leukemia 5 (trithorax homolog, Drosophila) (MLL5) | 0.21 |
| NM_013974 | Dimethylarginine dimethylaminohydrolase 2 (DDAH2) | 0.23 |
| NM_001675 | Activating transcription factor 4 (tax-responsive enhancer element B67) (ATF4) | 0.27 |
| NM_003489 | Nuclear receptor interacting protein 1 (NRIP1) | 0.28 |
| BC049823 | Ribosomal protein L22-like 1 (RPL22) | 0.30 |
| NM_175623 | RAB3A interacting protein (rabin3) (RAB3IP), transcript variant alpha 2 | 0.32 |
| BC022217 | Chromosome 6 open reading frame 85(C6ORF85) | 0.33 |
| NM-005431 | X-ray repair complementing defective repair in Chinese hamster cells 2 (XRCC2) | 0.33 |
| NM_000689 | Aldehyde dehydrogenase 1 family, member A1 (ALDH1A1) | 0.36 |
| NM_002876 | RAD51 homolog C (S. cerevisiae) (RAD51C), transcript variant 2 | 0.37 |
| NM_001008712 | RNA binding protein with multiple splicing (RBPMS), transcript variant 3 | 0.38 |
| NM_139207 | Nucleosome assembly protein 1-like 1 (NAP1L1) | 0.39 |
| NM_000075 | Cyclin-dependent kinase 4 (CDK4) | 0.40 |
| NM_053277 | Chloride intracellular channel 6 (CLIC6) | 0.40 |
| NM-021141 | X-ray repair complementing defective repair in hamster cells 5 (XRCC5) | 0.41 |
| NM_001380 | Dedicator of cytokinesis 1 (DOCK1) | 0.42 |
| NM_014515 | CCR4-NOT transcription complex, subunit 2 (CNOT2) | 0.42 |
| NM_005655 | Transforming growth factor-beta inducible early growth response (TGFBI EGR) | 0.42 |
| NM-003852 | Transcriptional intermediary factor 1 (TIF 1) | 0.43 |
| NM_152721 | Docking protein 6 (DOK6) | 0.46 |
| NM_139207 | Nucleosome assembly protein 1-like 1 (NAP1L1) | 0.46 |
| NM_006312 | Nuclear receptor corepressor 2 (NCOR2) | 0.48 |
| NM_000538 | Regulatory factor X-associated protein (RFXAP) | 0.49 |
| NM_016545 | Immediate early response 5 (IER5) | 2.35 |
| NM-002129 | High-mobility group box 2 (HMGB2) | 2.41 |
| BC005107 | Chromosome 21 open reading frame 105, mRNA (C21ORF10) | 2.43 |
| NM-006608 | Putative homeodomain transcription factor 1 (PHTF 1) | 2.57 |
| NM_178820 | F-box protein 27 (FBXO27) | 2.61 |
| NM_000927 | ATP-binding cassette, subfamily B (MDR/TAP), member 1 (ABCB1) | 2.73 |
| NM_002592 | Proliferating cell nuclear antigen (PCNA) | 2.85 |
| NM_001924 | Growth arrest and DNA-damage-inducible, alpha (GADD45A) | 2.91 |
| NM_012153 | Ets homologous factor (EHF) | 3.13 |
| NM_005845 | ATP-binding cassette, subfamily C (CFTR/MRP), member 4 (ABCC4) | 3.24 |
| NM_199242 | Unc-13 homolog D (C. elegans) (UNC13D) | 3.24 |
| NM_172366 | F-box protein 16 (FBXO16) | 3.28 |
| NM_000107 | Damage-specific DNA binding protein 2 (DDB2) | 3.35 |
| NM_000392 | ATP-binding cassette, subfamily C (CFTR/MRP), member 2 (ABCC2) | 3.38 |
| NM_002439 | MutS homolog 3 (MSH3) | 3.42 |
| NM_020165 | RAD18 homolog (S. cerevisiae) (RAD18) | 3.45 |
| NM_006332 | Interferon, gamma-inducible protein 30 (IFI30) | 3.57 |
| NM_018965 | Triggering receptor expressed on myeloid cells 2 (TREM2) | 4.35 |
| BC009507 | Interferon, alpha-inducible protein (clone IFI-15K) | 6.59 |
Notes: Total RNA from PC-2 cells treated with 10 nmol/L RPM for 48 hours was extracted for microarray analysis. Representative genes were significantly upregulated or downregulated by RPM in PC-2 cells.
Abbreviation: RPM, rapamycin.
Gene expression validation by RT-PCR
To validate the microarray results, DDB1, RAD51, XRCC5, PCNA, and ABCC4 were chosen from Table 2 as representative DNA-damage repair and transcription genes for RT-PCR analysis. The RT-PCR validation results showed that the mRNA expression ratios of DDB1, RAD51, XRCC5, PCNA, and ABCC4 in the RPM group and control group were 0.25, 0.37, 0.45, 3.03, and 3.86, respectively, as shown in Figure 7A. DDB1, RAD51, and XRCC5 were downregulated, whereas PCNA and ABCC4 were upregulated. Moreover, the RT-PCR results conformed to the results of cDNA microarray (Figure 7B).
Figure 7.
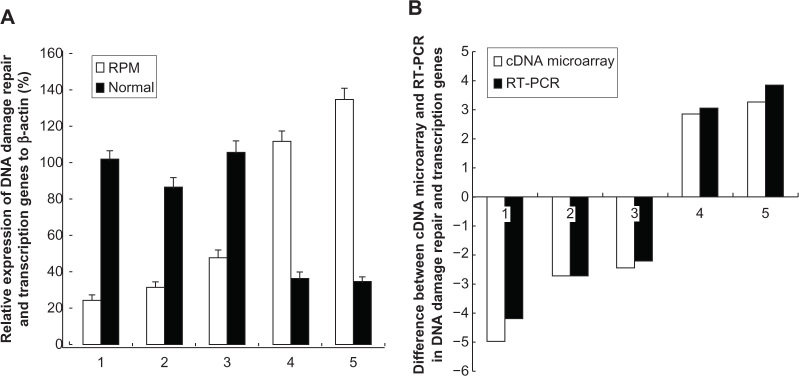
(A) RT-PCR analysis for the DNA-damage repair and transcription genes after the treatment with RPM; (B) Difference between the cDNA microarray and RT-PCR in DNA-damage repair and transcription genes.
Notes: The genes examined here are: (1) DDB1(NM_001923); (2) RAD51 (NM_002876); (3) XRCC5 (NM_021141); (4) PCNA (NM_002592); and (5) ABCC4 (NM_005845). The values represent the mean ± SD of the data from three independent experiments.
Abbreviations: RPM, rapamycin; RT-PCR, reverse transcription polymerase chain reaction; SD, standard deviation.
Double-strand breaks (DSB) are the most common injury in tumor cells after chemotherapy and irradiation. The DSB repair process has two major pathways: homologous recombination repair and nonhomologous recombination repair.49 RAD51 plays a critical role in the homologous recombination repair. Abnormal RAD51 expression often causes recombination repair runaway development, which is closely related to tumorigenesis. The RAD51 overexpression in tumor cells can lead to resistance to radiotherapy and chemotherapy.50 In this study, the RAD51 expression in the RPM-treated group was significantly downregulated, which suggests that RAD51 is involved in the RPM radiosensitization.
Radioactive DNA-damage repair causes resistance to the killing effect of radiotherapy; hence, genome stability has an important role in radiotherapy.51 X-ray repair cross-complementing genes (XRCC) are genes related to DNA-damage repair. Human XRCC5 gene is located on chromosome 2q33-35. XRCC5 encodes Ku80 and XRCC6 encodes Ku70 protein, which form the Ku heterodimer, a conserved DNA adhesion protein involved in repairing DSBs, for maintaining the integrity of genome function and repairing the damage caused by extrinsic factors.52 Ku80, the XRCC5 gene product, can be positioned coupled to the free end of DSBs; thus, it has an important role in the repair process of DSB recombinants.53 Evans et al54 reported that the lack of XRCC5 gene expression in hamster mutant XR-5 cells prevented the encoding of Ku 80, resulting in functional defects in DSB repair and increased radiation sensitivity. However, transfecting XR-5 cells with Ku80 cDNA restored their DSB recombination repair function, a mark of cells with radiation tolerance. In this study, the XRCC5 downregulation in the PC-2 cells after RPM treatment enhanced the radiosensitivity of PC-2 cells, which is consistent with the literature.
Proliferating cell nuclear antigen (PCNA) is a gene that reflects important indicators of cell proliferation that are involved in DNA replication and DNA repair synthesis.55 Previous studies suggest that PCNA is correlated with the histologic grade, clinical stage, and radiosensitivity of tumors; thus, it could be used as an indicator for radiosensitivity.56 The gene chip and the RT-PCR results in the experiment showed significantly upregulated PCNA expression, which suggests that increased PCNA gene expression may be one of the mechanisms of RPM in radiosensitization.
Conclusion
In conclusion, we have demonstrated that RPM can enhance the radiosensitivity of pancreatic cancer cells. Low doses of RPM can induce autophagy. The FCM assay indicated that RPM induced the cell cycle arrest of the PC-2 cells in the G2/M phase. The cDNA microarray results demonstrated that gene-expression patterns are influenced by RPM treatment in vitro, in a pancreatic cancer cell line. Furthermore, two overexpressed genes and three low-expressed genes were validated. However, we were unable to determine whether changes in gene expression are involved in the radiosensitization of pancreatic cancer cells, as well as the underlying mechanisms. Therefore, further protein level and functional studies are required to confirm the relevance of these gene expression changes.
Acknowledgments
This work was supported by the National Natural Science Foundation of China, No 81102711; the Science and Technology Foundation of Shannxi Province, People’s Republic of China, No 2010 K01-138; and the Sci-tech Program of Xi’an City, People’s Republic of China, No HM1117.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumont FJ, Bischoff P. Disrupting the mTOR signaling network as a potential strategy for the enhancement of cancer radiotherapy. Curr Cancer Drug Targets. 2012;12(8):899–924. doi: 10.2174/156800912803251243. [DOI] [PubMed] [Google Scholar]
- 5.Beuvink I, Boulay A, Fumagalli S. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120(6):747–759. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 6.Mabuchi S, Altomare DA, Cheung M. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13(14):4261–4270. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 7.Houghton PJ, Morton CL, Gorlick R. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010;9(1):101–112. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris RE. Rapamycins: Antifungal, antitumor, antiproliferative, and immunosuppressive macrolides. Transplant Rev. 1992;6(1):39–87. [Google Scholar]
- 9.Svirshchevskaya EV, Mariotti J, Wright MH. Rapamycin delays growth of Wnt-1 tumors in spite of suppression of host immunity. BMC Cancer. 2008;8:176. doi: 10.1186/1471-2407-8-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Q, Kiguchi K, Kawamoto T. Therapeutic effect of rapamycin on gallbladder cancer in a transgenic mouse model. Cancer Res. 2007;67(8):3794–3800. doi: 10.1158/0008-5472.CAN-06-3214. [DOI] [PubMed] [Google Scholar]
- 11.Butzal M, Loges S, Schweizer M. Rapamycin inhibits proliferation and differentiation of human endothelial progenitor cells in vitro. Exp Cell Res. 2004;300(1):65–71. doi: 10.1016/j.yexcr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Morikawa Y, Koike H, Sekine Y. Rapamycin enhances docetaxel-induced cytotoxicity in a androgen-independent prostate cancer xenograft model by survivin downregulation. Biochem Biophys Res Commun. 2012;419(3):584–589. doi: 10.1016/j.bbrc.2012.02.089. [DOI] [PubMed] [Google Scholar]
- 13.Li R, Wang R, Zhai R, Dong Z. Targeted inhibition of mammalian target of rapamycin (mTOR) signaling pathway inhibits proliferation and induces apoptosis of laryngeal carcinoma cells in vitro. Tumori. 2011;97(6):781–786. doi: 10.1177/030089161109700616. [DOI] [PubMed] [Google Scholar]
- 14.Di Paolo S, Teutonico A, Ranieri E, Gesualdo L, Schena PF. Monitoring antitumor efficacy of rapamycin in Kaposi sarcoma. Am J Kidney Dis. 2007;49(3):462–470. doi: 10.1053/j.ajkd.2006.11.037. [DOI] [PubMed] [Google Scholar]
- 15.Guba M, von Breitenbuch P, Steinbauer M. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8(2):128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 16.Albert JM, Kim KW, Cao C, Lu B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol Cancer Ther. 2006;5(5):1183–1189. doi: 10.1158/1535-7163.MCT-05-0400. [DOI] [PubMed] [Google Scholar]
- 17.No M, Choi EJ, Kim IA. Targeting HER2 signaling pathway for radiosensitization: alternative strategy for therapeutic resistance. Cancer Biol Ther. 2009;8(24):2351–2361. doi: 10.4161/cbt.8.24.10131. [DOI] [PubMed] [Google Scholar]
- 18.Cao C, Subhawong T, Albert JM. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66(20):10040–10047. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 19.Nagata Y, Takahashi A, Ohnishi K. Effect of rapamycin, an mTOR inhibitor, on radiation sensitivity of lung cancer cells having different p53 gene status. Int J Onco. 2010;37(4):1001–1010. doi: 10.3892/ijo_00000751. [DOI] [PubMed] [Google Scholar]
- 20.Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62(24):7291–7297. [PubMed] [Google Scholar]
- 21.Dai ZJ, Gao J, Ma XB. Up-regulation of hypoxia inducible factor-1á by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J Exp Clin Cancer Res. 2012;31:28. doi: 10.1186/1756-9966-31-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang B, Kong D, Liu Y. Autophagy inhibition plays the synergetic killing roles with radiation in the multi-drug resistant SKVCR ovarian cancer cells. Radiat Oncol. 2012;7:213. doi: 10.1186/1748-717X-7-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai ZJ, Gao J, Ma XB. Antitumor effects of rapamycin in pancreatic cancer cells by inducing apoptosis and autophagy. Int J Mol Sci. 2012;14(1):273–285. doi: 10.3390/ijms14010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Wang Y, Pang X. Enhanced radiosensitivity of EC109 cells by inhibition of HDAC1 expression. Med Oncol. 2012;29(1):340–348. doi: 10.1007/s12032-010-9559-3. [DOI] [PubMed] [Google Scholar]
- 25.Zsippai A, Szabó DR, Tömböl Z. Effects of mitotane on gene expression in the adrenocortical cell line NCI-H295R: a microarray study. Pharmacogenomics. 2012;13(12):1351–1361. doi: 10.2217/pgs.12.116. [DOI] [PubMed] [Google Scholar]
- 26.Guan HT, Xue XH, Dai ZJ, Wang XJ, Li A, Qin ZY. Down-regulation of survivin expression by small interfering RNA induces pancreatic cancer cell apoptosis and enhances its radiosensitivity. World J Gastroenterol. 2006;12(18):2901–2907. doi: 10.3748/wjg.v12.i18.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer. 2011;10:138. doi: 10.1186/1476-4598-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y, Wang X, Xia W. Antiproliferative and overadditive effects of rapamycin and FTY720 in pancreatic cancer cells in vitro. Transplant Proc. 2008;40(5):1727–1733. doi: 10.1016/j.transproceed.2008.03.150. [DOI] [PubMed] [Google Scholar]
- 29.Garrido-Laguna I, Tan AC, Uson M. Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer. 2010;103(5):649–655. doi: 10.1038/sj.bjc.6605819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shafer A, Zhou C, Gehrig PA, Boggess JF, Bae-Jump VL. Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int J Cancer. 2010;126(5):1144–1154. doi: 10.1002/ijc.24837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjelogrlić SK, Srdić T, Radulović S. Mammalian target of rapamycin is a promising target for novel therapeutic strategy against cancer. J BUON. 2006;11(3):267–276. [PubMed] [Google Scholar]
- 32.Saito K, Matsumoto S, Yasui H. Longitudinal imaging studies of tumor microenvironment in mice treated with the mTOR inhibitor rapamycin. PLoS ONE. 2012;7(11):e49456. doi: 10.1371/journal.pone.0049456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mauceri HJ, Sutton HG, Darga TE. Everolimus exhibits efficacy as a radiosensitizer in a model of non-small cell lung cancer. Oncol Rep. 2012;27(5):1625–1629. doi: 10.3892/or.2012.1666. [DOI] [PubMed] [Google Scholar]
- 34.Myers AL, Orr WS, Denbo JW. Rapamycin-induced tumor vasculature remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. J Pediatr Surg. 2012;47(1):183–189. doi: 10.1016/j.jpedsurg.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuwahara Y, Oikawa T, Ochiai Y. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2011;2:e177. doi: 10.1038/cddis.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson EN, Bristol ML, Di X. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Horm Cancer. 2011;2(5):272–285. doi: 10.1007/s12672-011-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy. 2009;5(8):1232–1234. doi: 10.4161/auto.5.8.9896. [DOI] [PubMed] [Google Scholar]
- 38.Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy. 2008;4(5):659–668. doi: 10.4161/auto.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eum KH, Lee M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Haras-transformed NIH 3T3 cells. Mol Cells. 2011;31(3):231–238. doi: 10.1007/s10059-011-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biederbick A, Kern HF, Elsässer HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66(1):3–14. [PubMed] [Google Scholar]
- 41.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928–942. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Pauwels B, Wouters A, Peeters M, Vermorken JB, Lardon F. Role of cell cycle perturbations in the combination therapy of chemotherapeutic agents and radiation. Future Oncol. 2010;6(9):1485–1496. doi: 10.2217/fon.10.104. [DOI] [PubMed] [Google Scholar]
- 43.Ree AH, Stokke T, Bratland A. DNA damage responses in cell cycle G2 phase and mitosis – tracking and targeting. Anticancer Res. 2006;26(3A):1909–1916. [PubMed] [Google Scholar]
- 44.Choi EJ, Ryu YK, Kim SY. Targeting epidermal growth factor receptor-associated signaling pathways in non-small cell lung cancer cells: implication in radiation response. Mol Cancer Res. 2010;8(7):1027–1036. doi: 10.1158/1541-7786.MCR-09-0507. [DOI] [PubMed] [Google Scholar]
- 45.Schiewer MJ, Den R, Hoang DT. mTOR is a selective effector of the radiation therapy response in androgen receptor-positive prostate cancer. Endocr Relat Cancer. 2012;19(1):1–12. doi: 10.1530/ERC-11-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SM, Li ML, Tse YC. Paeoniae Radix, a Chinese herbal extract, inhibit hepatoma cells growth by inducing apoptosis in a p53 independent pathway. Life Sci. 2002;71(19):2267–2277. doi: 10.1016/s0024-3205(02)01962-8. [DOI] [PubMed] [Google Scholar]
- 47.Yin X, Zhou J, Jie C, Xing D, Zhang Y. Anticancer activity and mechanism of Scutellaria barbata extract on human lung cancer cell line A549. Life Sci. 2004;75(18):2233–2244. doi: 10.1016/j.lfs.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Wang F, Tan W, Guo D, Zhu X, Qian K, He S. Altered expression of signaling genes in Jurkat cells upon FTY720 induced apoptosis. Int J Mol Sci. 2010;11(9):3087–3105. doi: 10.3390/ijms11093087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karagiannis TC, El-Osta A. Double-strand breaks: signaling pathways and repair mechanisms. Cell Mol Life Sci. 2004;61(17):2137–2147. doi: 10.1007/s00018-004-4174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choudhury A, Cuddihy A, Bristow RG. Radiation and new molecular agents part I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Semin Radiat Oncol. 2006;16(1):51–58. doi: 10.1016/j.semradonc.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Jenner TJ, Fulford J, O’Neill P. Contribution of base lesions to radiation-induced clustered DNA damage: implication for models of radiation response. Radiat Res. 2001;156(5 Pt 2):590–593. doi: 10.1667/0033-7587(2001)156[0590:cobltr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 52.Taccioli GE, Gottlieb TM, Blunt T. Ku80: product of the XRCC5 gene and its roles in DNA repair and V(D)J recombination. Science. 1994;265(5177):1442–1445. doi: 10.1126/science.8073286. [DOI] [PubMed] [Google Scholar]
- 53.Bryntesson F, Regan JC, Jeggo PA, Taccioli GE, Hubank M. Analysis of gene transcription in cells lacking DNA-PK activity. Radiat Res. 2001;156(2):167–176. doi: 10.1667/0033-7587(2001)156[0167:aogtic]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 54.Evans JW, Liu XF, Kirchgessner CU, Brown JM. Induction and repair of chromosome aberrations in scid cells measured by premature chromosome condensation. Radiat Res. 1996;145(1):39–46. [PubMed] [Google Scholar]
- 55.Hashiguchi K, Matsumoto Y, Yasui A. Recruitment of DNA repair synthesis machinery to sites of DNA damage/repair in living human cells. Nucleic Acids Res. 2007;35(9):2913–2923. doi: 10.1093/nar/gkm115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee BJ, Wang SG, Roh HJ, Goh EK, Chon KM, Park DY. Changes in expression of p53, proliferating cell nuclear antigen and bcl-2 in recurrent laryngeal cancer after radiotherapy. J Laryngol Otol. 2006;120(7):579–582. doi: 10.1017/S0022215106001150. [DOI] [PubMed] [Google Scholar]