

Abstract
Background:
Hemophagocytic lymphohistiocytosis (HLH) has been reported to complicate fulminant tropical infections but data on severe HLH with multi-organ dysfunction (MODS) are scant.
Materials and Methods:
Retrospective review of medical electronic records of our intensive care unit (ICU) over a 2-year period.
Results:
We describe 10 adult patients with HLH and MODS. Patients had short symptom duration prior to presentation and had rapid deterioration during their hospitalization course. Fever, organomegaly, neurologic abnormalities, hepatic abnormalities, and cutaneous signs were very common. No patient had diagnosed HLH at ICU admission (median 4 criteria [Inter Quartile Range 2-4.25]). All patients required mechanical ventilation and 80% required vasopressors. Infection-associated HLH (IAHS) was the most common etiology (80%). Seventy percent (7/10) of patients were treated with steroids and 20% received intravenous immunoglobulin. Etoposide and/or cyclosporine were administered in 20% (2/10). Nosocomial infections occurred in 40% and the ICU mortality was 70%.
Conclusions:
Severe HLH with MODS has a very high mortality. Data on adult cohorts with IAHS in the tropics with defined treatment protocols are urgently needed.
Keywords: Hemophagocytic lymphohistiocytosis, hemophagocytic syndrome, multi-organ dysfunction, tropics
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a catastrophic syndrome of unrestrained immune activation.[1] HLH may be genetic in origin or arise secondary to infectious, rheumatologic, malignant or metabolic disorders.[2] Irrespective of the cause, the unrestrained immune hyperactivation leads to host tissue damage and a characteristic clinical syndrome.[3] Evaluation and management of HLH in the tropics, especially in adults, is complicated by tropical infections which can trigger HLH and mimic its symptoms and signs, delayed clinician recognition, absence of prognostic data from prospective cohorts of infection-associated HLH (IAHS), lack of widespread availability of laboratory investigations to confirm HLH and lack of data on frequency of genetic mutations predisposing to familial HLH (FLH). Management of HLH associated with multi-organ dysfunction (MODS) is particularly challenging.[4] We report a retrospective analysis of our computerized database over a 2-year period on our experience with severe HLH managed in an intensive care unit (ICU).
Materials and Methods
The computer records of patients admitted between January 2007 and December 2008 to our ICU with a diagnosis of HLH were reviewed. Starting from January 2009, data collection was modified and hence excluded from this study. Patients with a final diagnosis of HLH fulfilling HLH-2004 criteria were selected [Table 1].[1] If at least five criteria were not met, cases were eligible for review if clinical findings were strongly suggestive of HLH and autopsy findings showed prominent hemophagocytosis in at least two organs. The original case records of the patients were then retrieved from the central registration department. Demographic information such as age and gender, clinical status at admission to ICU including the details of organ failure were abstracted. Details of the clinical manifestations and investigations like liver and renal function tests, arterial blood gases were recorded. Details of their stay in ICU, Acute Physiology and Chronic Health Evaluation APACHE II scores, treatment with steroids, etoposide and intravenous immunoglobulin and outcomes were noted. The study was approved by the institutional ethics board.
Table 1.
Revised diagnsotic criteria for hemophagocytic lymphohistiocytosis (Henter et al.)

Statistical analysis
The statistical package SPSS 14 was used to perform the statistical analysis. Continuous variables are presented in a descriptive fashion (Mean ± SD or median with range).
Results
We found 10 cases over a 2-year period with HLH in our ICU database, yielding a 10/960 (1.04%) prevalence of diagnosed cases over the same time period. Two of these cases have been previously described.[5,6] Most of the patients were young (median 25.5 years IQR 22.75-34.75) and without any co-morbidities [80%, Table 2]. Patient had short symptom duration prior to presentation (median 11 days IQR 9.25-30) and had a rapid deterioration during their hospitalization course (median 2 days, [5 h-3.75 days]). The indications for ICU admission included ARDS [Figure 1], fulminant hepatic failure (FHF) with encephalopathy, shock and seizures [Table 1]. Neurologic (90%), hepatic abnormalities (80%) and cutaneous (60%) signs were common in our cohort. At ICU admission, organomegaly (90%), lymphadenopathy (30%) and ascites (30%) were frequently observed. At least 90% had one cytopenia at ICU admission and 40% had bicytopenia. A diagnosis of HLH was strongly considered in 3/10 (30%) prior to ICU admission and HLH criteria were not met in most patients at ICU admission (median 4 criteria [IQR 2-4.25], N = 6) with at least 5/8 of HLH-2004 criteria required for definite diagnosis. The admission diagnosis was FHF (3/10, 30%), disseminated tuberculosis (10%), leptospirosis (20%) or malaria (20%) in the other patients [Table 3]. Patients had rapid deterioration after ICU admission with acute kidney injury (6/10, 60%), hypotension (vasopressors 8/10, 80%), status epilepticus (SE) (3/10, 30%) and acute renal failure requiring dialysis (30%). All patients required mechanical ventilation for pulmonary edema [acute respiratory distress syndrome (ARDS), 9/10 (90%), cardiogenic pulmonary edema (CPE) 1/10 (10%)]. The apparent short duration of ventilation (mean duration of 4.52 ± 0.98 days) was because of the high mortality associated with the syndrome. All patients were treated with intravenous antibiotics (a third generation cephalosporin and a macrolide or fluoroquinolone). Other antimicrobial agents administered included empiric anti-tuberculosis treatment (2/10, 20%), anti-viral (acyclovir 2/10, 20% for EBV), empiric anti-malarial therapy with artesunate (4/10, 40%) and amphotericin (1/10, 10% for leishmaniasis). The mean number of blood units (packed red blood concentrates, fresh frozen plasma, single donor apheresis platelet concentrates) was 8.22 ± 1.83 units per person for severe bleeding, anemia or facilitation of invasive procedures. A diagnosis of HLH was made ante-mortem in 60% [6/10, Figure 2] and strongly suspected [criteria not satisfied] in another patient. In 3/10 (30%) patients, the diagnosis was only made post-mortem because of the fulminant clinical course (case four and seven) or an alternate diagnosis being strongly considered [case ten, Figure 3]. In the three patients’ diagnosed post-mortem, clinical findings and hemophagocytosis in multiple organs strongly favored a diagnosis of HLH syndrome even though biochemically supportive investigations were unavailable. Seventy percent (7/10) of patients were treated with steroids for suspected or proven HLH ante-mortem. Intravenous immunoglobulin (IVIG) was administered in 2/10 (20%) and HLH-94 protocol (10%) and cyclosporine A (CSA) with steroids in another patient. Reasons for withholding administration of etoposide despite a diagnosis of HLH included infection, including septic shock, in 4/10 (40%) and the absence of a confirmed diagnosis (40%) of HLH. Clinically recognized nosocomial infections complicated 40% (4/10) of the patient's course. In another patient, a diagnosis of invasive mucormycosis was made post-mortem. Severe HLH with shock and ARDS had high ICU (7/10, 70%) and hospital mortality (8/10, 80%).
Table 2.
Clinical presentations, investigations, hospital course and outcome of 10 patients with severe hemophagocytic lymphohistiocytosis admitted to intensive care unit

Figure 1.
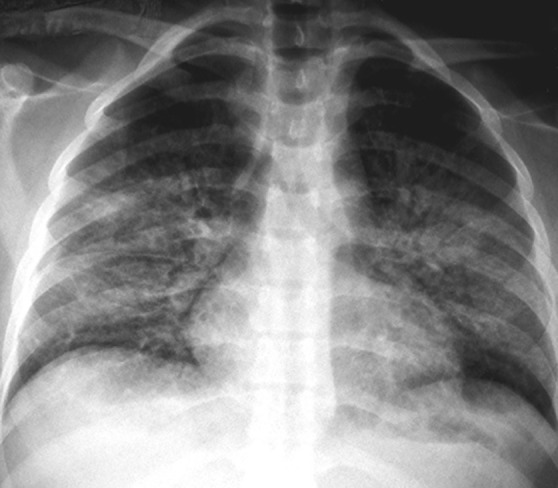
Chest radiograph showing bilateral perihilar consolidation characteristic of acute respiratory distress syndrome (ARDS) from Case 5. Ninety percent had findings of ARDS
Table 3.
Clinical details of individual patients with hemophagocytic lymphohistiocytosis
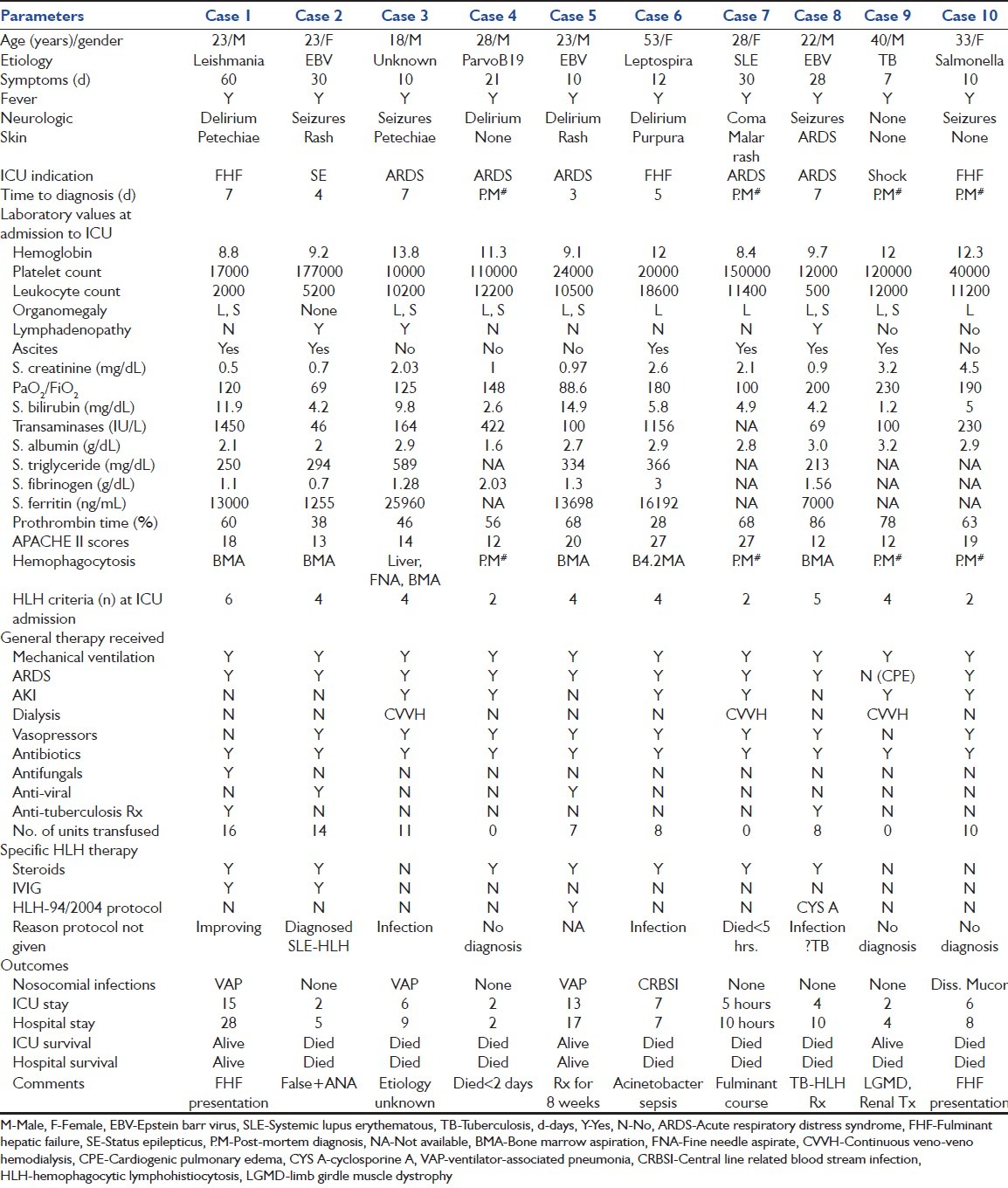
Figure 2.
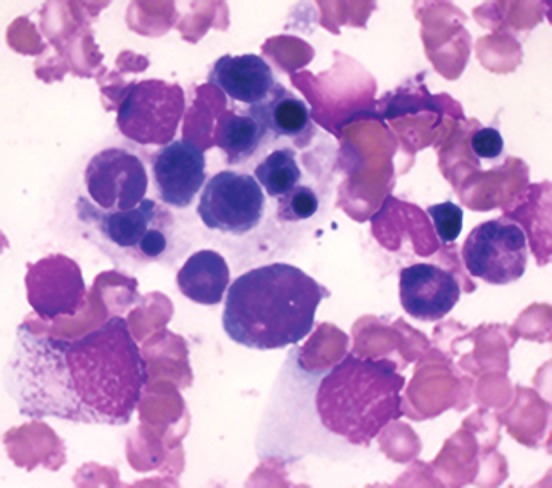
Bone marrow aspirate from Case 2 showing prominent hemophagocytosis. Both, anti-nuclear antibody and IgM anti-VCA (EBV) ELISA were reported positive. Post-mortem examination confirmed EBV-related HLH
Figure 3.
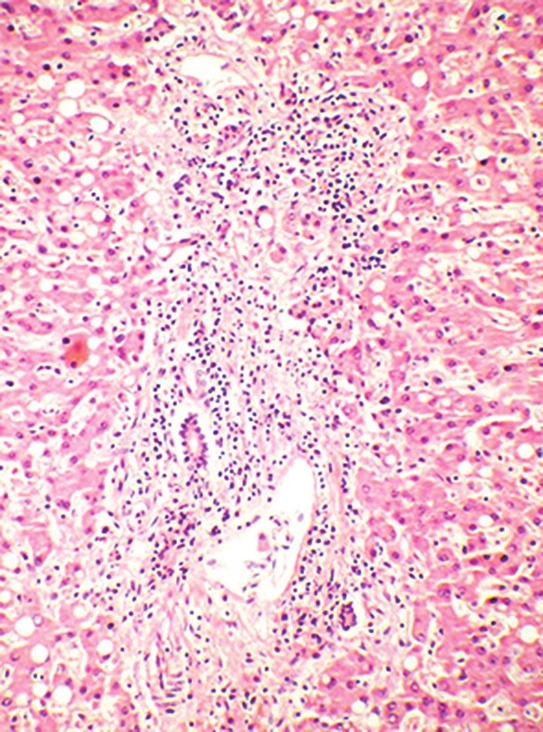
Photomicrograph from post-mortem liver specimen of Case 10 showing prominent peri-portal mononuclear infiltration suggestive of chronic hepatitis. Liver biopsies show this picture in 50% of patients and may be misleading if presentation is with jaundice and raised transaminases and HLH is not suspected
Discussion
HLH is a fulminant syndrome of unrestrained immune activation triggered by a variety of infectious, metabolic or malignant disorders.[7] The diagnosis of HLH is currently made by fulfilling at least five out of the eight criteria of HLH [Table 1].[1] Failure of apoptosis of activated mononuclear cells due to defect in the perforin-granzyme pathway leads to failure of immune down-regulation.[8] Defects in this critical immune regulatory pathway lead to hypercytokinemia and organ infiltration by phagocytizing histiocytes, with a subsequent characteristic clinical syndrome.[3]
HLH is an entity with major diagnostic and therapeutic challenges. The clinical features of HLH secondary to unrestrained immune activation are not specific and mimic sepsis with MODS, tropical infections [visceral leishmaniasis, disseminated tuberculosis, leptospirosis, scrub typhus and severe malaria], hematological malignancy and auto-immune disease in adults.[6] The criteria for diagnosis of HLH were primarily designed to select enrollment into clinical trials; the sensitivity of this criteria for early HLH is unknown given the lack of a gold standard test. Importantly, the clinical picture might be aggressive and diagnostic criteria might not be fulfilled at the onset, making management extremely challenging. In particular, the finding of bone marrow hemophagocytosis is neither sensitive nor required for the diagnosis of early HLH.[8,9] Also, the finding of isolated marrow hemophagocytosis has been noted in 0.8-4% of patients with sepsis and cytopenia in the absence of HLH syndrome and the importance of this finding is unclear.[10]
Patients with FLH and viral-related HLH (VAHS) are treated with the HLH-2004 protocol.[1] Briefly, the protocol specifies the use of CSA, etoposide and dexamethasone for 8 weeks; intrathecal methotrexate is administered to patients with neurological signs, persistent active Central nervous system (CNS) disease or CNS reactivation of HLH.[11] Patients with FLH, known mutations in perforin pathway and those with severe and persistent non-familial disease (VAHS) or relapsed HLH are then treated with continuation phase etoposide, dexamethasone, and CSA for 24 weeks. Hematopoietic stem cell transplantation (HSCT) is performed as early as possible, when an acceptable donor is available.[1] Delay in administration of etoposide to patients with EBV-HLH may increase mortality and this syndrome does not subside with anti-viral (acyclovir) alone.[12] In contrast, prognostic data on cohorts of IAHS are scant.[13] Systematic reviews and small series of HLH related to visceral leishmaniasis,[6] tuberculosis,[14] dengue[15] and scrub typhus suggest[16] that HLH triggered by these tropical infections is fulminant but resolves with specific anti-microbial therapy alone. The main determinant of mortality is the delay in administration of appropriate anti-microbials.[14] IVIG with steroids, administered early during severe hyperferritinemia, have been used in patients with infection-related or connective-tissue related HLH.[17] Although, no trials in IAHS have compared IVIG to etoposide, it is considered a less efficacious regimen.[18] Data from pediatric cohorts[9,19] [median age 24 and 33 months] reported from India suggest that infectious triggers are the most common etiology of HLH (42%, 14/33).[9] Steroids and IVIG are the first agents employed, followed by cyclosporine and etoposide for refractory disease.
Even less is known about adult patients with HLH and MODS in the medical ICU.[3] We are aware of only one previous single-center experience over a 10-year period which reported 56 adult patients with confirmed HLH.[4] In contrast to our series, the trigger was mostly hematological malignancy (43/56, 76.8%) and the diagnosis of HLH had been made prior to ICU admission in a large proportion (median number of HLH criteria at ICU admission 6, IQR 5-7). Severity of illness was lesser than our cohort with 57% (32/56) requiring mechanical ventilation, 53.6% developing (30/56) hypotension and 34% (19/56) requiring dialysis. Etoposide and/or steroids were administered in 80.35% (45/56). The hospital mortality rate was 51.8% (29/56); mortality increased with the presence of shock (O.R 4.33, 95% C.I 1.1-16.9) and etiology other than lymphoma or Castleman's disease (O.R 3.94, 95% C.I 1.25-12.73, P = 0.007).[4] Our series was characterized by the frequency of infection-related HLH (80%, short symptom interval, fulminant course and severe MODS [Table 3]. The presence of suspected bacterial infection and/or hypotension, suspected tropical triggers (tuberculosis, Leishmaniasis, dengue, salmonella, malaria and leptospirosis), severe thrombocytopenia, poor sensitivity of marrow examination, turnaround time of specific biochemical investigations (ferritin, SCD25) and fulminant clinical course were the major deterrents to early administration of etoposide-based regimen in our adult series.
In conclusion, HLH is a fulminant syndrome of immune dysregulation and occurs after a variety of triggers. Intensivists in the tropics must be aware of the possibility of HLH when managing fulminant tropical infections, malignancy and connective tissue diseases. Severe HLH with MODS has a very high mortality rate. Data on cohorts with HLH in the tropics with defined treatment protocol are urgently required.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 2.Janka GE. Hemophagocytic lymphohistiocytosis. Hematology. 2005;10:104–7. doi: 10.1080/10245330512331390087. [DOI] [PubMed] [Google Scholar]
- 3.Créput C, Galicier L, Buyse S, Azoulay E. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med. 2008;34:1177–87. doi: 10.1007/s00134-008-1111-y. [DOI] [PubMed] [Google Scholar]
- 4.Buyse S, Teixeira L, Galicier L, Mariotte E, Lemiale V, Seguin A, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med. 2010;36:1695–702. doi: 10.1007/s00134-010-1936-z. [DOI] [PubMed] [Google Scholar]
- 5.Bal A, Mishra B, Singh N, Das A, Jindal SK. Fulminant parvovirus B19-associated pancarditis with haemophagocytic lympho-histiocytosis in an immunocompetent adult. APMIS. 2009;117:773–7. doi: 10.1111/j.1600-0463.2009.02528.x. [DOI] [PubMed] [Google Scholar]
- 6.Rajagopala S, Dutta U, Chandra KS, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis-case report and systematic review. J Infect. 2008;56:381–8. doi: 10.1016/j.jinf.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008;28:293–313. doi: 10.1016/j.iac.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Henter JI. Biology and treatment of familial hemophagocytic lymphohistiocytosis: Importance of perforin in lymphocyte-mediated cytotoxicity and triggering of apoptosis. Med Pediatr Oncol. 2002;38:305–9. doi: 10.1002/mpo.1340. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran B, Balasubramanian S, Abhishek N, Ravikumar KG, Ramanan AV. Profile of hemophagocytic lymphohistiocytosis in children in a tertiary care hospital in India. Indian Pediatr. 2011;48:31–5. doi: 10.1007/s13312-011-0020-2. [DOI] [PubMed] [Google Scholar]
- 10.Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients–a postmortem clinicopathologic analysis. Crit Care Med. 2004;32:1316–21. doi: 10.1097/01.ccm.0000127779.24232.15. [DOI] [PubMed] [Google Scholar]
- 11.Imashuku S. Advances in the management of hemophagocytic lymphohistiocytosis. Int J Hematol. 2000;72:1–11. [PubMed] [Google Scholar]
- 12.Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44:259–72. doi: 10.1016/s1040-8428(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 13.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–22. doi: 10.1016/S1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis. 2006;6:447–54. doi: 10.1016/S1473-3099(06)70524-2. [DOI] [PubMed] [Google Scholar]
- 15.Jain D, Singh T. Dengue virus related hemophagocytosis: A rare case report. Hematology. 2008;13:286–8. doi: 10.1179/102453308X316095. [DOI] [PubMed] [Google Scholar]
- 16.Gopal GK, Anugrah C, Boorugu H. Scrub typhus associated macrophage activation syndrome. Trop Doct. 2010;40:249–50. doi: 10.1258/td.2010.100056. [DOI] [PubMed] [Google Scholar]
- 17.Emmenegger U, Frey U, Reimers A, Fux C, Semela D, Cottagnoud P, et al. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol. 2001;68:4–10. doi: 10.1002/ajh.1141. [DOI] [PubMed] [Google Scholar]
- 18.Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–3. [PubMed] [Google Scholar]
- 19.Mathew LG, Cherian T, Sudarshanam A, Korah I, Kumar NK, Raghupathy P. Hemophagocytic Lymphohistiocytosis: A Case Series. Indian Pediatr. 2000;37:526–31. [PubMed] [Google Scholar]