

Abstract
The signaling mediated by the chemokine receptor CXC chemokine receptor 2 (CXCR2) plays an important role in promoting the progression of many cancers, including pancreatic cancer, one of the most lethal human malignancies. CXCR2 possesses a consensus PSD-95/DlgA/ZO-1 (PDZ) motif at its carboxyl termini, which might interact with potential PDZ scaffold/adaptor proteins. We have previously reported that CXCR2 PDZ motif-mediated protein interaction is an important regulator for neutrophil functions. Here, using a series of biochemical assays, we demonstrate that CXCR2 is physically coupled to its downstream effector phospholipase C-β3 (PLC-β3) that is mediated by PDZ scaffold protein Na+/H+ exchange regulatory factor 1 (NHERF1) into a macromolecular signaling complex both in vitro and in pancreatic cancer cells. We also observe that disrupting the CXCR2 complex, by gene delivery or peptide delivery of exogenous CXCR2 C-tail, significantly inhibits the biologic functions of pancreatic cancer cells (i.e., proliferation and invasion) in a PDZ motif-dependent manner. In addition, using a human pancreatic tumor xenograft model, we show that gene delivery of CXCR2 C-tail sequence (containing the PDZ motif) by adeno-associated virus type 2 viral vector potently suppresses human pancreatic tumor growth in immunodeficient mice. In summary, our results suggest the existence of a physical and functional coupling of CXCR2 and PLC-β3 mediated through NHERF1, forming a macromolecular complex that is critical for efficient and specific CXCR2 signaling in pancreatic cancer progression. Disrupting this CXCR2 complex could represent a novel and effective treatment strategy against pancreatic cancer.
Introduction
Pancreatic cancer, the most lethal malignancy of the gastrointestinal tract with 5-year survival rates of less than 5%, is the fourth leading cause of cancer-related deaths in both men and women in the United States [1]. The most common type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC). Chemoresistance, early metastases, and late clinical presentation in this incurable human malignancy result in no effective methods for early prognosis as well as a lack of effective systemic therapies with reduced side effects [2,3]. Therefore, a more comprehensive understanding of PDAC biology and the mechanisms/factors that promote tumor growth and metastasis may help identify new molecular targets for the development of diagnostics and/or therapeutics of pancreatic cancer.
CXC chemokine receptor 2 (CXCR2) is the cognate receptor for the CXC chemokines CXCL1 to CXCL3 and CXCL5 to CXCL8 [4]. The CXC chemokine/CXCR2 signaling has been reported to promote malignant cancer progression in many cancer types including pancreatic cancer [5–9]. It has been documented that the elevated expression of CXCL5 and CXCL8 is correlated with poor differentiation, histopathologic grade, and advanced clinical grade pancreatic adenocarcinomas in patients [10,11]. Recent studies also suggest that CXCR2 is expressed in various PDAC cell lines [12–15] and is primarily involved in enhancing the proliferation and survival of cancer cells through the autocrine and/or paracrine effect [11,12,15]. More importantly, increased expression of CXCR2 and its ligands has been shown in higher grades and stages of pancreatic adenocarcinomas in patients [10,16], indicating that CXCR2 is involved in the exacerbation of tumors and could be a promising target for developing selective and effective treatments for pancreatic cancer. As a G protein-coupled receptor (GPCR), CXCR2 couples to the pertussis toxin-sensitive Gi proteins to stimulate phosphatidylinositide-specific phospholipase C (PLC) activities [17]. Agonist-induced activation of PLC-β, one of the six families of PLC isozymes, catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate, generating 1,2-diacylglycerol and inositol 1,4,5-trisphosphate, which activates protein kinase C (PKC) isoforms and triggers the release of Ca2+ from internal sources, respectively.
PSD-95/DlgA/ZO-1 (PDZ) domains are ubiquitous protein-protein recognition modules that form peptide-binding pockets and generally mediate physical interaction with the carboxyl termini of a wide variety of proteins (such as membrane receptors, ion channel, and so on) that terminate in consensus binding motifs (referred to as PDZ motif) [18,19]. A variety of PDZ domain-containing proteins (also referred to as PDZ scaffold/adaptor proteins) have been reported to nucleate the formation of compartmentalized multiprotein complexes that are critical for efficient and specific cell signaling [20–24]. Some PDZ scaffold proteins, such as Na+/H+ exchange regulatory factor 1 (NHERF1) and NHERF2 and PDZ domain containing 1 (PDZK1), preferentially associate with the surface membranes of epithelial cells and interact with membrane receptors and their downstream effectors. PLC-β is one of the downstream effectors for GPCR signaling, and it has been reported to specifically bind with certain PDZ scaffold proteins through PDZ-based interaction since all PLC-β isoforms possess consensus PDZ motifs, -X-S/T-X-L/V-COOH (X represents any amino acid), at their carboxyl termini [25–28]. Therefore, the specificity of agonist-induced PLC-β activation and subsequent intracellular signaling might be dependent on the specific interactions of PLC-β with particular PDZ scaffold proteins [29]. Similar to PLC-β isoforms, CXCR2 also possesses a consensus PDZ motif (-S-T-T-L-COOH) at its carboxyl termini. Previous studies by us and others have demonstrated that the PDZ motif of CXCR2 is involved in the regulation of intracellular signaling and cell functions in neutrophils [30] as well as post-endocytic sorting and cellular chemotaxis in CXCR2-overexpressing human embryonic kidney 293 (HEK293) cells [31]. Hence, the PDZ motif of CXCR2 can, theoretically, mediate potential interaction with certain PDZ scaffold proteins, which subsequently binds relevant downstream effectors, forming multiprotein macro-molecular complexes. However, the molecular mechanism(s) as to how this potential CXCR2 macromolecular complex are formed and/or regulated, as well as what role the CXCR2 complex might play in PDAC growth and progression, have not been determined.
In our present work, we applied a series of biochemical assays and cell functional studies, as well as human PDAC xenograft animal model, to explore the molecular mechanisms and functional significance of the PDZ-based CXCR2 macromolecular complex in pancreatic tumor growth. Our data show that PDZ scaffold protein NHERF1 clusters CXCR2 and its downstream effector enzyme PLC-β3 into a macromolecular complex in PDAC cells. Moreover, we demonstrated that disrupting the CXCR2-NHERF1-PLC-β3 macromolecular complex significantly inhibited malignant progression of pancreatic tumor in vitro and in vivo. Our results suggest that targeting CXCR2 macromolecular complex may be a novel and effective therapeutic strategy for pancreatic cancer.
Materials and Methods
Antibodies and Reagents
Anti-human CXCR2, NHERF1, PLC-β3, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant human chemokines, CXCL8/IL-8, CXCL5/ENA-78, and CXCL1/GROα, were obtained from ProSpec (East Brunswick, NJ). 3-(4,5-Dimethylthiazol2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (St Louis, MO). Growth factor-reduced Matrigel matrix, glutathione agarose beads, and Transwell inserts were purchased from BD Biosciences (San Jose, CA). Human CXCR2 C-tail peptides (biotin-conjugate at N terminus), wild-type “WT” (biotin-FVGSSSGHTSTTL), PDZ motif deletion mutant “ΔTTL” (biotin-FVGSSSGHTS), and PDZ motif mutant “AAA” (biotin-FVGSSSGHTSAAA), were synthesized by Genemed Synthesis, Inc (San Antonio, TX) and used as reported before [30]. Chariot peptide/protein delivery reagent was purchased from Active Motif (Carlsbad, CA). S-protein agarose and streptavidin beads were purchased from Novagen/EMD Millipore (Billerica, MA). The plasmid pcDNA3.1(+)-FLAG-PLC-β3 used for construct generation was described before [32].
Plasmid Construction, Mutagenesis, and Protein Purification
C-terminal tail fragments of human CXCR2 (last 45 amino acids; i.e., amino acids 316–360) or human PLC-β3 (last 100 amino acids; i.e., amino acids 1135–1234) were generated by polymerase chain reaction cloning into pTriEx4 (encoding a His-S double tags at N terminus) or pET30 (His-S tag; Novagen). Various C-tail PDZ motif mutants (PDZ motif deletion or mutation) for either CXCR2 or PLC-β3 were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) and also cloned into pTriEx4 or pET30 vectors for plasmid construction and protein purification. Glutathione S-transferase (GST) PDZ fusion proteins (GST-NHERF1, NHERF2, or PDZK1) were purified by using glutathione agarose beads (BD Biosciences) and eluted with 50 mM glutathione. His-S-tagged fusion proteins for CXCR2 C-tail or PLC-β3 C-tail fragments (wild-type and PDZ mutants) were purified using Cobalt resin (Thermo Scientific, Hennigsdorf, Germany) and eluted with 200 mM imidazole. These affinity-purified fusion proteins (full length and/or C-terminal tail fragments) were used in the subsequent biochemical assays (such as pull-down, pairwise binding, and macromolecular complex assembly).
Cell Culture and Transfection
Human PDAC cell lines (PANC-1, MIA PaCa-2, and HPAC) were obtained from American Type Culture Collection (Manassas, VA). Normal human pancreatic duct epithelial (HPDE) cells, Colo357 and L3.6pl cells were obtained from Dr Paul J Chiao at the University of Texas MD Anderson Cancer Center (Houston, TX). PDAC cells were cultured in Dulbecco's modified Eagle's medium (Thermo Scientific Hyclone) containing 4.5 g/l d-glucose and l-glutamine supplemented with 10% FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin at 37°C in humidified air with 5% CO2. HPDE cells were cultured in keratinocyte serum-free medium (Invitrogen/Life Technologies, Carlsbad, CA) supplied with 5 ng/ml epidermal growth factor and 50 µg/ml bovine pituitary extract. In some experiments, cells were cultured or incubated in serum-free or antibiotic-free media wherever indicated. The PDAC cells were transfected with plasmids encoding CXCR2 C-tail fragments (WT, PDZ motif deletion ΔTTL, or PDZ motif mutation AAA) using Lipofectamine 2000 (Invitrogen/ Life Technologies) according to the manufacturer's instructions. After transfection, the PDAC cells were used for proliferation assays and invasion assays.
Western Blot Analysis
Cells were lysed in lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS)] supplemented with a mixture of protease inhibitors (containing 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, 1 µg/ml pepstatin, and 1 µg/ml leupeptin). Protein concentration of the cleared supernatant (17,000g, 15 minutes) was estimated by Bradford protein assay (Bio-Rad Laboratories, Redmond, WA). Proteins were eluted in Laemmli sample buffer containing β-mercaptoethanol, separated by SDS-polyacrylamide gel electrophoresis (PAGE; 7.5% or 4–15%), and immunoblotted using indicated antibodies. The blots were visualized and recorded using a BioSpectrum 500 Imaging System (UVP, Upland, CA).
Pull-down Assay
GST pull-down assay was performed as previously described [30]. Briefly, fresh PDAC cells were lysed in binding buffer [phosphatebuffered saline (PBS) + 0.2% Triton X-100, supplemented with a mixture of protease inhibitors], and the cleared supernatant (17,000g, 15 minutes) was equally mixed with GST alone or various GST-PDZ fusion proteins (GST-NHERF1, GST-NHERF2, or GST-PDZK1) at 4°C for 2 hours. The mixture was pulled down by glutathione agarose beads at 4°C overnight, washed three times with binding buffer, and eluted in Laemmli sample buffer containing β-mercaptoethanol. The eluents were separated by SDS-PAGE and immunoblotted with anti-CXCR2 or anti-PLC-β3 antibodies.
Pairwise Binding Assay
Purified GST-NHERF1 was mixed with various purified His-S- PLC-β3 C-tail fragments (WT or PDZ motif mutants ΔTQL and AAA) or CXCR2 C-tail peptides (biotin-conjugate at N terminus; WT or PDZ motif mutants ΔTTL and AAA) in binding buffer (PBS + 0.2% Triton X-100 + protease inhibitors) at 22 to 24°C for 1 hour. The mixtures were incubated with S-protein agarose (for His-S-tagged fusion proteins) or streptavidin beads (for biotin-conjugated peptides) for 2 hours. The beads were washed three times with binding buffer and eluted with Laemmli sample buffer containing β-mercaptoethanol. The eluents were resolved by SDS-PAGE and immunoblotted with anti-NHERF1 antibody.
Macromolecular Complex Assembly
Purified His-S-tagged CXCR2 C-tail fragments (WT and PDZ motif mutants ΔTTL or AAA) or His-S-tagged PLC-β3 C-tail fragments (WT and PDZ motif mutants ΔTQL or AAA) were mixed with GST-NHERF1 (or GST alone) in 200 µl of binding buffer (PBS + 0.2% Triton X-100 + protease inhibitors), and the complex was pulled down with S-protein agarose. This step is also referred to as pairwise binding as described above. The dimeric complex was then mixed with the lysates of PDAC cells expressing endogenous full-length PLC-β3 and CXCR2 for 3 hours at 4°C and washed extensively with binding buffer. The bound proteins were then eluted and immunoblotted using anti-PLC-β3 or anti-CXCR2 antibodies.
Co-immunoprecipitation
A Co-immunoprecipitation Kit (Thermo Scientific/Pierce) was used to immobilize the normal IgG control and anti-CXCR2 IgG to the resin according to the manufacturer's instruction. PDAC cells were lysed in binding buffer (PBS + 0.2% Triton X-100 + protease inhibitors), and cleared cell lysates (17,000g, 15 minutes) were processed for co-immunoprecipitation (co-IP) as reported before [22,30]. The co-precipitated protein complex was resolved by SDS-PAGE and probed for NHERF1 and PLC-β3. For the reverse co-IP, anti-PLC-β3 IgG was used to immunoprecipitate the complex. The co-precipitated protein complex was separated by SDS-PAGE and probed for NHERF1 and CXCR2. The signal was detected by SuperSignal West Pico (or Femto) substrate (Thermo Scientific).
Cell Proliferation Assay
Cell proliferation was assessed by MTT assay as reported before [11]. In brief, PDAC cells were seeded in 96-well plates (7 x 103 cells per well) and allowed to adhere overnight. Then, the cells were fed with serum-free fresh media with or without 100 ng/ml CXCR2 ligands (CXCL1, CXCL5, or CXCL8). After indicated growth periods, cells were incubated with 20 µl of MTT solution (1 mg/ml) at 37°C for 3.5 hours and then incubated with MTT solvent (4 mM HCl, 0.1% NP-40 in isopropanol) under constant mixing protected from light for 15 minutes at 22 to 24°C. Spectrophotometric absorbance of the samples at 590 nm was determined by a microplate reader (Bio-Rad). In parallel, Colo357 and HPAC cells were transfected with various pTriEx4 plasmids (vector alone, CXCR2 C-tail ΔTTL, or CXCR2 C-tail WT) or delivered with CXCR2 C-tail peptides (WT or ΔTTL) for 24 to 48 hours before the MTT assay.
Invasion Assay
PDAC cells were plated at a density of 1.5 x 105 cells per well in 24-well plate. Cells were transfected with various pTriEx4 plasmids (vector alone, CXCR2 C-tail ΔTTL, or CXCR2 C-tail WT). After a 48-hour incubation, in vitro invasion assay was performed using Transwell inserts with 8.0-µm pore size. Briefly, 1 x 105 transfected PDAC cells were suspended in serum-free medium and seeded onto the Transwell inserts pre-coated with diluted (1:3) Matrigel. The Transwell inserts were then placed into 24-well plates filled with the same medium containing 100 ng/ml CXCL8. After a 16-hour incubation, the upper surface of the Transwell inserts was wiped with a cotton swab and the invaded cells were fixed and stained with Diff-Quick stain (IMEB, San Marcos, CA). The number of invading cells was counted under an inverted microscope (x50) in three randomly selected fields per well. In separate experiments, PDAC cells were delivered with CXCR2 C-tail peptides (WT or ΔTTL) for 24 hours, and the cell invasion was assessed as described above.
Human PDAC Xenografts in Immunodeficient Mice
Three green fluorescent protein (GFP)-tagged adeno-associated virus type 2 (AAV2) constructs, AAV2/2CMV-GFP, AAV2/2CMV-GFP-CXCR2 C-tail ΔTTL, and AAV2/2CMV-GFP-CXCR2 C-tail WT (customized by the Gene Transfer Vector Core, University of Iowa), were used to transduce HPAC cells. CB17 severe combined immunodeficient (CB17-SCID) mice (female, 4–6 weeks old) were randomly divided into four groups (n=10–12), and each mouse received 200 µl of serum-free Dulbecco's modified Eagle's medium containing 3 x 106 HPAC cells (transduced or non-transduced) subcutaneously in the unilateral flank area. The mice were subjected to measurement of subcutaneous tumors every other day and monitored for changes in body weight and other side effects. Tumor volume was calculated by the formula (L x W2)/2, where L and W are the tumor length and width (in mm), respectively. To avoid severe discomfort in the control group, animals were killed after 4 weeks. Tumor tissues were harvested for histologic analysis and immunohistochemical staining. Tumor volume in SCID mice was plotted against time, and the final tumor weights were measured after the mice were killed. All the animal studies were accomplished under the protocol approved by Wayne State University Institutional Animal Care and Use Committee.
Ki-67 Proliferation Index
Tumor tissue from the xenografts was fixed by paraformaldehyde and embedded within paraffin. Paraformaldehyde-fixed and paraffin-embedded sections (5 µm) were stained with Ki-67 antibody (Ventana Medical Systems, Tucson, AZ) as reported [33]. Results were expressed as percentage of Ki-67-positive cells per 200x magnification. A total of 10 sections from each experimental group were examined by Zeiss Axiophot epifluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) and analyzed by Image-Pro Plus 6.0 software (Media Cybernetics, Berkshire, United Kingdom).
Statistical Analysis
Data are presented as means ± SEM of at least three independent experiments. Statistical significance of differences was assessed with the Student's t test. A value of P < .05 was considered statistically significant.
Results
CXCR2 Is Overexpressed in PDAC Cells
The expression of CXCR2 in normal HPDE cells and several PDAC cell lines (HPAC, Colo357, L3.6pl, PANC-1, and MIA PaCa-2) was examined and compared by Western blot analysis. All the five PDAC cell lines tested in our study showed significantly increased CXCR2 expression compared to HPDE cells (Figure 1), which is in agreement with the previous clinical study that reported an up-regulation of interleukin-8 (IL-8)/CXCL8 and its receptors in both pancreatic adenocarcinomas and neuroendocrine tumors [10].
Figure 1.
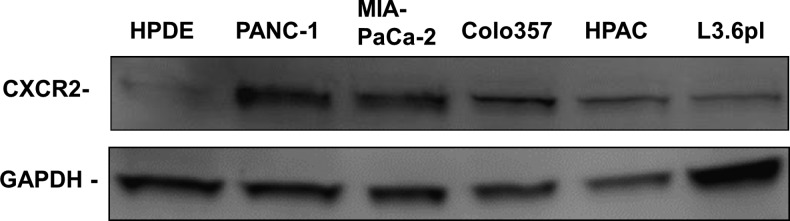
CXCR2 is overexpressed in human pancreatic cancer cells. Expression levels of CXCR2 in normal HPDE cells and several PDAC cell lines (HPAC, Colo357, L3.6pl, PANC-1, and MIA PaCa-2) were examined by Western blot analysis. Blot analysis of GAPDH was used as internal control.
CXCR2 and PLC-β3 in PDAC Cells Preferentially Interact with NHERF1
In a recent study, we demonstrated that the consensus PDZ motif at the carboxyl terminus of CXCR2 mediates PDZ-based interactions with certain PDZ scaffold proteins (such as NHERF1 and NHERF2) in neutrophils [30]. To investigate if endogenous CXCR2 in PDAC cells binds to any PDZ scaffold proteins, we performed a pull-down assay [30] as described in Materials and Methods section. As shown in Figure 2A, we observed interactions between CXCR2 and the membrane-associated PDZ proteins NHERF1 and NHERF2 in Colo357, L3.6pl, and HPAC cells, among which NHERF1 has a higher binding affinity for CXCR2 as compared with NHERF2. However, neither GST nor PDZK1 was found to bind to endogenous CXCR2 in these PDAC cell lines (Figure 2A).
Figure 2.

CXCR2 and PLC-β3 in PDAC cells preferentially interact with NHERF1. (A) Endogenous CXCR2 (from indicated PDAC cells) was pulled down by PDZ scaffold protein (NHERF1, NHERF2, or PDZK1). The membrane was blotted with anti-CXCR2 antibody. (B) Endogenous PLC-β3 (from indicated PDAC cells) was pulled down by PDZ scaffold protein (NHERF1, NHERF2, or PDZK1). The membrane was blotted with anti-PLC-β3 antibody. (C) Endogenous PLC-β3 was pulled down by NHERF1 in a dose-dependent manner. (D) Pairwise binding between GST-NHERF1 and His-S- tagged PLC-β3 C-tails (containing the last 100 amino acids) WT, PDZ motif deletion (ΔTQL), or PDZ motif mutant (AAA). The complex was pulled down by S-protein agarose and immunoblotted with anti-NHERF1 antibody.
CXCR2 couples to the pertussis toxin-sensitive Gi to stimulate phosphatidylinositide-specific PLC activities [17]. Similar to CXCR2, all human PLC-β isoforms possess consensus class I PDZ motifs at their carboxyl termini [29]. Our previous study has demonstrated that PLC-β3 (containing a PDZ motif -TQL-COOH) overexpressed in HEK293 cells interact with NHERF1 and NHERF2 [30]. Here, we explored the potential interactions between PDZ scaffold proteins (NHERF1, NHERF2, and PDZK1) and endogenous PLC-β3in PDAC cell lines. By similar GST pull-down experiments, we observed that endogenous PLC-β3 in PDAC cells bind to both NHERF1 and NHERF2; however, it did not bind to PDZK1 or to GST alone (Figure 2B). Moreover, in comparison to NHERF2, NHERF1 appears to interact with PLC-β3 with a higher affinity in most of the PDAC cell lines we tested (Figure 2B). In addition, the binding between endogenous PLC-β3 in PDAC cells and NHERF1 increased with increasing amounts of NHERF1 in a dose-dependent manner (Figure 2C).
PLC-β3 Interacts with NHERF1 in a Direct and PDZ Motif-Dependent Manner
The data from the GST pull-down studies (Figure 2B) does not reveal that whether the interaction between PLC-β3 and NHERF1 is direct, since there exists a possibility that PLC-β3 might bind NHERF1 through other intermediary proteins in the cell lysates. We performed a pairwise binding assay [30] that detects a direct interaction between PLC-β3and NHERF1 in vitro. We observed that PLC-β3 C-tail (containing the PDZ motif) directly binds to NHERF1 in a PDZ motif-dependent manner, as the interaction between NHERF1 and PLC-β3 C-tail lacking the PDZ motif (ΔTQL) or PLC-β3 C-tail with PDZ motif mutation (AAA) was almost completely abolished compared to wild-type (WT) PLC-β3C-tail (Figure 2D).
NHERF1 Clusters CXCR2 and PLC-β3 into a Macromolecular Complex In Vitro and in PDAC Cells
Results from the above GST pull-down experiments demonstrated that both endogenous CXCR2 and PLC-β3 in PDAC cells preferentially interact with NHERF1 (Figure 2, A and B), and PLC-β3 binds to NHERF1 in a direct and PDZ motif-dependent manner (Figure 2D), similar to the interaction between CXCR2 and NHERF1 as we previously reported [30]. Hence, we hypothesized that NHERF1 might nucleate CXCR2 and PLC-β3 forming a macromolecular complex in a PDZ motif-dependent manner and this complex might be critical for efficient and specific signaling mediated by CXC chemokine/CXCR2 biologic axis in PDAC cells. Toward this end, we sought to determine if we could detect a macromolecular complex containing CXCR2, NHERF1, and PLC-β3 in vitro. Using an in vitro macromolecular complex assembly assay (Figure 3A) [30], we observed the existence of a complex composed of CXCR2 C-tail, NHERF1, and endogenous PLC-β3 in PDAC cells (Figure 3B). Furthermore, we demonstrated that CXCR2 C-tail did not bind to PLC-β3 directly, and the macro-molecular complex was not formed by CXCR2 C-tail PDZ motif mutants (ΔTTL or AAA), indicating that the formation of the CXCR2 macromolecular complex is PDZ motif-dependent (Figure 3B). In addition, we detected a similar complex consisting of PLC-β3 C-tail (containing the PDZ motif TQL), NHERF1, and endogenous CXCR2 from PDAC cells (data not shown).
Figure 3.

NHERF1 clusters CXCR2 and PLC-β3 into a macromolecular complex in vitro and in PDAC cells. (A) Pictorial representation of in vitro macromolecular complex assembly (refer to Materials and Methods section for details). (B) In vitro macromolecular complex assembly of CXCR2 C-tails (WT, ΔTTL, and AAA), GST-NHERF1, and endogenous PLC-β3 (from Colo357 and HPAC cells). (C) Endogenous PLC-β3 and NHERF1 were co-immunoprecipitated with CXCR2 from Colo357 and HPAC cells. (D) Endogenous CXCR2 and NHERF1 were co-precipitated with PLC-β3 from Colo357 and HPAC cells.
Results from the above demonstrated that a CXCR2 macromolecular complex exists in vitro; however, it did not provide the evidence whether this complex exists in native membranes of PDAC cells that endogenously express all the relevant interacting proteins. To address this issue, co-IP was performed using either anti-CXCR2 or anti- PLC-β3 antibodies as described in Materials and Methods section. We observed that NHERF1 and PLC-β3 in PDAC cells were coimmunoprecipitated with CXCR2 (Figure 3C). Similarly, CXCR2 and NHERF1 were also co-precipitated with PLC-β3 from PDAC cells (Figure 3D), indicating that there is likely to be a macromolecular complex composed of endogenous CXCR2, NHERF1, and PLC-β3 on the surface membranes of PDAC cells.
CXC Chemokine/CXCR2 Biologic Axis Promotes PDAC Cell Proliferation
To examine the effect of CXCR2 signaling on PDAC cell proliferation, PDAC cells were treated with CXCR2 ligands followed by the MTT cell proliferation assay. As illustrated in Figure 4A, HPAC cells showed significantly elevated cell proliferative activities in response to both CXCL8 and CXCL1 (P < .05), and CXCL5 also promoted HPAC proliferation, though without statistical significance (P = .07). Proliferation of Colo357 cells was significantly augmented by CXCL5 (P < .05) and also by CXCL8 and CXCL1 though without statistical significance (P = .09 and .2, respectively; Figure 4B). It has been reported that Colo357 cells showed a significant high level of CXCL5 secretion [34], underpinning the autocrine effects of CXCL5 on Colo357 cell proliferation. However, the normal pancreatic duct HPDE cells did not demonstrate significantly increased growth stimulated by the treatment of either of these CXCR2 ligands (CXCL1, CXCL5, or CXCL8; Figure 4C).
Figure 4.

Disrupting the CXCR2 macromolecular complex inhibits PDAC cell proliferation. (A) HPAC, (B) Colo357, and (C) HPDE cell proliferation in response of CXCL1/CXCL5/CXCL8 (100 ng/ml) was assessed and quantified by MTT assay and expressed as relative to the initial time point (0 hour). Cell proliferation in response to indicated chemokines (CXCL5 or CXCL8; 100 ng/ml) in Colo357 and HPAC cells, which were transfected with plasmids expressing CXCR2 C-tail WT or ΔTTL (D, E) or pre-delivered with CXCR2 C-tail-specific peptide WT or ΔTTL (F, G). (H) Pairwise binding between GST-NHERF1 and various biotin-conjugated CXCR2 C-tail-specific peptide (containing last 13 amino acids) WT, ΔTTL, or AAA. The complex was pulled down by streptavidin agarose and immunoblotted with anti-NHERF1 antibody. Columns/dots, means of quintuplicates; bars, SEM; *P < .05. NT, cells not transfected or pre-delivered with peptides.
Disrupting the CXCR2 Macromolecular Complex Inhibits PDAC Cell Proliferation
Since the CXC chemokine/CXCR2 biologic axis plays an important role in PDAC cell proliferation (growth; Figure 4, A and B), and here we also demonstrated that the PDZ motif of CXCR2 is essential for the physical coupling of CXCR2 to PLC-β3 mediated by NHERF1 into a macromolecular signaling complex (Figure 3B), therefore, it is possible that perturbation of the CXCR2 macromolecular complex might affect CXCR2 ligand-induced PDAC cell proliferation. In a recent study, we used a CXCR2 C-tail peptide (containing the PDZ motif ) to disrupt the CXCR2 PDZ motif-mediated interaction with NHERF1 and observed a functional inhibition of CXCR2 ligand- induced cell migration in neutrophils [30]. We went on to evaluate the functional significance of this CXCR2 macromolecular complex in the CXCR2 ligand-induced PDAC cell proliferation. We transfected HPAC and Colo357 cells with plasmids encoding CXCR2 C-tail (WT or PDZ deletion ΔTTL) and evaluated cell proliferative activities. As shown in Figure 4D, in response to CXCL5, Colo357 transfected with plasmid containing CXCR2 C-tail WT showed significantly reduced proliferative activities compared to cells transfected with the vector alone or CXCR2 C-tail ΔTTL, suggesting that disrupting CXCR2 macromolecular complex inhibits CXCR2 ligand-induced Colo357 growth and PDZ motif of CXCR2 is important for PDAC cell proliferation. HPAC transfected with CXCR2 C-tail WT showed significantly decreased cell proliferation in response to CXCL8 (Figure 4E). In addition, we delivered the CXCR2 C-tail peptides (WT and ΔTTL) used in our previous studies [30] to Colo357 and HPAC cells, and we also observed significantly reduced cell proliferation stimulated by CXCL5 (Figure 4F) and CXCL8 (Figure 4G). Furthermore, using the pairwise binding assay, we also demonstrated that the CXCR2 C-tail peptide interacts with NHERF1 in a direct and PDZ motif-dependent manner, because the CXCR2 WT peptide interacts with NHERF1, while the PDZ deletion peptide (ΔTTL) or mutation peptide (AAA) failed to bind to NHERF1 (Figure 4H).
Disrupting the CXCR2 Macromolecular Complex Blocks PDAC Cell Invasion
We then examined the effect of disrupting CXCR2 signaling complex on the invasive capability of PDAC cells. PDAC cells were transfected with plasmids encoding CXCR2 C-tail (WT or ΔTTL) and used to evaluate the invasive potency by an in vitro invasion assay as reported before [34]. As illustrated in Figure 5, gene delivery of CXCR2 C-tail WT sequence, but not the ΔTTL PDZ deletion sequence, significantly inhibited invasion of HPAC cells through Matrigel induced by CXCL8, implicating that the PDZ motif of CXCR2 is important for PDAC cell invasion.
Figure 5.

Gene delivery of CXCR2 C-tail sequence significantly inhibits malignant invasion of pancreatic cancer cell. HPAC cells were transfected with pTriEx4 vector alone, pTriEx4 CXCR2 C-tail PDZ motif deletion (ΔTTL), or pTriEx4 CXCR2 C-tail WT. Invasion through the Transwells pre-coated with Matrigel of transfected HPAC cells was initiated by CXCL8 (100 ng/ml) and quantified by microscopy. Columns, means of triplicates; bars, SEM; **P < .01.
Disrupting the CXCR2 Macromolecular Complex Inhibits Pancreatic Tumor Growth In Vivo
To analyze the functional significance of the CXCR2 macromolecular complex in PDAC growth in vivo, a subcutaneous xenograft induced by HPAC cells in CB17-SCID mice was developed to determine whether disrupting the CXCR2 signaling complex could lead to inhibition of tumor growth in vivo. HPAC cells that were transduced with AAV2 viruses expressing GFP-human CXCR2 C-tails (WT or ΔTTL) or GFP alone, or non-transduced cells, were injected subcutaneously into CB17-SCID mice. Tumor volume was measured every other day starting at day 12. At day 28, all mice were sacrificed, and tumors were excised and final tumor weights were determined. We found that HPAC cells expressing GFP alone or GFP-CXCR2 C-tail ΔTTL grew into sizable tumors underneath the skin that were comparable to non-transduced cells (Figure 6A). However, HPAC cells expressing GFP-CXCR2 C-tail WT grew into significantly smaller tumors compared with the cancer cells expressing GFP vector alone or GFP-CXCR2 C-tail ΔTTL or non-transduced cells (Figure 6A). Furthermore, transduction with GFP-CXCR2 C-tail WT significantly reduced final tumor weight compared to the three other groups (Figure 6B). This result suggests an inhibitory effect of exogenous CXCR2 C-tail (containing the PDZ motif) on human pancreatic tumor development and progression in vivo. We then demonstrated that the proliferation index, determined by Ki-67 expression, in the group of mice injected with HPAC cells expressing GFP-CXCR2 C-tail WT was significantly lower (P < .01) compared to the three other groups (non-transduction group, AAV2-GFP group, and AAV2-GFP- CXCR2 C-tail ΔTTL group; Figure 6, C and D). Collectively, our date indicates the potential therapeutic effect of disrupting the CXCR2 macromolecular complex on the primary tumor of PDAC in vivo.
Figure 6.

Gene delivery of CXCR2 C-tail sequence inhibits PDAC tumor growth in vivo. Tumor volume (A) was measured according to the formula (1/2 x L x W2) every other day; and final tumor weights (B) were measured, averaged, and compared after 4 weeks. Proliferation index (D) was expressed as percentages of Ki-67+ cells (see details in Materials and Methods section); and representative Ki-67+ immunohistochemical images (200x) are shown in (C). *P < .05; **P < .01. NT, non-transduced cells.
Discussion
CXC chemokines (such as GRO-α/CXCL1, IL-8/CXCL8, ENA-78/ CXCL5) and their cognate receptor CXCR2 have been reported to play a critical role in tumor growth and angiogenesis, as blockade of the CXC chemokines/CXCR2 biologic axis reduced tumorigenesis and angiogenesis in many human cancers including pancreatic cancer [16,34–36]. However, most of the interventional approaches have been conducted by systemic blockade or depletion of CXCR2 and/ or its ligands, which might cause global undesired effects on other vital functions, as CXCR2 had also been reported in many cellular functions, such as in preservation of oligodendrocyte function and myelinization of neural tissues [37]. This issue warrants the necessity of a more comprehensive understanding of the molecular mechanisms of CXCR2 and its signaling, on the basis of which, selective and cell-specific therapeutic targets could be identified.
CXCR2 possesses a consensus PDZ motif at their carboxyl termini, and the PDZ motif has been reported to modulate cellular chemotaxis [31]. Recently, we demonstrated that the PDZ motif of CXCR2 plays an important role in regulating neutrophil functions as disrupting the interaction mediated by PDZ motif through using an exogenous peptide mimic (mapping CXCR2 PDZ motif) significantly inhibited CXCR2-mediated calcium mobilization and neutrophilic trans-epithelial migration [30]. In the present study, we identified the PDZ scaffold protein NHERF1 as an interacting partner for CXCR2 in PDAC cells, and we also demonstrated the existence of a PDZ-based CXCR2 macromolecular signaling complex containing endogenous CXCR2, NHERF1, and PLC-β3 in PDAC cells. Furthermore, we provided functional evidence showing that disrupting the CXCR2 complex significantly inhibited the malignant cellular functions (i.e., proliferation and invasion) in vitro and pancreatic tumor growth in vivo.
Controversy exists regarding the expression of CXCR2 in human pancreatic cancer cell lines. Despite some studies reported that CXCR2 was not detected in some human PDAC cell lines (such as PANC-1, MIA PaCa-2, AsPC-1, BxPC-3, and HPAF-II) [33,34,38], other groups reported the detection of CXCR2 and/or the autocrine effect of CXCR2 ligands in various human PDAC cells (such as PANC-1, MIA PaCa-2, Capan-1, Capan-2, SUIT-2, HuP-T4, Bx-PC-3, and Panc03.27) [11–15,39] and pancreatic tumors specimens from patients [10,16]. In the present study, we also detected the expression of CXCR2 in PANC-1, MIA PaCa-2, HPAC, Colo357, and L3.6pl cells in Western blot analysis (Figure 1) by using the same antibody Frick et al. used to detect the CXCR2 expression in patient specimens [16]. Reverse transcription-polymerase chain reaction results from our study also confirmed the expression of CXCR2 in these cell lines (data not shown). One possible reason for the failure of some groups to detect CXCR2 in PDAC cell lines might be that expression level of CXCR2 varies in different conditions. Hussain et al. reported that the expression level of CXCR2 correlates with tumor grade and stage in pancreatic adenocarcinomas [10]. Yamamoto et al. also reported that CXCR2 is upregulated in an orthotopic colon cancer model compared to the subcutaneous model [40].
PLC-β is the major isozyme that has been well studied to participate in GPCR-mediated signaling and modulate the physiological responses, such as promoting cell growth in cancer [41–43]. Each PLC-β subtype has its distinct expression pattern and physiological relevance [41]. Among the four subtypes of PLC-β,PLC-β3is expressed in a wide range of cells and tissues [43] and exhibits the highest affinity to Gβγ subunits and subsequent activation by Gβγ subunits [44]. A growing body of evidence suggests that PDZ scaffold proteins are involved in the modulation of PLC-β isoforms in the PDZ motif-dependent manner. PLC-β3, with its PDZ motif (-STQL-COOH), was reported to bind to PDZ domains of NHERF2 and Shank2 through its PDZ motif at its carboxyl termini, in mouse small intestine [28] and in the postsynaptic density region in neuronal cells [25], respectively. It has also been shown that PLC-β3 was downregulated in jejuna villus cells in NHERF1-knockout mice [27]. Results from our present study revealed that PLC-β3 preferentially binds to NHERF1 in PDAC cells in a direct and PDZ motif-dependent pattern. Therefore, the specificity and diversity of agonist-induced PLC-β activity and its downstream signaling may be regulated by the specific interactions of PLC-β isoforms and certain PDZ scaffold proteins.
There is accumulating evidence suggesting that the formation of particular macromolecular signaling complexes beneath the plasma membrane enables the membrane receptors to transduce signals into cell interior and thereafter influence cell behavior with higher specificity and efficiency [20–23,30,45]. Our present study revealed a PDZ motif-dependent CXCR2 macromolecular signaling complex, in which CXCR2 and PLC-β3 were bridged by NHERF1, in the PDAC cells. We also demonstrated the functional importance of the CXCR2 complex in pancreatic cancer cell functions in vitro and in vivo, as disturbing the CXCR2 complex by using an exogenous CXCR2 C-tail sequence (containing the PDZ motif ) significantly attenuated malignant cell proliferation and cell invasion of PDAC cells and tumor growth in vivo. By investigating a network of protein complexes rather than CXCR2 alone in pancreatic cancer, our results reveal a novel molecular target for the development of specific therapeutic strategies and agents that could combat pancreatic cancer.
Footnotes
This work was supported in part by American Heart Association grant 0765185B (to C.L.), the Elsa U. Pardee Foundation research grant (to C.L.), and Wayne State University intramural startup fund and Cardiovascular Research Institute Isis Initiative award (to C.L.). The authors disclose no potential conflicts of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Huang ZQ, Saluja AK, Dudeja V, Vickers SM, Buchsbaum DJ. Molecular targeted approaches for treatment of pancreatic cancer. Curr Pharm Des. 2011;17:2221–2238. doi: 10.2174/138161211796957427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Li C. Tumor microenvironment and pancreatic cancer. J Mol Biol. 2012;1:e104. [Google Scholar]
- 4.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–R28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 5.Xie K, Wei D, Huang S. Transcriptional anti-angiogenesis therapy of human pancreatic cancer. Cytokine Growth Factor Rev. 2006;17:147–156. doi: 10.1016/j.cytogfr.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Xiong HQ, Abbruzzese JL, Lin E, Wang L, Zheng L, Xie K. NF-κB activity blockade impairs the angiogenic potential of human pancreatic cancer cells. Int J Cancer. 2004;108:181–188. doi: 10.1002/ijc.11562. [DOI] [PubMed] [Google Scholar]
- 7.Shi Q, Abbruzzese JL, Huang S, Fidler IJ, Xiong Q, Xie K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin Cancer Res. 1999;5:3711–3721. [PubMed] [Google Scholar]
- 8.Zhu VF, Yang J, Lebrun DG, Li M. Understanding the role of cytokines in glioblastoma multiforme pathogenesis. Cancer Lett. 2012;316:139–150. doi: 10.1016/j.canlet.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 10.Hussain F, Wang J, Ahmed R, Guest SK, Lam EWF, Stamp G, El-Bahrawy M. The expression of IL-8 and IL-8 receptors in pancreatic adenocarcinomas and pancreatic neuroendocrine tumours. Cytokine. 2010;49:134–140. doi: 10.1016/j.cyto.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Li AH, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, Li G, Lu XY, Strieter RM, Burdick M, et al. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am J Pathol. 2011;178:1340–1349. doi: 10.1016/j.ajpath.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takamori H, Oades ZG, Hoch OC, Burger M, Schraufstatter IU. Autocrine growth effect of IL-8 and GROalpha on a human pancreatic cancer cell line, Capan-1. Pancreas. 2000;21:52–56. doi: 10.1097/00006676-200007000-00051. [DOI] [PubMed] [Google Scholar]
- 13.Hidaka H, Ishiko T, Furuhashi T, Kamohara H, Suzuki S, Miyazaki M, Ikeda O, Mita S, Setoguchi T, Ogawa M. Curcumin inhibits interleukin 8 production and enhances interleukin 8 receptor expression on the cell surface: impact on human pancreatic carcinoma cell growth by autocrine regulation. Cancer. 2002;95:1206–1214. doi: 10.1002/cncr.10812. [DOI] [PubMed] [Google Scholar]
- 14.Kuwada Y, Sasaki T, Morinaka K, Kitadai Y, Mukaida N, Chayama K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int J Oncol. 2003;22:765–771. [PubMed] [Google Scholar]
- 15.Miyamoto M, Shimizu Y, Okada K, Kashii Y, Higuchi K, Watanabe A. Effect of interleukin-8 on production of tumor-associated substances and autocrine growth of human liver and pancreatic cancer cells. Cancer Immunol Immunother. 1998;47:47–57. doi: 10.1007/s002620050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frick VO, Rubie C, Wagner M, Graeber S, Grimm H, Kopp B, Rau BM, Schilling MK. Enhanced ENA-78 and IL-8 expression in patients with malignant pancreatic diseases. Pancreatology. 2008;8:488–497. doi: 10.1159/000151776. [DOI] [PubMed] [Google Scholar]
- 17.Wu D, Jiang H, Katz A, Simon MI. Identification of critical regions on phospholipase C-β1 required for activation by G-proteins. JBiol Chem. 1993;268:3704–3709. [PubMed] [Google Scholar]
- 18.Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CY, Naren AP. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr Biol (Camb) 2010;2:161–177. doi: 10.1039/b924455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naren AP, Cobb B, Li CY, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, et al. A macromolecular complex of β2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci USA. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li CY, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li CY, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, et al. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell. 2007;131:940–951. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Schuetz JD, Naren AP. Tobacco carcinogen NNK transporter MRP2 regulates CFTR function in lung epithelia: implications for lung cancer. Cancer Lett. 2010;292:246–253. doi: 10.1016/j.canlet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, Wang S, Li C. In vitro analysis of PDZ-dependent CFTR macromolecular signaling complexes. J Vis Exp. 2012;66:e4091. doi: 10.3791/4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang JI, Kim HS, Lee JR, Kim E, Ryu SH, Suh PG. The interaction of phospholipase C-β3 with Shank2 regulates mGluR-mediated calcium signal. J Biol Chem. 2005;280:12467–12473. doi: 10.1074/jbc.M410740200. [DOI] [PubMed] [Google Scholar]
- 26.Choi JW, Lim S, Oh YS, Kim EK, Kim SH, Kim YH, Heo K, Kim J, Kim JK, Yang YR, et al. Subtype-specific role of phospholipase C-β in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell Signal. 2010;22:1153–1161. doi: 10.1016/j.cellsig.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Donowitz M, Singh S, Singh P, Salahuddin FF, Chen Y, Chakraborty M, Murtazina R, Gucek M, Cole RN, Zachos NC, et al. Alterations in the proteome of the NHERF1 knockout mouse jejunal brush border membrane vesicles. Physiol Genomics. 2010;42A:200–210. doi: 10.1152/physiolgenomics.00001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang JI, Heo K, Shin KJ, Kim E, Yun CHC, Ryu SH, Shin HS, Suh PG. Regulation of phospholipase C-β3 activity by Na+/H+ exchanger regulatory factor 2. J Biol Chem. 2000;275:16632–16637. doi: 10.1074/jbc.M001410200. [DOI] [PubMed] [Google Scholar]
- 29.Suh PG, Hwang JI, Ryu SH, Donowitz M, Kim JH. Breakthroughs and views—the roles of PDZ-containing proteins in PLC-beta-mediated signaling. Biochem Biophys Res Commun. 2001;288:1–7. doi: 10.1006/bbrc.2001.5710. [DOI] [PubMed] [Google Scholar]
- 30.Wu Y, Wang S, Farooq SM, Castelvetere MP, Hou Y, Gao JL, Navarro JV, Oupicky D, Sun F, Li C. A chemokine receptor CXCR2 macro-molecular complex regulates neutrophil functions in inflammatory diseases. JBiol Chem. 2012;287:5744–5755. doi: 10.1074/jbc.M111.315762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baugher PJ, Richmond A. The carboxyl-terminal PDZ ligand motif of chemokine receptor CXCR2 modulates post-endocytic sorting and cellular chemotaxis. J Biol Chem. 2008;283:30868–30878. doi: 10.1074/jbc.M804054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Vogel WK, McCullar JS, Greenwood JA, Filtz TM. Phospholipase C-β3 and -β1 form homodimers, but not heterodimers, through catalytic and carboxyl-terminal domains. Mol Pharmacol. 2006;70:860–868. doi: 10.1124/mol.105.021923. [DOI] [PubMed] [Google Scholar]
- 33.Guha S, Eibl G, Kisfalvi K, Fan RS, Burdick M, Reber H, Hines OJ, Strieter R, Rozengurt E. Broad-spectrum G protein-coupled receptor antagonist, [D-Arg1,D-Trp5,7,9,Leu11]SP: a dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer Res. 2005;65:2738–2745. doi: 10.1158/0008-5472.CAN-04-3197. [DOI] [PubMed] [Google Scholar]
- 34.Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong ZM, Burdick MD, Yang ZJ, Strieter RM, Hoffman RM, et al. CXC-chemokine/ CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer. 2009;125:1027–1037. doi: 10.1002/ijc.24383. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, Takeyama H, Tong Z, Guha S. CXCL8/IL-8 and CXCL12/SDF-1α cooperatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer. 2009;124:853–861. doi: 10.1002/ijc.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wente MN, Keane MP, Burdick MD, Friess H, Buchler MW, Ceyhan GO, Reber HA, Strieter RM, Hines OJ. Blockade of the chemokine receptor CXCR2 inhibits pancreatic cancer cell-induced angiogenesis. Cancer Lett. 2006;241:221–227. doi: 10.1016/j.canlet.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 37.Hosking MP, Tirotta E, Ransohoff RM, Lane TE. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS One. 2010;5:e11340. doi: 10.1371/journal.pone.0011340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill KS, Gaziova I, Harrigal L, Guerra YA, Qiu SM, Sastry SK, Arumugam T, Logsdon CD, Elferink LA. Met receptor tyrosine kinase signaling induces secretion of the angiogenic chemokine interleukin-8/CXCL8 in pancreatic cancer. PloS One. 2012;7:e40420. doi: 10.1371/journal.pone.0040420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M, Zhang Y, Feurino LW, Wang H, Fisher WE, Brunicardi FC, Chen C, Yao Q. Interleukin-8 increases vascular endothelial growth factor and neuropilin expression and stimulates ERK activation in human pancreatic cancer. Cancer Sci. 2008;99:733–737. doi: 10.1111/j.1349-7006.2008.00740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto M, Kikuchi H, Ohta M, Kawabata T, Hiramatsu Y, Kondo K, Baba M, Kamiya K, Tanaka T, Kitagawa M, et al. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res. 2008;68:9754–9762. doi: 10.1158/0008-5472.CAN-08-1748. [DOI] [PubMed] [Google Scholar]
- 41.Kim JK, Lim S, Kim J, Kim S, Kim JH, Ryu SH, Suh PG. Subtype-specific roles of phospholipase C-β via differential interactions with PDZ domain proteins. Adv Enzyme Regul. 2011;51:138–151. doi: 10.1016/j.advenzreg.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Kim CG, Park D, Rhee SG. The role of carboxyl-terminal basic amino acids in Gqα-dependent, activation, particulate association, and nuclear localization of phospholipase C-β1. J Biol Chem. 1996;271:21187–21192. doi: 10.1074/jbc.271.35.21187. [DOI] [PubMed] [Google Scholar]
- 43.Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee CW, Lee KH, Lee SB, Park D, Rhee SG. Regulation of phospholipase C-beta 4 by ribonucleotides and the alpha subunit of Gq. J Biol Chem. 1994;269:25335–25338. [PubMed] [Google Scholar]
- 45.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]