

Abstract
For patients with solid tumors, the tolerance of surrounding tissues often limits the dose of radiation that can be delivered. Thus, agents that preferentially increase the cytotoxic effects of radiation toward tumor cells would significantly alter the therapeutic ratio and improve patient survival. Using a high-throughput, unbiased screening approach, we have identified 4′-bromo-3′-nitropropiophenone (NS-123) as a radiosensitizer of human glioma cells in vitro and in vivo. NS-123 radiosensitized U251 glioma cells in a dose-dependent and time-dependent manner, with dose enhancement ratios ranging from 1.3 to 2.0. HT-29 colorectal carcinoma and A549 lung adenocarcinoma cells were also radiosensitized by NS-123 in vitro, whereas NS-123 did not increase the radiation sensitivity of normal human astrocytes or developmental abnormalities or lethality of irradiated Zebrafish embryos. In a novel xenograft model of U251 cells implanted into Zebrafish embryos, NS-123 enhanced the tumor growth-inhibitory effects of ionizing radiation (IR) with no apparent effect on embryo development. Similar results were obtained using a mouse tumor xenograft model in which NS-123 sensitized U251 tumors to IR while exhibiting no overt toxicity. In vitro pretreatment with NS-123 resulted in accumulation of unrepaired IR-induced DNA strand breaks and prolonged phosphorylation of the surrogate markers of DNA damage H2AX, ataxia telangiectasia mutated protein, DNA-dependent protein kinase, and CHK2 after IR, suggesting that NS-123 inhibits a critical step in the DNA repair pathway. These results show the potential of this cell-based, high-throughput screening method to identify novel radiosensitizers and suggest that NS-123 and similar nitrophenol compounds may be effective in antiglioma modalities.
Introduction
The prognosis for patients with glioblastoma multiform has improved only slightly over the last few decades. Randomized trials have shown that radiotherapy improves outcomes (1) and results in a median survival of 12 months (2, 3). A recent prospective clinical trial found that radiotherapy combined with temozolomide significantly improved survival, albeit by a modest 2.5 months (4). Attempts have been made at increasing the radiation dose, either with additional external beam radiotherapy (5), brachytherapy (6), or stereotactic radiosurgery (7); no significant improvement in survival has been shown. Thus, an agent which preferentially enhances the cytotoxic effects of radiation on glioma cells, but has minimal effects on the survival of normal brain cells, has the potential to improve the therapeutic result for these patients (8).
The major cellular pathways leading to sensing and repairing radiation-induced damage have been extensively studied, and the mechanisms involved have become attractive targets for agents that have the potential to increase therapeutic gain when combined with ionizing radiation (IR). This approach has identified some promising candidates (9–11), which are currently being investigated at the preclinical stage. Another approach to identify tumor-specific radiosensitizers is to use a cell-based assay, in which the target may not be immediately known but a functional end point (e.g., killing transformed but not normal cells) is used to screen for and identify promising candidates. This approach has the added benefits of being able to screen a much larger pool of compounds that have been preselected to conform to desirable pharmacologic properties, as well as the opportunity to ultimately identify novel targets for radiosensitization.
We have used a stepwise, cell-based screening approach that entails a simple end point, cell survival, to identify a novel radiosensitizing compound for glioblastoma multiform. Using the U251 human glioma cell line, we initially investigated the radiosensitizing capability of 870 compounds within a commercially available chemical library (Nanosyn, Inc.). We present here the preclinical evaluation of a compound identified in this initial screen, 4′-bromo-3′-nitropropiophenone (NS-123). Our results show that NS-123 enhances the cytotoxic effects of radiobiologically relevant doses of IR in tumor cells without any measurable normal tissue toxicity in vitro or in vivo by a mechanism that involves inhibition of DNA repair pathways and primarily double-strand break(DSB) repair.
Materials and Methods
Chemicals
A library consisting of 10,000 compounds was obtained from Nanosyn. Eighty-seven percent of these compounds conform to four Lipinski criteria and 98% to three Lipinski criteria, suggesting that they have desirable pharmacologic properties (12). NS-123 (500 mg) was synthesized both via a contract from Exclusive Chemistry Ltd. and as a generous gift from Dr. Robert Mach (Washington University, St. Louis, MO). Chemical structure and purity of the synthesized product was confirmed by nuclear magnetic resonance and mass spectroscopy.
Cell culture
All cell lines (American Type Culture Collection) were incubated at 37 °C with 5% CO2. U251 glioma, HT-29 colorectal cancer, and A549 non–small cell lung cancer cells were grown in RPMI 1640, McCoy 5A, and Ham's/F12 medium, respectively, supplemented with 10% fetal bovine serum (FBS), L-glutamine, and penicillin/streptomycin. Nontransformed human astrocytes (NHA, Cambrex) were cultured according to the manufacturer's instructions in astrocyte basal medium (Cambrex) supplemented with ascorbic acid, recombinant human epidermal growth factor, GA-1000, insulin, L-glutamine, and 3% FBS. In all of the tissue culture experiments, NS-123 was readily dissolved in DMSO and was added to cells such that the maximum concentration of DMSO was 0.1% v/v.
Chemical library screen
Six hundred U251 cells were plated per well in 96-well plates. Cells in each well were then treated with a different compound from the Nanosyn library for 8 h at 2.5 μmol/L concentration. Treatment was done with paired 96-well plates; one plate was exposed to 4 Gy, and the other plate was an unirradiated control. One hour after irradiation, the medium was replaced with fresh medium alone. Four days later, the MTS assay was used to evaluate cell survival. Absorbance was measured at 490 nm with a microplate reader (Sprectramax, Molecular Devices). Radiosensitivity factors (RSF) were calculated by for each compound tested with the assay.
Survival assays
Clonogenic survival assays were done as described previously (13). U251, HT-29, or A549 cells were plated at different densities in triplicate, treated with NS-123 or DMSO (alone) and irradiated 4, 8, or 12 h later with a range of doses using 137Cs γ-rays at a dose rate of ∼4 Gy/min. One hour after irradiation, the medium was replaced with fresh medium without NS-123 or DMSO. The cells were fixed 7 to 14 days after IR and stained with crystal violet. Colonies greater than 50 cells were then counted and normalized against the nonirradiated controls for each treatment. Because normal human cells do not grow clonogenically, MTS assays were used to assess survival. In previous studies, we showed that MTS assays, when done on subconfluent cultures of tumor cells for >5 days after IR, yield results that are comparable with those of clonogenic survival assays (Naczki and Koumenis, unpublished results). NHA cells were plated in 24-well plates in quadruplicate at equal density, treated with equal volumes of NS-123 or DMSO (alone), and irradiated as described above. One hour after irradiation, the medium was replaced with fresh astrocyte basal medium. Five days later, the MTS assay was used to evaluate cell survival as above.
Immunoblotting
After treatments, whole-cell and nuclear extracts were prepared. Equal lysate volumes were loaded on 15% and 12% SDS-PAGE gels to assay for the histone variant H2AX (γ-H2AX) and phosphorylated CHK2 (P-CHK2) proteins, respectively. To assay for phosphorylated ataxia telangiectasia mutated (P-ATM) and phosphorylated DNA-dependent protein kinase catalytic subunit (P-DNA-PKcs) protein, 30 to 50 μg of nuclear protein were diluted with an equal volume of 2× SDS and resolved with 6% SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride membranes and then incubated with either anti–γ-H2AX antibody (1:5,000; Upstate), anti–P-CHK2 antibody (1:1,000; Cell Signaling), anti–P-ATM (pSer1981) antibody (1:300; Calbiomchem), or anti–P-DNA-PKcs (T2609) antibody (1:500; Abcam). To confirm equivalent loading and protein transfer, membranes were probed with mouse anti-human β-actin monoclonal antibody (Sigma) or anti–DNA-PKcs monoclonal antibody (25-4 at 1:5,000; NeoMarkers). All membranes were then incubated with horseradish peroxidase–conjugated anti-mouse or anti-rabbit secondary antibodies (Santa Cruz Biotechnology). Immunoreactive bands were detected using ECL Plus chemiluminescence (Amersham Biosciences).
DSB measurement using pulsed field gel electrophoresis and number average length analysis
Cells were treated with 30 μmol/L NS-123 or DMSO alone. Four hours later, the cells were either sham-irradiated or irradiated with 4 Gy of 137Cs γ-rays (Radiation Facility, Brody School of Medicine, ECU) at the dose rate of 0.57 Gy/min and placed immediately at 37°C in fresh medium without NS-123 and harvested at different repair times up to 24 h (14). No significant changes in the percentage of dead cells were detected by trypan blue exclusion as a result of irradiation. After mixing of cells into low melting point agarose (Bio-Rad) plugs (∼ 200,000 cells per plug; ref. 15), each plug was placed in 10 mL of TE buffer for 1 h, followed by treatment with 1 mg/mL proteinase K (Promega) for 2 h, and then placed at 37°C for lysis. Proteinase K solution was replaced every 24 h along with a daily 0.5% reduction in N-lauroylsarcosine concentration. Lysed cell plugs were washed in NTE buffer [150 mmol/L NaCl, 10 mmol/L Tris-HCl, 0.1 mmol/L EDTA (pH 8.0) supplemented with 40 ng/mL phenylmethylsulfonyl fluoride] and stored in fresh TE buffer. For the AscI (New England Biolabs) restriction enzyme treatment, plugs were washed using 1-mL AscI reaction buffer [20 mmol/L Tris-acetate, 10 mmol/L magnesium acetate, and 50 mmol/L potassium acetate (pH 7.9) ] for 1 h, replaced with fresh buffer every 20 min, and then incubated for 24 h at 4°C. Plugs were incubated for 1 h on ice and then moved to 37°C for 16 h. After incubation, AscI enzyme solution was removed and replaced with 1 mL of ice-cold native stop solution [70 mmol/L HEPES-KOH, 100 mmol/L KCl, and 100 mmol/L EDTA (pH 7.6) ]. Plugs were washed with TE buffer (six times, 1 h each), and then equilibrated into 0.5 × Tris-borate EDTA [TBE; 45 mmol/L Tris base, 45 mmol/L boric acid, and 1 mmol/L Na2-EDTA (pH 7.8) ]. Samples and molecular length standards were electrophoresed in a 0.85% neutral gel prepared in 0.25 × TBE in a Bio-Rad CHEF DR-II apparatus and electrophoresed using a dual pulsed-field gel electrophoresis (PFGE) pulsing regimen, optimized for separation of DNA fragments ranging from 5.7 Mbp to 9.4 kbp. Gels were stained with ethidium bromide (1 μg/mL) for 1 h and destained overnight, and an electronic image was obtained using a FluorChem 8800 imaging system (Alpha Innotech). Images were processed using QuantiScan (BioSoft), and a densitogram was obtained for each gel lane. A DNA dispersion curve, relating DNA length to electrophoretic mobility based on all length standards, was determined from an analytic mobility function as previously described using number average length analysis (15).
Zebrafish maintenance, embryo irradiation, and drug exposure
Zebrafish were raised in accordance with previously established protocols (16) at the University of Pennsylvania School of Medicine. Embryos were maintained at 30°C after transplantation procedures. The age of embryos is indicated as the hours postfertilization (hpf) and days postfertilization (dpf) for all experimental data shown. Embryos were either irradiated with 10 Gy or sham-irradiation, as previously described (16), immediately after photography at 24 hpf. Selected embryos were incubated with NS-123 at a concentration of 30 μmol/L for 14 h before either irradiation or sham-irradiation.
U251 xenograft Zebrafish model
U251 cells were stably transfected with a red fluorescent protein (RFP) construct (pDsRed2-C1; Clontech). Using a Nanoject II microinjector (Drummond Scientific), 1 to 100 cells were transplanted into each Zebrafish embryo still in the chorion, from the high stage of their development (∼ 3.5 hpf) to the oblong-sphere stage (∼ 4.5 hpf). The transplantation site was localized either to the blastodisc approximately halfway between the margin and the animal pole or the center of the embryonic yolk sac. Morphologic analysis was done as previously described (16). Transplanted embryos were examined under a 100 × PlanNeofluor objective mounted on a Nikon TE-200 epifluorescence microscope. Images of embryos bearing RFP-positive cells were captured with a Hammamatsu CCD camera controlled with IP LabSpectrum v2.0.1 software (Scanalytics, Inc.). Image analysis and pseudocoloring was done with Kodak Molecular Imaging software v. 4.0.5 (Kodak). Two images were taken in the same focal plane in bright field and in transmitted light passing through RFP filters and processed by Adobe Photoshop CS2 (Adobe).
Nu/nu mouse maintenance and NS-123 toxicity evaluation
Female nu/nu (nude) mice (6–8 weeks old; Charles River, Wilmington, MA) were maintained at the Wake Forest University School of Medicine animal facility under conditions approved by the Animal Care and Use Committee. NS-123 in DMSO was mixed with 30% propylene glycol (DMSO concentration, <1%) and given, via an i.p. injection at a dose of 50 mg/kg, to three mice on 3 consecutive days. The mice were monitored over 30 days for signs of drug-related toxicity, lethality, and weight loss. Mice were euthanized by CO2 asphyxiation, and necropsy was done by a veterinary pathologist (N. D. Kock). The heart, lung, liver, spleen, kidney, brain, salivary gland, gastrointestinal tract, ovary, uterus, adrenal gland, and thyroid gland were preserved in 10% neutral buffered formalin for at least 48 h. The tissues were embedded in paraffin, processed routinely for histology, cut at 4 to 6 μm, stained with H&E, and examined by light microscopy.
U251 xenograft mouse model
U251 cells (5 × 106) were suspended in 0.1 mL of PBS and then injected into the flanks of nu/nu female nude mice. After tumors formed (45 ± 10 mm3), mice were randomly assigned into one of four groups: DMSO + Sham IR, DMSO + 5 Gy IR, NS-123 + Sham IR, and NS-123 + 5 Gy IR. NS-123 was given as described above. Groups not receiving NS-123 were treated with the same volume of DMSO alone dissolved in 30% propylene glycol. Mouse flanks were irradiated 4 h after the second injection with 5 Gy (Precision X-ray, Inc.) given as one fraction or Sham IR. Tumor volumes were measured every other day based on the formula Volume = length2 × width, wherein length was always the longest dimension. Data are presented as average relative tumor volume (RTV): RTV [dayX] = volume [dayX]/volume [day0]. The measurement of tumor regrowth was calculated by the time needed to grow to 10 × the treatment volume. As per Seo et al. (17), the in vivo radiosensitizer enhancement ratio was defined as the ratio TNS-123+IR/TDMSO+IR, wherein T is tumor regrowth delay (days).
Results
Initial radiosensitizer screen and identification of NS-123
The small chemical compound chemical library (Nanosyn) was replicated in a 96-well format, and compounds were dissolved in DMSO. Using the U251 glioma cell line, we initially investigated the radiosensitizing capability of 870 compounds within the library. The assay was optimized in terms of the number of cells, radiation dose used, and the number of days incubated so that a decrease in survival could be assessed. A picture of a pair of sample plates at 0 and 4 Gy is shown in Fig. 1A. NS-123 exhibited the highest average RSF of 4.62 among the 870 compounds and was selected for further investigation. The structure for NS-123 is shown in Fig. 1B.
Figure 1.

Screening a chemical library identifies the potent radiosensitizer NS-123. A, MTS assays were used to assay the lethality of the compounds with 4 Gy of IR and normalized against the lethality of the compound alone (Control). The absorbance (purple) of each well is proportional to cell survival. Green square, compound that shows potential cytotoxicity to cells, both with and without radiation; red squares, compounds of potential agents of interest as radiation sensitizers. The screening assay was done thrice to eliminate false positives with the resultant RSFs averaged. B, the compound shown NS-123 exhibited an average RSF of 4.62 and was selected for further experiments. C and D, dose-dependent and time-dependent effects of NS-123 on U251 cell survival. U251 cells were treated with the indicated doses of NS-123 for 12 h (C) or for the indicated time with 5.0 μmol/L NS-123 (D). Columns, averages of three experiments (n = 3); bars, SE.
NS-123 radiosensitizes U251 glioma tumor cells
We first determined the effects of micromolar doses of NS-123 on clonogenic survival of U251 cells without IR. As shown in Fig. 1C, treatment with 2.5 μmol/L NS-123 for 12 h did not cause any significant decrease in survival, which is in agreement with the results from the screening assay. Treatment with a 5 μmol/L dose for 12 h caused a modest (30%), but statistically significant, decrease in survival. In time-response experiments, a 5 μmol/L dose of NS-123 did not induce any significant loss of viability unless it was given for 12 h as seen in Fig. 1D.
Next, we investigated the dose-dependent radiosensitizing effects of NS-123 (2.5 and 5.0 μmol/L concentration) in U251 cells (Fig. 2A). Radiosensitization was evident at 2.5 μmol/L, a dose at which NS-123 did not cause any significant loss of viability, and at the radiobiologically relevant IR dose of 2 Gy used in fractionated radiation schemes. Therefore, NS-123 can act as a true radiosensitizer at low micromolar concentrations. Corresponding dose enhancement ratios (DER) at 0.1 survival were 1.3 (2.5 μmol/L NS-123) and 2.0 (5.0 μmol/L NS-123). We then investigated the time-dependent radiosensitizing effects of 5.0 μmol/L NS-123 in U251 cells. As shown in Fig. 2B, NS-123 induced a dose-dependent increase in radiosensitization. Corresponding DER at 0.1 survival were 1.2 (4 h preincubation with NS-123), 1.4 (8 h), and 1.9 (12 h).
Figure 2.

NS-123 radiosensitizes U251 glioma, HT-29 colorectal carcinoma, and A549 lung adenocarcinoma cells in a dose-dependent and time-dependent manner. U251 cells were treated with NS-123 for 12 h at the indicated doses (A) or for the times indicated with 5 μmol/L (B) before irradiation. Calculated DER at 0.1 survival are 1.3 for 2.5 μmol/L and 2.0 for 5.0 μmol/L (A) and 1.2 for 4 h, 1.4 for 8 h, and 1.9 for 12 h pretreatment (B). C, HT-29 colorectal cancer cells were treated with DMSO (control) or 5 μmol/L NS-123 for 12 h before IR. DER at 0.5 survival is 3.0. D, A549 lung tumor cells were treated with DMSO (control) or 5 μmol/L NS-123 for 12 h before IR. DER at 0.1 survival is 1.4. Points, averages of three experiments (n = 3); bars, SE.
NS-123 radiosensitizes HT-29 colorectal cancer cells and A549 lung tumor cells
To exclude the possibility that the radiosensitizing effect of NS-123 was specific to a single cell line, we investigated if NS-123 could radiosensitize additional cell lines of different tissue origin. Thus, we did clonogenic survival assays. Pretreatment of HT-29 colorectal cancer cells and A549 non– small cell lung cancer cells with NS-123 induced significant radiosensitization in both cell lines (Fig. 2C and D), with DER values at 0.1 survival of 3.0 and 1.4, respectively. These results show that the radiosensitization of NS-123 is not restricted to only one cell line.
NS-123 does not radiosensitize normal cells
The objective of any combination of therapeutic agents is to achieve an improved therapeutic result, which is a function of both tumor response and normal tissue damage (8). No improvement in the therapeutic result of IR is gained using a drug that increases the sensitivity of both tumor and normal cells to the same extent. Thus, we investigated if NS-123 radiosensitizes normal tissues in two models. First, we did an MTS assay using NHA, which represents the normal cell counterpart of the U251 glioma cell lines. As shown in Fig. 3A, there was no significant increase in radiosensitization seen with either 8 or 12 h pretreatment with 5 μmol/L NS-123 compared with DMSO-treated cells.
Figure 3.

A, NS-123 does not radiosensitize normal human glial cells. Modified MTS assays done on normal human glial cells in the presence or absence of NS-123 after IR. Results are the average of two independent experiments, and each experiment was done in quadruplicate (n = 4 wells). B, NS-123 does not potentiate adverse effects of IR on Zebrafish development. Zebrafish embryos were pretreated for 8 h with 5 μmol/L NS-123 before 6 Gy of IR at 16 hpf. Pretreatment for 8 h with 1 μmol/L staurosporine (SRS), a known radiosensitizer of both normal and tumor cells, was used as a positive control. Points, average of 60 to 140 embryos. C, morphologic effects of IR in the developing Zebrafish. The effects of NS-123 on morphology were also evaluated for each group with representative photographs shown. NS-123 + IR induced minimal changes. When Zebrafish were irradiated in the presence of staurosporine, morphologic changes, consistent with increased toxicity, were shown, including shorten body length, spine curvature, and pericardial edema.
Effects of NS-123 on embryonic development and survival
The radiosensitizing effects of NS-123 on human cancer cells, but not normal cells, prompted us to examine the potential of NS-123 in vivo. Zebrafish (Danio rerio) embryos provide a unique vertebrate model that is ideal for the screening of therapeutic agents because of their close genetic and physiologic homology to upper level vertebrates, such as mammals, their rapid embryonic development, optical clarity, and suitability for investigations regarding the effects of radiation and radiosensitizers (16, 18). These features allow the efficient screening of novel drugs and observation of effects on specific organs without the need for euthanization and necroscopy. The Zebrafish embryos were pretreated for 8 h with 5 μmol/L NS-123 before 6 Gy of radiation. As shown in Fig. 3B and C, NS-123 did not appreciably impede normal embryonic development or viability. In contrast, staurosporine, a potent radiosensitizer but with effects too general to be used clinically, greatly diminished the viability and development of the Zebrafish embryos.
NS-123 radiosensitizes U251 cells transplanted into Zebrafish embryos
Human cancer cells can be transplanted into Zebrafish embryos through microinjection (19–21). These human cancer cells will then proliferate, nourished by the nutrients circulating through the embryos. We transplanted U251 cells expressing RFP to be distinguished from the background tissues of the embryos (Fig. 4A, left). The transparency and ex utero development of the Zebrafish embryos facilitate the tracking and counting in real time of the transplanted human cells, again without the need for euthanization and necroscopy (Fig. 4A, middle and right). The U251 cells were transplanted into the embryonic yolk sac of 4.5 hpf embryos, treated with NS-123, and irradiated at 24 hpf. The embryos were subsequently examined for fluorescence on sequential days after irradiation. As shown in Fig. 4B, treatment of the embryos with NS-123 alone did not have any significant effect on the size of the U251 xenografts at 3 or 5 dpf. The U251 cells irradiated in the absence of NS-123 showed some regression at 3 and 5 dpf, whereas markedly fewer cells irradiated in the presence of NS-123 are survived at 5 dpf. Quantitation of these results using 10 embryos/condition/experiment and assessing the relative fluorescence showed a faster decrease in luminescence compared with the irradiated-only control, indicating the more rapid extinction of fluorescence in the embryo irradiated in the presence of NS-123 (Fig. 4C). These data show the radiosensitizing potential of NS-123 in vivo.
Figure 4.

NS-123 radiosensitizes U251 cells xenografts in Zebrafish (ZF) embryos. A, U251-RFP cells in culture at 30°C for 7 d (left). Fluorescently labeled cells in a Zebrafish embryo at 3 dpf (middle). Lateral merged phase and fluorescent image view of an embryo at 18 hpf, 2 h after transplantation, showing ∼150 human glioma cells transplanted into the yolk sac observed at the same magnification (right). Scale bar: 75 μm (middle), 100 μm (right). B, irradiation in the presence of NS-123 further decreases the proportion of surviving human glioma cells in Zebrafish embryos. Lateral views of live Zebrafish embryos after transplantation of fluorescently labeled human glioma cells. Top, 1 dpf, Zebrafish embryos are photographed immediately before irradiation with 10 Gy; middle, at 3 dpf; bottom, at 5 dpf. C, histograms showing the relative levels of fluorescence from A to C displayed in arbitrary units of luminescence from triplicate experiments.
NS-123 is well tolerated in nu/nu (nude) mice
The Zebrafish results suggest that NS-123 has low toxicity in higher order vertebrates. To test this hypothesis, we gave 50 mg/kg (i.p.) to nude mice for 3 consecutive days. Clinical signs of drug toxicity and lethality were not observed over the following 30 days. As seen in Fig. 5A, all mice gained weight. Furthermore, both gross and histopathologic examination did not reveal any evidence suggestive of NS-123-associated toxicity. With these results, we considered 50 mg/kg NS-123 × 3 days to be an acceptable treatment regimen for further investigation to test the radiosensitization of NS-123 in mice.
Figure 5.

NS-123 is well tolerated in nu/nu nude mice and induces radiosensitization in tumor xenografts. A, three nu/nu nude female mice were injected for 3 consecutive days with 50 mg/kg of NS-123 (↓). No NS-123 associated toxicity was observed over 30 d after treatment. The plots of each animal weight were fit with a first-order linear regression; all mice slightly increased in weight. B, effect of NS-123 on tumor growth in vivo. Mice were implanted with U251 cells and tumors were allowed to grow. The mice were then randomly assigned to one of four groups: DMSO + Sham IR (n = 4), DMSO + 5 Gy IR (n = 5), NS-123 + Sham IR (n =4), and NS-123 + 5 Gy IR (n = 7). The tumors were treated with 50 mg/kg of NS-123 or a similar volume of DMSO for 3 consecutive days (↓). Four hours after the second injection, the mice were treated with Sham IR or 5 Gy IR (
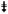 ). NS-123 combined with 5 Gy IR induced the greatest growth delay with an in vivo sensitization enhancement ratio of 2.0.
). NS-123 combined with 5 Gy IR induced the greatest growth delay with an in vivo sensitization enhancement ratio of 2.0.
NS-123 induces in vivo radiosensitization of U251 tumors implanted in nu/nu (nude) mice
To investigate in a more sophisticated model the radiosensitization observed by NS-123 in Zebrafish, we did a growth delay assay of U251 tumors xenografts in nu/nu nude mice. Mice were treated for 3 consecutive days with either NS-123 or DMSO (i.p.) and received either 5 Gy IR or Sham IR 4 h after the second injection. As seen in Fig. 5B, the mean time to reach RTV = 10 was 10 days in the DMSO group, 12 days in the NS-123 group, 14 days in the DMSO+IR group, and 20 days in the NS-123 + IR group, which translates in an in vivo radiosensitizer enhancement ratio for NS-123 of 2.0. The results show that NS-123 can significantly increase the IR-induced tumor growth delay without significant toxicity in vivo. Furthermore, they provide additional support for the use of the novel Zebrafish xenograft model as a faster and more economical screening tool for potential in vivo radiosensitizing activity of lead compounds.
NS-123 inhibits dsDNA break repair and prolongs dsDNA damage-dependent signaling after radiation
As a first step in understanding the mechanism of the radiosensitizing activity of NS-123, we measured the processing of DSBs in U251 cells treated with 30 μmol/L NS-123 or DMSO (control) using an adaptation of PFGE. We found that when plated at high density (required to obtain amount of sufficient protein), a 4-h 30 μmol/L dose of NS-123 induced comparable radiosensitization as a 12-h 5 μmol/L dose of preincubation on low density–plated cells (data not shown). Therefore, we used a 4-h 30 μmol/L NS-123 treatment for experiments analyzing the effects of NS-123 on DSB repair and immunoblotting (see below) which require a significant quantity of cells. As shown in Fig. 6A, the initial number of dsDNA breaks was not affected by NS-123 (NS-123–treated, 88.5 ± 5.3 DSBs/Gbp; DMSO-treated, 84.8 ± 3.8 DSBs/Gbp). However, the processing of DSBs in NS-123–treated U251 cells was significantly compromised. The NS-123–treated U251 cells showed not only slower repair kinetics but also incomplete repair of DSBs even after 24 h. Specifically, the DMSO-treated cells show almost complete repair of DSBs at 24 h (∼9% of their initial damage), whereas the NS-123– treated U251 cells show ∼25% residual damage compared with their initial levels.
Figure 6.

A, NS-123 inhibits dsDNA break repair. DSB processing was measured using PFGE and NALA after exposure to 4 Gy of γ-rays. Points, values normalized to the initial maximum yields for each cell line and averages from three independent irradiation experiments; bars, SE (in some cases are smaller than the corresponding symbol). B, NS-123 prolongs dsDNA damage-dependent signaling after IR. Cells were treated with DMSO (control) or 30 μmol/L NS-123 for 4 h before IR. Immunoblotting was done with an anti–γ-H2AX antibody, an anti–P-CHK2 antibody, anti–P-ATM, or anti–P-DNA-PKcs antibody, followed by incubation with secondary anti–mouse-IgG-horseradish peroxidase or anti–rabitt-IgG-HRP antibody. Immunoblotting for β-actin or DNA-PKcs was used as a loading control.
In response to DNA DSB, inactive ATM protein undergoes autophosphorylation, including at serine 1981 (P-ATM; refs. 22, 23). The active ATM protein migrates to the sites of damage and phosphorylates γ-H2AX at serine 139. The levels of γ-H2AX decrease after repair of the DNA damage (24–26), and therefore, γ-H2AX can serve as a surrogate quantitative measure of DSBs (27–30). CHK2 is a protein kinase that targets two critical effectors operating in distinct branches of the G1 checkpoint: the Cdc25A phosphatase and the p53 transcription factor, both of which prevent entry of cells into S phase with damaged DNA (31). P-ATM phosphorylates CHK2 at threonine 68 (termed P-CHK2). Both P-CHK2 and γ-H2AX increase immediately after IR and gradually decrease as DSBs become repaired (32, 33). Similar to ATM, the DNA-dependent protein kinase catalytic subunit (termed P-DNA-PKcs) can be phosphorylated following DNA DSB resulting from IR and will also phosphorylate both γ-H2AX and P-CHK2 (34, 35).
We therefore assayed the levels of γ-H2AX, P-CHK2, P-ATM, and P-DNA-PKcs in U251 cells by immunoblotting at various time points after IR. Cells were treated with DMSO (control) or 30 μmol/L NS-123 for 4 h before IR. Figure 6B shows representative results. In the absence of NS-123, IR caused a significant increase in γ-H2AX, P-CHK2, P-DNA-PKcs, and P-ATM at 15 min, which decreased and returned to baseline by 6 h. In contrast, in cells treated with NS-123 at 6 h after IR (a time at which >90% of DSBs should have been repaired), U251 cells exhibited substantially higher levels of γ-H2AX, P-CHK2, P-DNA-PKcs, and P-ATM, suggesting that unrepaired DSBs accumulate in the presence NS-123. Although the precise target of NS-123 is currently not known, these results strongly point toward an inhibition of DNA damage signaling pathway as a potential mechanism for radiosensitization.
Discussion
High-throughput screening (HTS) of small molecule libraries is a powerful and rapid method for identifying promising chemotherapeutic candidates. We note that the vast majority of current HTS screening methods are designed predominantly to search for single compounds that have antiproliferative or other antitumor properties. However, few clinically used drugs are effective or given as single agents against malignancies. Because radiotherapy is prescribed for the majority of solid tumors, often in combination with chemotherapy, there is a critical need for improving the therapeutic efficacy of such combined modalities. Therefore, we designed our HTS to specifically identify agents with minimal or low activity but would substantially increase the effectiveness of radiation. The identification of NS-123 as such a compound from only a partial screen of the library offers “proof of principle” for the power of our screening assay. It is conceivable that screens of much larger chemical libraries would identify numerous compounds with radiosensitizing properties. We believe that this approach has the potential, with further refinement and scale-up processes, to identify a large number of similarly promising candidates.
Other target-dependent screens for radiosensitizers have produced promising results. Hickson et al. identified a specific inhibitor of the ATM, KU-55933 (36). Zhao et al. identified a small molecule inhibitor of DNA-PK, LY294002, which they used as a lead to develop another more specific and more potent inhibitor, NU7026 (11). Small molecule inhibitors of DNA repair pathways, such as NS-123, that seem to inhibit the repair of the DNA rather than the signaling of damage, may lead to the development of a distinct, but complementary, class of therapeutics; they may also be important as useful probes for elucidating the mechanisms of action and function of proteins and pathways.
Our findings that NS-123 significantly enhanced the in vitro cell killing induced by radiation in three different cell lines, coupled with the lack of radiosensitization within the two normal tissue models (NHA and Zebrafish) we used, strongly suggested that NS-123 may be clinically effective at doses that cause little or no toxicity. These findings were supported by the finding that radiotherapy delivered in conjunction with NS-123 resulted in the greatest control of U251 tumors implanted in Zebrafish xenografts or in nude mice again without any significant developmental or toxic effects to the embryos or the animals. To our knowledge, the Zebrafish xenograft model is the first demonstration of radioenhancing properties in this vertebrate model system. Although use of Zebrafish to monitor the potential normal tissue toxicity of chemotherapeutics and radiation is rather novel, this model is currently used in several large screening facilities (37, 38). Its value as a relatively inexpensive and HTS-amenable system is gaining ground. Importantly, the results obtained with the Zebrafish xenograft model were corroborated by demonstrating radiosensitization in a mouse xenograft model. Clearly, NS-123 requires further preclinical testing (especially in terms of pharmacokinetics and toxicology) before use in humans. Nevertheless, our results, together with previous studies (16), support the value of Zebrafish as an intermediate-level screening tool of radiosensitizers. For example, one could envision a scenario where potential drug candidates resulting from an in vitro screen could be prioritized for testing in expensive and time-consuming rodent tumor models based on their tumor cell radiosensitization versus developmental side effects profile in Zebrafish.
The classic and emerging agents frequently enhance the effect of radiation through diverse mechanisms, including modification of DNA damage, interference with DNA repair processes, cytokinetic cooperation caused by cell cycle redistribution, inhibition of proliferation, and enhancement of apoptosis (39, 40). Our findings that pretreatment of NS-123 does not induce any significant change in cell cycle distribution up to 8 h after IR (data not shown) suggests that it is unlikely that NS-123 radiosensitizes U251 cells by arresting them at the G2-M interface. Undoubtedly, one of the most biologically significant consequences of IR is the induction of DSB. Unrepaired DSBs can lead to cell killing or chromosomal aberrations (41). Based on our PFGE experiment, we conclude that NS-123 does not increase the initial damage, which is similar, with and without NS-123 (Fig. 6A), but decreases significantly the ability of cells to efficiently process DSBs. These DSBs are expected to be primarily repaired by nonhomologous end joining (NHEJ) but the contribution of homologous recombination cannot be excluded (42). However, the presence of residual DSB, even after 24 h, does suggest a significant inhibition of NHEJ. These data are further supported by the prolonged phosphorylation of the surrogate markers of dsDNA damage, P-ATM, P-DNA-PKcs, γ-H2AX, and P-CHK2 (43).
ATM plays a central role in DNA damage checkpoint signal transduction. γ-H2AX phosphorylation is among the earliest ATM-dependent responses to DSB (25, 44). The number and resolution of γ-H2AX foci over time is an indicator of global DSB repair and correlates with tumor cell radiosensitivity under oxic and hypoxic conditions (45). CHK2 is a signal-transducing kinase, sometimes called an effector kinase, which is activated mainly by ATM in response to DSB from IR. The role of P-CHK2 is to amplify the signal in response to DNA damage and thus maintain genomic integrity (46). DNA-PKcs is not thought to function in signal transduction damage response pathways but rather participate in DNA NHEJ, a pathway for DSB repair. There is extensive redundancy in the functions of activated ATM and DNA-PKcs, but the precise role of DNA-PKcs in DNA damage checkpoint signaling is not yet clear. In our study, the levels of γ-H2AX, P-CHK2, P-ATM, and P-DNA-PKcs increase immediately after irradiation and, in the absence of NS-123, return to baseline levels by 6 h. In cells irradiated after a 4-h pretreatment with NS-123, the phosphorylation of all these proteins continues to increase or remains constant 6 h after IR, indicating that the majority of DSB have not been repaired. These results are temporally consistent with our PFGE results. An inhibitory effect of NS-123 on one or more components of the dsDNA repair machinery could delay complete repair of DNA after radiation and, thus, be responsible for the radiosensitization exhibited by NS-123. However, further investigation is needed to identify the precise component of the DNA repair machinery affected by NS-123 and to understand why this mechanism seems to be selective for tumor cells.
Whereas NS-123 is a previously uncharacterized compound, we have noted a degree of structural similarity with bromodeoxyuridine (47). Bromodeoxyuridine substitutes for thymidine, increasing radiation–induced DNA DSBs (48). More recent studies (49) have shown that radiation of a single-stranded oligonucleotide brominated at a single site results in up to 20-fold more strand breaks than for a nonbrominated oligonucleotide. Whereas NS-123 is unlikely to be incorporated into newly synthesized DNA, it is tempting to speculate that it may intercalate or embed near the DNA helix. Upon irradiation, NS-123 may participate in similar anionic complex formation assays, blocking access of DNA repair proteins to the damaged site. Further analyses of cellular localization and potential interaction with free DNA in solution are required to validate this hypothesis.
In summary, our investigation has yielded a novel and potent chemical compound, NS-123, as a tumor cell–specific radiosensitizer in vitro and in vivo. The mechanism responsible for NS-123 radiosensitization seems to involve inhibition of DSB repair. We have further established the anticancer radiosensitizing effects and lack of adverse normal tissue and developmental effects of NS-123 via a novel Zebrafish-based system that incorporates the many compelling advantages of this vertebrate. Further preclinical evaluation of this drug, including evaluation of its chemosensitizing potential (in particular in combination with genotoxic chemotherapeutic agents) and further elucidation of its molecular targets, are under way.
Acknowledgments
Grant support: NIH grant T32CA113267 (B.E. Lally).
We thank Dr. William Blackstock for his helpful discussions, Dr. Lori Hart, Prashanthi Javvadi, Jiangbin Ye, Christine Naczki, and Diane Fels for expert technical assistance, and Dr. Jennifer Olson, Dr. J. Daniel Bourland, Weili Fu, and Valerie Payne for their assistance with the experiments involving animals.
References
- 1.Walker MD, Green SB, Byar DP, et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303:1323–9. doi: 10.1056/NEJM198012043032303. [DOI] [PubMed] [Google Scholar]
- 2.Curran WJ, Jr, Scott CB, Horton J, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst. 1993;85:704–10. doi: 10.1093/jnci/85.9.704. [DOI] [PubMed] [Google Scholar]
- 3.Scott CB, Scarantino C, Urtasun R, et al. Validation and predictive power of Radiation Therapy Oncology Group (RTOG) recursive partitioning analysis classes for malignant glioma patients: a report using RTOG 90-06. Int J Radiat Oncol Biol Phys. 1998;40:51–5. doi: 10.1016/s0360-3016(97)00485-9. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Chan JL, Lee SW, Fraass BA, et al. Survival and failure patterns of high-grade gliomas after three-dimensional conformal radiotherapy. J Clin Oncol. 2002;20:1635–42. doi: 10.1200/JCO.2002.20.6.1635. [DOI] [PubMed] [Google Scholar]
- 6.Tatter SB, Shaw EG, Rosenblum ML, et al. An inflatable balloon catheter and liquid 125I radiation source (GliaSite Radiation Therapy System) for treatment of recurrent malignant glioma: multicenter safety and feasibility trial. J Neurosurg. 2003;99:297–303. doi: 10.3171/jns.2003.99.2.0297. [DOI] [PubMed] [Google Scholar]
- 7.Regine WF, Patchell RA, Strottmann JM, et al. Preliminary report of a phase I study of combined fractionated stereotactic radiosurgery and conventional external beam radiation therapy for unfavorable gliomas. Int J Radiat Oncol Biol Phys. 2000;48:421–6. doi: 10.1016/s0360-3016(00)00688-x. [DOI] [PubMed] [Google Scholar]
- 8.Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys. 1979;5:85–91. doi: 10.1016/0360-3016(79)90044-0. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Takeda Y, Wragg S, et al. Modification of the ionizing radiation response in living cells by an scFv against the DNA-dependent protein kinase. Nucleic Acids Res. 2003;31:5848–57. doi: 10.1093/nar/gkg775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smart DK, Ortiz KL, Mattson D, et al. Thioredoxin reductase as a potential molecular target for anticancer agents that induce oxidative stress. Cancer Res. 2004;64:6716–24. doi: 10.1158/0008-5472.CAN-03-3990. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y, Thomas HD, Batey MA, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–62. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 12.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 13.Koumenis C, Naczki C, Koritzinsky M, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol. 2002;22:7405–16. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgakilas AG, Bennett PV, Wilson DM, III, Sutherland BM. Processing of bistranded abasic DNA clusters in γ-irradiated human hematopoietic cells. Nucleic Acids Res. 2004;32:5609–20. doi: 10.1093/nar/gkh871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutherland BM, Georgakilas AG, Bennett PV, Laval J, Sutherland JC. Quantifying clustered DNA damage induction and repair by gel electrophoresis, electronic imaging and number average length analysis. Mutat Res. 2003;531:93–107. doi: 10.1016/j.mrfmmm.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Geiger GA, Parker SE, Beothy AP, et al. Zebrafish as a “biosensor”? Effects of ionizing radiation and amifostine on embryonic viability and development. Cancer Res. 2006;66:8172–81. doi: 10.1158/0008-5472.CAN-06-0466. [DOI] [PubMed] [Google Scholar]
- 17.Seo Y, Yan T, Schupp JE, Radivoyevitch T, Kinsella TJ. Schedule-dependent drug effects of oral 5-iodo-2-pyrimidinone-2′-deoxyribose as an in vivo radiosensitizer in U251 human glioblastoma xenografts. Clin Cancer Res. 2005;11:7499–507. doi: 10.1158/1078-0432.CCR-05-1138. [DOI] [PubMed] [Google Scholar]
- 18.McAleer MF, Davidson C, Davidson WR, et al. Novel use of zebrafish as a vertebrate model to screen radiation protectors and sensitizers. Int J Radiat Oncol Biol Phys. 2005;61:10–3. doi: 10.1016/j.ijrobp.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 19.Lee LM, Seftor EA, Bonde G, Cornell RA, Hendrix MJ. The fate of human malignant melanoma cells transplanted into zebrafish embryos: assessment of migration and cell division in the absence of tumor formation. Dev Dyn. 2005;233:1560–70. doi: 10.1002/dvdy.20471. [DOI] [PubMed] [Google Scholar]
- 20.Haldi M, Ton C, Seng WL, McGrath P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis. 2006;9:139–51. doi: 10.1007/s10456-006-9040-2. [DOI] [PubMed] [Google Scholar]
- 21.Mizgireuv IV, Revskoy SY. Transplantable tumor lines generated in clonal zebrafish. Cancer Res. 2006;66:3120–5. doi: 10.1158/0008-5472.CAN-05-3800. [DOI] [PubMed] [Google Scholar]
- 22.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 23.Kozlov SV, Graham ME, Peng C, et al. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–14. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bassing CH, Chua KF, Sekiguchi J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 26.Siino JS, Nazarov IB, Zalenskaya IA, et al. End-joining of reconstituted histone H2AX-containing chromatin in vitro by soluble nuclear proteins from human cells. FEBS Lett. 2002;527:105–8. doi: 10.1016/s0014-5793(02)03176-9. [DOI] [PubMed] [Google Scholar]
- 27.Olive PL, Banath JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;58:331–5. doi: 10.1016/j.ijrobp.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 28.Nazarov IB, Smirnova AN, Krutilina RI, et al. Dephosphorylation of histone γ-H2AX during repair of DNA double-strand breaks in mammalian cells and its inhibition by calyculin A. Radiat Res. 2003;160:309–17. doi: 10.1667/rr3043. [DOI] [PubMed] [Google Scholar]
- 29.Macphail SH, Banath JP, Yu TY, et al. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–8. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 30.Bonner WM. Low-dose radiation: thresholds, bystander effects, and adaptive responses. Proc Natl Acad Sci U S A. 2003;100:4973–5. doi: 10.1073/pnas.1031538100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 2004;3:997–1007. doi: 10.1016/j.dnarep.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 33.Pandita TK, Lieberman HB, Lim DS, et al. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19:1386–91. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–50. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 35.Stiff T, O'Driscoll M, Rief N, et al. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 36.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 37.Milan DJ, Jones IL, Ellinor PT, MacRae CA. In vivo recording of adult zebrafish electrocardiogram and assessment of drug-induced QT prolongation. Am J Physiol Heart Circ Physiol. 2006;291:H269–73. doi: 10.1152/ajpheart.00960.2005. [DOI] [PubMed] [Google Scholar]
- 38.Fleming A, Sato M, Goldsmith P. High-throughput in vivo screening for bone anabolic compounds with zebrafish. J Biomol Screen. 2005;10:823–31. doi: 10.1177/1087057105279952. [DOI] [PubMed] [Google Scholar]
- 39.Lawrence TS, Blackstock AW, McGinn C. The mechanism of action of radiosensitization of conventional chemotherapeutic agents. Semin Radiat Oncol. 2003;13:13–21. doi: 10.1053/srao.2003.50002. [DOI] [PubMed] [Google Scholar]
- 40.Wilson GD, Bentzen SM, Harari PM. Biologic basis for combining drugs with radiation. Semin Radiat Oncol. 2006;16:2–9. doi: 10.1016/j.semradonc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Iliakis G. The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells. Bioessays. 1991;13:641–8. doi: 10.1002/bies.950131204. [DOI] [PubMed] [Google Scholar]
- 42.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–20. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 43.Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 2004;3:1323–34. doi: 10.1016/j.dnarep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 44.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 45.Banath JP, Macphail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res. 2004;64:7144–9. doi: 10.1158/0008-5472.CAN-04-1433. [DOI] [PubMed] [Google Scholar]
- 46.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 47.Dewey WC, Humphrey RM. Increase in radiosensitivity to ionizing radiation related to replacement of thymidine in mammalian cells with 5-bromodeoxyuridine. Radiat Res. 1965;26:538–53. [PubMed] [Google Scholar]
- 48.Limoli CL, Ward JF. A new method for introducing double-strand breaks into cellular DNA. Radiat Res. 1993;134:160–9. [PubMed] [Google Scholar]
- 49.Cecchini S, Girouard S, Huels MA, Sanche L, Hunting DJ. Single-strand-specific radiosensitization of DNA by bromodeoxyuridine. Radiat Res. 2004;162:604–15. doi: 10.1667/rr3267. [DOI] [PubMed] [Google Scholar]