

Abstract
In thyrocytes, cell polarity is of crucial importance for proper thyroid function. Many intrinsic mechanisms of self-regulation control how the key players involved in thyroid hormone (TH) biosynthesis interact in apical microvilli, so that hazardous biochemical processes may occur without detriment to the cell. In some pathological conditions, this enzymatic complex is disrupted, with some components abnormally activated into the cytoplasm, which can lead to further morphological and functional breakdown. When iodine intake is altered, autoregulatory mechanisms outside the thyrocytes are activated. They involve adjacent capillaries that, together with thyrocytes, form the angiofollicular units (AFUs) that can be considered as the functional and morphological units of the thyroid. In response to iodine shortage, a rapid expansion of the microvasculature occurs, which, in addition to nutrients and oxygen, optimizes iodide supply. These changes are triggered by angiogenic signals released from thyrocytes via a reactive oxygen species/hypoxia-inducible factor/vascular endothelial growth factor pathway. When intra- and extrathyrocyte autoregulation fails, other forms of adaptation arise, such as euthyroid goiters. From onset, goiters are morphologically and functionally heterogeneous due to the polyclonal nature of the cells, with nodules distributed around areas of quiescent AFUs containing globules of compact thyroglobulin (Tg) and surrounded by a hypotrophic microvasculature. Upon TSH stimulation, quiescent AFUs are activated with Tg globules undergoing fragmentation into soluble Tg, proteins involved in TH biosynthesis being expressed and the local microvascular network extending. Over time and depending on physiological needs, AFUs may undergo repetitive phases of high, moderate, or low cell and tissue activity, which may ultimately culminate in multinodular goiters.
Introduction
The Thyroid Gland: The Result of a Long Evolutionary Journey
-
Thyroid Hormone Synthesis: A Dangerous Process
Iodine: intracellular journey and transformation
Colloid Tg: an easily accessible reserve
The TPO-DUOX couple: the heart of the synthesis complex
ROS: key factors in thyroid function and growth
-
The Angiofollicular Unit: From Concept to Demonstration
The role of the local microvasculature in the maintenance of endocrine function
ID: the trigger of an immediate TSH-independent microvascular response
ID-induced microvascular response: which molecular mechanism?
AFUs: a heterogeneous population of three-dimensional structures that protect the thyroid gland against functional failure
-
Angiofollicular Heterogeneity, Nodulogenesis, and Multinodular Goiter: Why and How?
The polyclonal and mutation theory
The stem cell theory
Concluding Remarks
I. Introduction
The thyroid hormones (THs) T3 and T4 are essential for energy metabolism and embryonic development, especially for brain maturation. THs influence all anabolic and catabolic pathways involved in intermediary and structural metabolism (1–3). Claiming that the main role of the thyroid gland is the synthesis and secretion of THs seems, at first sight, a truism. Nevertheless, this task represents a continuous challenge, because the gland must not only deal with the scarcity of a trace element, iodine, but also perform within a potentially highly toxic biochemical environment. The goal of this review is to present the most recent and innovative data about TH synthesis and to explain how thyrocytes, within three-dimensional structures known as angiofollicular units (AFUs), adapt to iodine deficiency (ID). The existence of corrective mechanisms, including cross talk between epithelial and endothelial compartments, and the role of reactive oxygen species (ROS) under physiological and pathological conditions will be described to illustrate the continuous adaptation of thyroid cells to their ever-changing environment.
II. The Thyroid Gland: The Result of a Long Evolutionary Journey
Iodine (the 47th most abundant element in the earth's crust) is a nonmetallic element of the halogen family, which also includes bromine, fluorine, and chlorine. It was accidentally discovered by Bernard Courtois in 1811, when he was searching for new raw materials for explosives during the Napoleonic wars (4, 5). Over a century later, the biological relevance of iodine was recognized when E. C. Kendall isolated and crystallized T4 in 1914 (6). The name iode, given by Gay-Lussac (thereafter anglicized to iodine), is derived from the Greek word iodes, which means violet-colored (7). The total mass of stable iodine on Earth is estimated to be 8.6 × 1015 kg (8). Although it is still expelled from hot water sources and rift faults, iodine was mainly generated early on in the earth's formation during the degasification of the asthenosphere. Since then, leaching from glaciation, flooding, and erosion have depleted surface soils of iodine, which explains why it is mostly found in oceanic sediments, the largest reservoir of iodine on Earth (8, 9).
From an evolutionary point of view, the thyroid gland in mammals is the most advanced system for storing and producing iodocompounds from an iodine-deficient milieu (5, 10). Mother Nature first used iodine for its potent antioxidant properties, for instance in algae, the first living cells that produced oxygen in the terrestrial milieu. Some marine seaweeds and algae can accumulate iodine to 4,000–30,000 times its seawater concentration, which results in levels of 0.8–4.5 mg of iodine per gram of dried material (7, 9, 11, 25). The human thyroid gland is the result of an endless adaptation that started millions of years ago when primitive marine animals left the sea, an iodine-enriched environment, to colonize the iodine-deficient terrestrial mainland. At that time, these organisms had to solve many problems, including the sequestration of iodine from an iodine-deficient ecosystem. Animal species have used different strategies to cope with this problem, from jellyfishes lacking a thyroid gland to chordates developing an organ called the endostyle on the floor of the pharynx, which concentrates iodine and produces THs (13, 14). In early vertebrates, such as lampreys, nonencapsulated follicles evolve from the larval endostyle during metamorphosis (15). It is worth noting that some endostyle cells produce thyroglobulin (Tg), display peroxidase activity, and incorporate iodine, which are all features of the thyroid gland in vertebrates (15–22). The homology between the endostyle and the thyroid gland of higher vertebrates is supported by the results of recent comparative analyses, which demonstrated that genes (Pax8 and Ttf1) involved in the expression of thyroid-specific transcription factors have similar expression patterns during the morphogenesis of the mouse thyroid and in the chordate endostyle (13, 22–24). In higher vertebrates, the organ kept on evolving, following a process that culminated in the thyroid gland (10, 25–28).
Over millions of years, T4 had no hormonal actions per se in invertebrates, but instead served to transport the ancient antioxidant iodide (I−), the reduced form of iodine (I2), into peripheral cells, which, in more recent vertebrates, started using the remaining T3 as an active hormone to control processes such as metamorphosis in amphibians, spawning changes in fishes, and thermogenesis. In the organs (stomach, salivary glands, and mammary tissue) of more recent mammals, I− still acts as an antioxidant in the presence of hydrogen peroxide (H2O2). In the thyroid gland, after disposing of its electrons, I− is instead transformed into oxidized forms that immediately iodinate tyrosyl residues of Tg, thereby neutralizing its own oxidant properties, while generating T4 (10, 25, 29–31).
Thus, from this endless adaptation of iodine-concentrating cells, a highly sophisticated system emerged in mammals, known as the follicle. Each follicle is composed of polarized cells, called thyrocytes or follicular cells, which are perfectly adapted to achieve a task that is not only demanding, due to the scarcity of iodine, but also somewhat perilous because it requires the production of potentially cytotoxic ROS.
III. Thyroid Hormone Synthesis: A Dangerous Process
A. Iodine: intracellular journey and transformation
The intrathyroidal journey of iodide is shown in Fig. 1. TH synthesis requires the incorporation of iodine into Tg (iodine organification). The main sources of iodine and products containing high amounts of iodine are fish, shellfish, kelp and seaweed, milk, iodinated salt, bakery products where iodates are used as conditioners for dough, preservatives, therapeutics (amiodarone, vitamins, and Lugol's solution), topical antiseptics, and contrast dyes (9, 32). Under normal conditions, dietary iodine, which is found in nature in a reduced state (I−), is nearly completely absorbed from the gastrointestinal tract to join the inorganic I− pool in the extracellular fluid (9, 33). As for the thyroid, the intestinal transport of I− occurs via the sodium-iodide symporter (NIS) [a 643-amino-acid, 13-transmembrane-domain glycoprotein encoded by the soluble carrier 5A5 (SLC5A5) gene on chromosome 19] (34). Under conditions of sufficient iodine intake, the body of a healthy adult contains 15–20 mg of iodine, of which 70–80% is found in the thyroid. This content may fall to less than 20 μg under conditions of chronic ID (33). In the thyroid, the local clearance of I− varies with iodine intake, ranging from 10% uptake in situations of adequate iodine supply to 80% uptake under conditions of chronic ID (33). To balance losses and preserve TH synthesis, an adult thyroid must trap at least 60 μg of I− per day (33). Under conditions of sufficient iodine nutrition, more than 90% of the ingested I− is excreted in the urine (9, 33, 35). Urinary iodine can therefore be used to assess iodine intake in human populations (33, 35).
Figure 1.
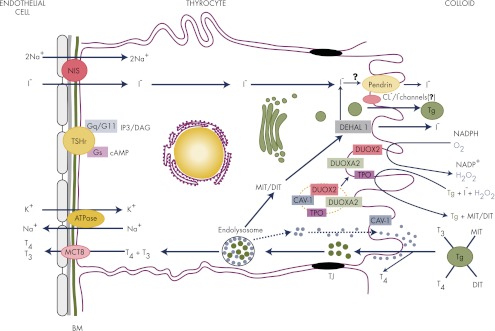
The intrathyroidal journey of iodide. Iodide (I−) entering the thyroid gland through fenestrated microvessels must sequentially cross the two basal laminae of endothelial and epithelial cells to be transported into the thyrocyte by NIS, which uses the energy provided by a Na+-K+-ATPase. Once inside the cell, I− rapidly goes to the apical surface (according to a process that still remains elusive) where it is transported across the apical membrane by pendrin, even though other apical transporters have been proposed to be part of this process. I− is readily oxidized by TPO in the presence of H2O2 and incorporated into tyrosine residues of Tg to form MIT and DIT, which are coupled, again by TPO, to form T3 and T4. H2O2 is produced by DUOX2 in coordination with DUOXA2. TH biosynthesis occurs at the interface with the colloid in a harmless environment for the cell. Iodinated Tg is stored in the colloid until use (see Fig. 2). T4 can be partly released extracellularly by cathepsins K and L. In the human, the uptake of Tg by thyrocytes occurs by micropinocytosis, which can be either nonspecific (fluid phase) or receptor mediated. After endocytosis, colloid droplets containing partly digested Tg are transported to the endolysosomal compartment for complete hydrolysis by lysosomal enzymes including cathepsins. THs are released into the circulation via basal TH transporters (including MCT8) and transported to their target tissues via binding transport proteins. Unused MIT and DIT are dehalogenated by DEHAL1, and most of the I− is recycled into TH synthesis. The binding of TSH to its receptor activates both Gs and Gq proteins. Green dots represent colloid droplets with partially degraded Tg, and blue dots represent cathepsins. BM, Basal membranes; DAG, diacylglycerol; Gq/G11, guanine nucleotide-binding protein αq and α11 subunits; Gs, guanine nucleotide-binding protein α-subunit; IP3, inositol triphosphate; TJ, tight junctions; TSHr, TSH receptor.
After entering the thyroid capillaries, I− is actively taken up by the thyrocytes. The active transport of I− into thyrocytes is mediated by NIS that is localized at the basolateral membrane and the energy required for transport is provided by a ouabain-sensitive Na+/K+-ATPase (33, 36–38). NIS couples the simultaneous transport of two Na+ ions with one I− ion by taking advantage of the favorable gradient concentration of Na+. This pushes I− into the cell against an electrochemical gradient, which results in intracellular concentrations that are 20–50 times higher than that of blood plasma. Thus, this transport is electrogenic, because two Na+ cations are transported for each I− anion. This is true for all NIS substrate anions (ClO3−, SCN−, SeCN−, NO3−, Br−, BF4−, IO4−, and BrO3−), with the exception of perchlorate (ClO4−, a pollutant occurring widely in food and water in the United States), whose transport is electroneutral (39, 40). This indicates that NIS translocates different substrates with different stoichiometries (39, 40). The side chain of the amino acid Gly at NIS position 93 is crucial in this matter, because it is used as a pivot during the change from an outward to an inward open conformation during the transport cycle. This controls the size and chemical characteristics of the ion cavities as well as the kinetics and the stoichiometry of transport (41). The basal expression of NIS is up-regulated by TSH (42) and down-regulated by I− (43–45), which is most likely caused by posttranslational modifications, including polyadenylation and mRNA destabilization (46). The significantly decreased levels of NIS mRNA and protein have been implicated in escape from the Wolff-Chaikoff effect (44), which corresponds to a blockade of I− organification and of TH biosynthesis and release in response to elevated I− plasma levels (47). Of note, TSH fully induces NIS expression at the basolateral membrane only when thyrocytes are organized as follicles, suggesting that a signal (of unknown nature) resulting from specific interactions between thyrocytes in follicles modulates TSH-induced NIS expression (48). This again emphasizes the importance of the follicle three-dimensional organization and of the cell polarization for a full expression and regulation of some thyroid-specific proteins. In models of cultured thyroid cells where TSH levels are kept constant, chronic ID does not influence NIS expression. This is in contrast to acute ID, which induces a clear up-regulation of NIS expression via a currently unknown mechanism (49, 50). Recently, other ionic channels were reported to play an important role in NIS-mediated I− transport. Thus, the potassium voltage-gated channel, KQT-like subfamily, member 1-potassium voltage-gated channel, Isk-related family, member 2 (KCNQ1-KCNE2) complex, which is probably localized to the basolateral membrane, was identified as a TSH-stimulated thyrocyte K+ channel that is critical for normal I− accumulation (51). KCNQ1 and KCNE2 are two potassium channel subunits that function in repolarizing cardiac myocytes. They have been suggested to form a constitutively active K+ channel in thyrocytes, which is required for normal TH biosynthesis (51, 52).
Because NIS mediates I− uptake in organs other than the thyroid, it is considered to be the master regulator of iodine metabolism. Besides the intestine (34), NIS has also been identified in lactating mammary glands, gastric mucosa, and salivary glands (reviewed in Refs. 53 and 54). In contrast to enterocytes where it is localized apically, NIS is found exclusively in the basolateral region of cells in other organs. In the breast, it is involved in I− transport from the bloodstream to the milk, thereby fulfilling the iodine requirement of newborn babies. In gastric mucosa and salivary glands, NIS transports I− in the gastric juice and in saliva as an antimicrobial and antioxidant substance before being reabsorbed by NIS located lower in the digestive tract at the apical surface of enterocytes. This reduces the loss of I− (34, 53, 54).
After entering the cell, I− crosses the apical membrane via a transporter [a candidate being the Cl−/I− exchanger, pendrin/Pendred syndrome (PDS); a 780-amino-acid, 12-transmembrane-domain, hydrophobic membrane protein encoded by the SLC26A4 (also called the PDS) gene on chromosome 7] (55–57). Pendrin translocates to the membrane in response to TSH and forskolin through the protein kinase A pathway in PCCL-3 rat thyroid cells (58). However, the transport mechanism of I− through the apical membrane remains a matter of debate. Other apical transporters, such as chloride channel protein 5 (ClC-5), a chloride/proton antiporter, and the chloride channel cystic fibrosis transmembrane conductance regulator (CFTR), have been proposed to mediate or comediate apical I− efflux (38, 56, 57, 59–63). Studies using heterologous systems have indicated that the product of the SLC5A8 gene, also known as sodium monocarboxylic acid transporter (SMCT1), human apical iodide transporter (AIT), or SLC5A8, is not involved in I− efflux (63–66), as previously held (67). After transport through the apical membrane, I− is covalently bound to the tyrosyl residues of Tg by thyroid peroxidase (TPO).
Tg has a relative molecular mass of 660,000 Da (660 kDa) and is a homodimeric glycoprotein protein. It is the product of a 270-kb gene with an 8.5-kb coding sequence divided into 48 exons on chromosome 8q24 (reviewed in Ref. 68). Each chain consists of 2748 amino acids, 67 of which are tyrosines (69). Eighty percent of the molecule has three regions (called regions I-II-III) with cysteine-rich repeat domains covalently bound by disulfide bonds. These are followed by the cholinesterase-like carboxyl-terminal domain (∼570 residues), which has two functions: 1) homodimerization and 2) binding to the I–II–III region to facilitate oxidative maturation required for intracellular protein transport (70). Correctly folded Tg homodimers are glycosylated in the Golgi and are secreted into the follicular lumen (71). It is worth noting that Tg is not only a scaffold protein for TH synthesis but also possibly a regulator of thyroid function. One research team repeatedly reported that the accumulation of Tg acts as a feedback suppressor of thyroid function, including the expression of important thyroid-specific transcription factors (Titf1/Nkx2-1, Foxe1, and Pax8), thereby decreasing the expression of Tg, TPO, and NIS (reviewed in Ref. 72). However, many questions concerning the mechanisms controlling Tg-induced negative regulation remain unanswered.
TPO is a 933-amino-acid, transmembrane-bound glycoprotein protein of the mammalian peroxidase superfamily (which includes lactoperoxidase and myeloperoxidase) with a short intracellular C-terminal and a large N-terminal extracellular region. It is encoded by a 150-kb gene with 17 exons and 16 introns on chromosome 2 (73). The protein possesses a heme-containing, extracellular catalytic domain covalently linked to the protein, which uses H2O2 as an oxidizing agent for I− (74, 75). TPO exists in two isoforms, TPO1 (110 kDa), which is active, and its alternatively spliced form, TPO2 (100 kDa). TPO2 is found only in intracellular compartments, has no prosthetic heme group, and is inactive (76, 77). Until recently, it was thought that only a small fraction (∼15%) of TPO was expressed in the microvilli of the apical plasmalemma; however, this notion has been called into question, at least in FRTL-5 cells (a rat thyroid cell line) where a large fraction of TPO is delivered to the plasma membrane (78).
The ability of the thyroid gland to produce H2O2 was first reported in 1971 (79, 80). Ten years later, it was demonstrated that H2O2 is generated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases located in the apical plasma membrane (81, 82). Another 10 yr passed before the cloning of the two human cDNAs encoding these NADPH oxidases [now called dual oxidase 1 (DUOX1) and DUOX2], which share 83% sequence similarity (83, 84). DUOX proteins belong to a family of adenine dinucleotide phosphate oxidase (NOX) enzymes and exhibit the characteristics of heme-bound NADPH oxidases, like NOX1–NOX5 (83–87). Both enzymes have a seventh transmembrane domain and possess an extracellular NH2 terminus peroxidase-like domain, also called the TPO-like N-terminal domain (43% similarity to TPO, even though no peroxidase activity is associated with human DUOX) (88, 89), which is able to associate with TPO (83, 84, 90). The C-terminal region contains the catalytic NADPH oxidase core, which is common to all NOX proteins (83, 84). DUOX proteins exist in two forms depending on their N-glycosylation states; only the fully N-glycosylated form (190 kDa) is expressed in the plasma membrane, whereas the high-mannose glycosylated immature form (180 kDa) is expressed in the endoplasmic reticulum (ER) in a nonfunctional state (91, 92). DUOX proteins need calcium to become fully activated (91, 93–95). Both proteins have EF-hand calcium-binding motifs in their first intracellular loop. EF-hand involvement in H2O2 production was demonstrated recently when mutations in this site were shown to block ionomycin-induced H2O2 production (96). In contrast to other genes involved in TH synthesis, DUOX expression is not influenced by TSH in human and mouse thyrocytes (84, 97). DUOX1 is responsive to cAMP and protein kinase A-mediated phosphorylation, whereas DUOX2 is stimulated by phospholipase cascade and protein kinase C-dependent phosphorylation (96). In the thyroid, the main source of H2O2 is DUOX2 (98), which, unlike other NOXs, does not produce the intermediate superoxide (O2·−), although the immature partially glycosylated form of DUOX2 is thought to generate O2·− (94). The ability of DUOX2 to produce H2O2 instead of O2·− depends also upon associated maturation proteins as recently suggested by a study that showed that the NH2-terminal tail of DUOX maturation factor 2 (DUOXA2) (one of the two maturation proteins as described below) favors the dismutation of O2·− to generate H2O2 (99). Congenital hypothyroidism due to an I− organification defect can be associated with inactivating mutations in the DUOX2 gene (100), even though rescue mechanisms by DUOX1 remain possible (101, 102). I− plays an important role in the control of H2O2 production, regulating DUOX activity in a dual fashion: at low concentrations, I− is stimulatory, thereby favoring the efficient iodination of Tg tyrosyl residues (103); however, when it is present in excess, H2O2 production is transiently blocked to protect the organism from overproduced TH (Wolff-Chaikoff effect) (47). The block may depend (at least partly) on the synthesis of iodolipids (6-iodo-δ-lactone and 2-iodo-hexadecanal) (104–107). I− must anyway be oxidized to inhibit H2O2 production. This oxidized form acts at a posttranscriptional level by reducing the availability of the mature DUOX2 protein (92). Blocking H2O2 production when iodine is present in excess is one of the many iodine-induced autoregulatory mechanisms that protect the body against excessive TH release. Other mechanisms include the inhibition of TH secretion, cAMP generation, Tg proteolysis, glucose and amino acid transport, and protein and mRNA synthesis (108) and (reviewed in Refs. 109 and 110). In the presence of sufficient amounts of I−, H2O2 generation becomes the limiting step for TH synthesis (111, 112).
In addition to I− organification, TPO also catalyzes H2O2-dependent head-to-tail coupling reactions of mono-iodotyrosine (MIT) and di-iodotyrosine (DIT) to form T4 (80% of TH) and T3 (111, 113) (reviewed in Ref. 5). Forty of the 140 TPO tyrosyl residues are iodinated in vivo, but only a few are involved in coupling reactions (111). Four hormonogenic acceptor tyrosines were identified at positions 5 (the most favored one), 1291, 2554, and 2747 in human Tg, whereas tyrosines at positions 130, 847, and 1448 were identified as potential outer ring donor sites (114). The hormonogenic residues are located close to the extreme ends of the molecule to facilitate proteolytic cleavage and the release of T4 and T3. The iodinated residues that are not involved in hormonogenesis are readily deiodinated to a NADPH-dependent specific iodotyrosine dehalogenase 1 (DEHAL1) for recycling. DEHAL1 is a 33-kDa, transmembrane, nitroreductase-related enzyme present in intracellular vesicles and in the apical plasma membrane, which is located close to the organification site (115–118). Two other variant gene products (DEHAL1B and DEHAL1C) that are probably functionally inactive were also cloned (119).
The way in which I− oxidation, iodination and coupling reactions occur in the thyroid gland remains controversial (5, 120). Concerning iodination, three mechanisms have been proposed: one includes a free radical scheme based on a one-electron transfer process (111, 121); the two others imply a two-electron transfer process with either iodinium cation (I+) (122–125), or hypoiodite anion (IO−) (126–128) as the iodinating species. Under conditions of normal iodine supply, H2O2 oxidizes the native ferric form (FeIII) of TPO to form compound I, a Fe(IV) porphyrin π-cation radical species that contains two oxidation equivalents above the ferric state of the free enzyme (129). According to the free radical model (which appears the less likely), I− and tyrosine undergo one-electron oxidation to produce the radicals I· and Tyr·, which readily combine to form MIT or DIT. In the I+ hypothesis, or the IO− hypothesis (the most widely accepted), I− is oxidized in a two-electron transfer process in which E-I+ or E-OI− acts as an intermediate. After this step, compound I reverts to its native state. As mentioned above, coupling reactions, which refer to the reaction between two DIT residues or between one MIT residue and one DIT residue to form T4 or T3, respectively, are also catalyzed by TPO (113, 130) (reviewed in Ref. 5). This reaction occurs within the Tg molecule matrix and comprises three steps. The first involves the oxidation of iodotyrosyl residues by TPO. According to the most accepted univalent scheme, the second step involves the oxidation of two iodotyrosine residues to free iodotyrosyl radicals that then couple to form a quinol ether intermediate. In the third step, this unstable intermediate undergoes fission, leading to the formation of T4 or T3 in the hormonogenic site, whereas the alanine side chain of the donor tyrosyl remains in the Tg polypeptide chain as dehydroalanine.
After endocytosis, iodinated Tg is hydrolyzed by proteases in the lysosomes, which contributes to release TH from the Tg backbone. The last enigma related to TH secretion from thyrocytes is perhaps being clarified, as this process is mediated in the basolateral membrane by TH transporters of which the monocarboxylate transporter 8 [MCT8 (SLC16A2)], a protein with 12 predicted transmembrane domains, is an important contributor (131, 132) (reviewed in Ref. 133). This does not exclude the possibility that other TH transporters will be discovered, as was suggested recently (132, 133). MCT8 mutations are associated with the Allan-Herndon-Dudley syndrome (AHDS), which has been identified in more than 45 families worldwide. The symptoms of AHDS include severe X-linked mental retardation, disrupted locomotor development due to the hindered access of T3 to target cells during development (because MCT8 is also involved in the transport of T3 in peripheral cells, as described below), and abnormal TH levels, in particular, high serum T3 levels (133–137). These anomalies are explained by the relative insensitivity of the pituitary to T3 and the associated alteration in peripheral deiodination (138–140). MCT8 is thought to play a role in the maintenance of thyroid integrity, as recently reported in a paper describing severe thyroid morphological alterations (hyperplastic nodules, microfollicular areas with stromal fibrosis, and nuclear features close to papillary thyroid carcinoma) in an AHDS patient and in Mct8-deficient mice (138).
T4 and T3 are transported to their target tissues after binding plasma proteins (T4-binding globulin, transthyretin, and albumin) (141). It was thought initially that THs passed through the plasma membrane of their target cells by passive transport. This simple view was challenged when it was discovered that MCT8 selectively transports T3 across the membranes of target cells (142), indicating that, in contrast with previous thoughts, THs do not cross cell membranes passively. Other TH transporters have been discovered, and more are likely to be discovered in the near future, which will generate new knowledge on TH peripheral actions (reviewed in Ref. 133). It is usually admitted that after T4 deiodination, T3, the active hormone, exerts genomic effects after binding TH receptors (TRs). In addition to this classic nuclear TH action, THs could also influence nongenomic pathways. TH interacts with αvβ3 integrins to induce ERK1/2 signaling-associated angiogenesis (143) and DNA synthesis (144). After binding TR, T3 may engage in nongenomic activation of the phosphatidylinositol-3 kinase/AKT/mammalian target of rapamycin pathway in different cell types (145–150). Furthermore, T4 and rT3 were reported to regulate actin polymerization and microfilament organization independently of T3 (151). Although it remains meaningful to evaluate the pathophysiological relevance of these TR-induced non-TH response element-dependent effects, the finding of nonclassical TH actions opens up new perspectives on TH biological actions in targets cells, as reviewed recently (152).
B. Colloid Tg: an easily accessible reserve
The colloid is not a homogeneous form of Tg storage (Fig. 2). It is rather heterogeneous with a more rapid Tg turnover at the periphery than in the center. This is because the iodination process first affects newly synthesized, soluble molecules at the periphery before affecting older molecules stored as compact Tg globules, as formulated in the last-come-first-served principle (153, 154). Thus, when Tg molecules escape rapid uptake, they proceed to the colloid where they are stored as globules of highly concentrated, insoluble, covalently cross-linked, multimerized 19 Svedberg Tg, which is not immediately available for TH synthesis but is made available upon follicle stimulation (155–157). Therefore, Tg globules store high amounts of Tg in follicles, while preventing osmotic injury (155, 156). The iodine content of isolated globules is high, although hormones are not detected (155, 158). A mouse monoclonal antibody against iodinated Tg containing T4 at the N-terminal hormonogenic site (TgI) (159) failed to detect globules (157), which could be due to the absence of TH (158) or to the limited access of the antibodies to epitopes buried in aggregates or covered by carbohydrate chains. However, the latter possibility seems unlikely, because the treatment of paraffin sections with neuraminidase, which breaks down the sialic acid-induced rigidity of the polysaccharide cover, did not restore the antigenicity (157). These globules are often observed in hypofunctioning follicles, but only very rarely during hyperactive endocytosis, as observed in Graves' disease and in autonomous adenomas (157).
Figure 2.

Proposed mechanism of hypofunctioning follicle reactivation and processing of Tg globules upon TSH stimulation. In the normal human thyroid, active follicles coexist with many hypofunctioning follicles that are surrounded by hypotrophic capillaries and that contain compact Tg globules. These globules correspond to condensed Tg in covalently cross-linked form, not immediately available for TH synthesis, but eventually usable in case of increased hormonal needs. When this occurs, hypofunctioning follicles serve as a functional reserve that can be reactivated upon TSH stimulation. TPO, DUOX, and pendrin expression then reemerges along with progressive dilution of Tg globules into soluble Tg, under the action of secreted cathepsins. This occurs along with the expansion of adjacent microvessels, which, besides TSH, depends also upon many locally secreted growth and vasoactive factors. Persistent TSH stimulation induces thyrocyte hypertrophy and proliferation resulting in goitrous follicles with no more colloid Tg reserves. The microvascular reaction keeps evolving with the proliferation of endothelial cells, the fusion of adjacent microvessels, and the formation of larger microvessels. At this stage, in addition to being secreted by thyrocytes, VEGF, as well as FGF at least, could also function as crinopexins that are locally synthesized molecules sequestered in pericellular structures (extracellular matrix, cell surface, and proteoglycans) under a latent form (305). According to this theory, early alterations in local blood flow and in microvessel shape may alter the extracellular matrix, thereby provoking the release and the activation of crinopexins that could, in turn, stimulate endothelial cell proliferation. This would amplify local changes in the microvasculature initially originated from thyrocytes. Of note, VEGF expression is observed even in thyrocytes of hypofunctioning follicles. ECM, Extracellular matrix; ET, endothelins; TSHr, TSH receptor.
TSH-stimulated thyrocytes metabolize globules in successive fragmentation steps from compact, insoluble Tg into soluble molecules (157). The fragments are then used for TH liberation. Several mechanisms may account for globule dissolution. Besides a first mechanism that involves protein disulfide isomerase (PDI)-induced reduction of intermolecular disulfide bridges (156) (very unlikely because PDI requires reducing and acidic conditions) and another involving H2O2-associated oxidative attacks of globule surface (158), a third mechanism involving lysosomal enzymes has received great attention over the last 15 yr (160–164). Tg proteolysis is a highly specific process, because Tg is resistant to proteolytic cleavage because of its size, its level of glycosylation, and the presence of many intra- and intermolecular bonds, which probably protects the protein from early degradation. The process starts in the colloid before endocytosis and involves the action of cysteine cathepsins in the pericellular space. These proteases are present in the follicle lumen after being released from the endolysosomal compartment. Before endocytosis, Tg undergoes a limited proteolysis in the follicular lumen by cathepsins B and L, which results in small fragments that are processed further by cathepsins K, L, and S (163, 164). T4, but not T3, is released to some extent by the extracellular proteolysis of Tg within the colloid, at the apical surface of thyrocytes (160). This form of proteolysis is not an easy process compared with bone resorption by osteoclasts. Although bone resorption occurs under acidic and reducing conditions, Tg proteolysis occurs at neutral pH and under oxidizing conditions, which are not ideal for maximum protease activity. However, evidence suggests that cathepsins conserve their proteolytic properties for awhile, even under oxidizing conditions (165). Upon TSH stimulation, the proteolytic process is activated as cathepsins are rapidly transported extracellularly through retrograde trafficking vesicles. TSH-induced cytosolic calcium release activates the fusion of apical vesicles with the plasma membrane within minutes. Thereafter, intracellular reserves of cathepsins are reconstituted, either from the extracellular colloid lumen or by de novo biosynthesis (165). A recent paper (166) reported that altered trafficking routes of cathepsins, for instance toward the basal membrane instead of the apical membrane, could have devastating consequences for malignant cell invasion because they degrade components of the extracellular matrix. This observation is in accord with another study published in the early 1990s (167).
In resting or moderately activated states, highly iodinated Tg is internalized via fluid-phase pinocytosis and receptor-mediated endocytosis (reviewed in Ref. 168). Under TSH stimulation, pseudopods form at the apical membrane of thyrocytes and incorporate Tg as colloid droplets (169); however, this process has not been observed in species (including humans) other than rodents (170). The multiligand endocytic receptor megalin, a member of the low-density lipoprotein receptor family, acts as a high-affinity receptor for Tg (171). Megalin mediates Tg transcytosis from the apical to the basolateral surface, especially under intense TSH stimulation, which prevents excessive hormone release (168). Megalin also transports Tg molecules with a low hormone content away from lysosomes, which promotes the lysosomal degradation of hormone-rich Tg molecules (172). Evidence suggests that immature Tg molecules are internalized and recycled through the Golgi compartment (173, 174), and although still controversial, it is thought that this may occur via PDI (175) or via the vacuolar protein sorting 10 protein family member sortilin, which is expressed in thyroid epithelial cells in a TSH-dependent manner (176).
C. The TPO-DUOX couple: the heart of the synthesis complex
The multiprotein complex involved in TH biosynthesis is shown in Fig. 3. TH synthesis depends on biochemical reactions that would be highly toxic if not perfectly controlled. H2O2, a key element in this biosynthesis, would be lethal if not used immediately and/or properly detoxified (177–182).
Figure 3.

The multiprotein complex involved in TH biosynthesis. A, Upon TSH stimulation, DUOX2 and TPO migrate into the plasma membrane where the complex is activated. TPO and DUOX2 are viewed as a producer-consumer unit that allows TPO to use H2O2 instantly to oxidize iodide (I−) into iodine, to iodinate tyrosyl residues of Tg, and to couple iodotyrosyl residues to form T3 and T4, while protecting the cell against H2O2 possible oxidative damage. DUOX2 is represented here as a seven-transmembrane-domain glycoprotein with an extracellular NH2-terminal peroxidase-like domain that appears essential for interaction with TPO, a long COOH-terminal region that contains the catalytic NADPH oxidase core along with the flavin adenine dinucleotide (FAD) and NADPH binding cavities, and two EF-hands in the first intracellular loop that are essential calcium binding sites. TPO is represented with a short intracellular COOH terminus and a long extracellular NH2 terminus (90% of its amino acids) that exhibits a catalase-like activity in certain circumstances. His407 is involved in the covalent binding of the heme prosthetic group that is essential for enzyme activity. The manner in which TPO and DUOX2 interact makes the apical multiprotein complex perfectly suited to detoxify ROS and avoid H2O2 spillages. The local H2O2 concentration is kept under control by the TPO catalase-like action (which protects DUOX2, as long as both proteins are closely associated). In addition, the complex contains the thioredoxin-related protein EFP1, which interacts with the two EF-hands of DUOX2 and detoxifies H2O2 that is not readily used in the biosynthesis process. EFP1 may also be involved in the maturation process of DUOX2. For this biochemical unit to be activated, DUOX2 must be fully glycosylated (Y, N-glycosylation sites) following successive maturation steps in the ER and the Golgi apparatus. DUOX2 must also be associated with the maturation factor DUOXA2, which is represented here as a five-transmembrane-domain protein with an extracellular loop between the second and the third transmembrane domains along with three glycosylation sites (Y). I− and H2O2 regulate the activity of the enzymatic complex, depending on their local relative concentration. When I− is present in adequate amounts, H2O2 is the limiting factor of the reaction and mediates the association between TPO and DUOX2. In thyroids with low intracellular I− content, I− instead exerts a stimulatory effect on H2O2 production. When I− is present in excess, H2O2 synthesis is blocked. Because I−-induced effects are inhibited by methimazole or propylthiouracil, this indicates that I− acts through oxidized species. B, In resting conditions, the complex TPO-DUOX2-DUOXA2 is kept inactive underneath the apical membrane. Cav-1 is required to keep DUOX2 quiescent, because it may interact with domains in the cytoplasmic region of the molecule that are putative sites for Cav-1 scaffolding domains (following a hypothesis that still needs to be proved). According to the most plausible explanation, Cav-1 would come off the enzyme complex after its incorporation into the apical membrane. The absence of Cav-1 is responsible for mislocalization and premature activation of the complex in the cytoplasm, creating potentially devastating consequences due to increased OS and/or lack of efficient antioxidant mechanisms naturally activated when I− organification and coupling reactions normally occur in apical microvilli.
For cells to be protected from life-long, cytotoxic free radical attacks, all oxidizing and coupling reactions must occur outside the cell, at the interface with the colloid, in a special biochemical environment that was called the thyroxisome by Song et al. (178). The thyroxisome cannot be considered as a true morphological entity or a cellular organelle because it has never been visualized as such. Rather, it should be viewed as a biochemical concept (that still needs to be further worked out) that helps to understand how TH biosynthesis occurs safely despite an a priori toxic environment. A harmless biochemical milieu is created in microvilli when the TPO-like extracellular N-terminal domain of DUOX interacts with TPO (178, 181, 182). How this specialized environment remains harmless needs to be further clarified, even though recent data, such as those reported below, brought new knowledge in this matter. Thus, both proteins are thought to be already closely related during their posttranslational maturation and transport across the Golgi apparatus, but in the absence of TSH, they are kept in an inactive state just below the apical membrane. One may admit that this assumption is still rather weak because it is based only on indirect arguments about location and activation of TPO and DUOX (84, 181–184). Under TSH-induced calcium signaling, DUOX is fully activated to form a competent H2O2-generating system after its integration into the apical membrane (178, 180–182).
In addition to the TPO-DUOX couple, other forms of autoregulation are required to make TH biosynthesis operate as a safe process. For instance, H2O2 favors the association between TPO and DUOX in human thyrocytes (181); however, when produced in the absence of I−, H2O2 instead decreases the activity of both TPO and DUOX (182). TPO protects DUOX activity against the inhibitory effects of H2O2. This can be ascribed to its catalase-like activity, which requires a close interaction with the N-terminal domain of DUOX2 (182). DUOXs also need partners to work properly. One such partner is the thioredoxin-related protein EF-hand binding protein 1 (EFP1), which interacts with the intracellular region of DUOX1 and DUOX2 that contains two EF-hand domains. EFP1 may also be involved in the destruction of H2O2 when H2O2 is not used as an oxidizing substance in TH synthesis and leaks away from the site of its production (177). Two other partners are the DUOX-specific quality controlling maturation proteins DUOXA1 and DUOXA2, which are required in overcoming the ER retention of DUOX enzymes (98, 185). The critical role of the DUOX2/DUOXA2 complex in hormonogenesis was confirmed recently by results showing that loss-of-function mutations in DUOX2 and DUOXA2 cause congenital hypothyroidism (100, 186–188). Further support for this role comes from a mouse model of inactivated Duoxa1 and Duoxa2, where DUOXA deficiency led to intracellular retention of DUOX subunits and the loss of calcium-inducible H2O2 release (189). Stable DUOX/DUOXA complexes at the apical membrane are necessary for proper H2O2 generation. Less stable combinations (such as DUOX2/DUOXA1) do not form complexes at the plasma membrane and produce O2·− instead of H2O2, which indicates that DUOX activators not only promote DUOX maturation but also take part directly in H2O2 production (185). Caveolin-1 (Cav-1) is another important member of the multiprotein complex involved in TH biosynthesis (183). Cav-1 is one of three caveolar proteins that are important components of caveolae (190). Caveolae are invaginated microdomains of the plasma membrane that influence endocytosis, transcytosis, lipid metabolism, signal transduction pathways, and cell proliferation in many eukaryotic cells (adipocytes, endothelial and epithelial cells, myocytes, and fibroblasts) (reviewed in Ref. 191). In the thyroid, caveolae are required for TH synthesis and homeostasis, as recently demonstrated (183, 192). For instance, an increased apoptotic rate was reported in the thyroid of Cav-1-knockout mice (183). In these mice, the organification process is abnormal because the synthesis enzymatic complex is mislocalized to, and active in, the cytoplasm instead of the apical pole of the cell (183). There are reasons to believe that this mislocalization is responsible for the increased apoptotic rate observed in this knockout model, because apoptosis is associated with high oxidative stress (OS). High OS is a consequence of the increased expression and activity of H2O2-generating DUOX, which occurs when Cav-1-induced DUOX inhibition is insufficient. Thus, in the absence of Cav-1, DUOXs are localized to the cytosol, where they are abnormally active. It has been proposed, but not yet formally proven, that the Cav-1-induced inhibition of DUOX is caused by the presence of three amino acid sequences that may act as putative binding sites for the Cav-1 scaffolding domain (183, 193). This negative regulation is reminiscent of the role played by caveolar proteins in the negative regulation of signal transduction pathways and cell proliferation (191). Accordingly, as recently demonstrated in autonomous adenomas, in a model of adenosine A2-receptor transgenic mice, and in Cav-1-knockout mice, reciprocal negative regulation occurs between TSH/cAMP-mediated proliferation and Cav-1 expression via mRNA destabilization (183, 192). The increased proliferation rate of thyrocytes in Cav-1-knockout mice provides further evidence for the antimitogenic role of Cav-1 in the thyroid. This would explain why the overall size and function of the Cav-1-knockout thyroid remain unchanged, because the OS-mediated increase in the apoptotic rate is likely compensated by the increase in the proliferation rate in the Cav-1-knockout mice. A pathological mechanism similar to that observed in Cav-1-knockout mice was described in a hypothyroid PDS patient with a large goiter (184). PDS is an autosomal recessive disorder caused by mutations in the SLC26A4 gene. The disease is characterized by sensorineural deafness, diffuse goiter, and a positive perchlorate discharge test (reviewed in Ref. 59). In this case, iodination processes were observed to also occur in the cytosol rather than in the apical membrane, suggesting that the disturbance of the TPO-DUOX molecular complex and its shift into the cytoplasm were, at least partly, responsible for the thyroid tissue destruction that is sometimes observed in PDS patients (184). This pathological mechanism cannot be generalized to all PDS patients because the phenotypes of PDS are highly variable and depend on nutritional iodine intake. Thus, in countries with a high iodine intake, PDS patients are usually euthyroid. This is in contrast to patients from iodine-deficient regions who may present with overt congenital hypothyroidism (59).
Thus, TPO and DUOX, the two main proteins involved in TH biosynthesis, interact with each other and are activated in microvilli under TSH stimulation. For TH biosynthesis occurring in safe conditions, additional proteins are required. How fine-tuned interactions physiologically occur between the different actors still requires further investigations, which opens new perspectives in this field of research. This sophisticated biochemical setting is responsible for the fact that H2O2 production is tightly controlled, because H2O2 is readily consumed during TPO-induced I− oxidation, tyrosyl iodination, and iodophenoxy-ether coupling, whereas any H2O2 intracellular leakage is strictly contained (178). Consequently, H2O2 can be produced in the micromolar range similar to that in stimulated leukocytes (103, 194) without being lethal to cells. The local H2O2 production in a restricted area of the cell (i.e. the apical membrane) is reminiscent of the recently proposed model of compartmentalized redox signaling, which infers that, in response to the activation of receptor tyrosine kinases, H2O2 does not diffuse freely across the cytoplasm, but instead localizes to microdomains that are located close to sites of production (plasma membrane, endosomal compartment, and ER membranes) (195).
D. ROS: key factors in thyroid function and growth
Increasing evidence indicates that ROS other than H2O2, such as OH· and O2·−, are produced during TH synthesis, but in very small amounts (130, 179, 196, 197). These ROS do not harm thyroid cells, but they do increase the level of basal DNA damage in thyrocytes, which may increase the spontaneous mutation rate with functional and/or tumoral consequences (198–203), as explained later in this review. In fact, beyond the protection provided by the aforementioned regulatory mechanisms operating during TH biosynthesis, toxicity due to ROS leakage is kept under tight control by many potent other intracellular antioxidant defense systems, including catalases, superoxide dismutase, peroxiredoxins, and glutathione peroxidases (178, 179, 203–207). Peroxiredoxins degrade H2O2 and reduce peroxidized membrane phospholipids. They are associated with enzymes of the thioredoxin reductase family, which are selenoenzymes as is glutathione peroxidase. This emphasizes the role of selenium in the redox protection of the thyroid, its beneficial action during the course of thyroid autoimmune diseases, and its involvement in myxoedematous cretinism in conditions of combined iodine and selenium deficiency (208) (reviewed in Refs. 179, 209, and 210). It was reported recently that the inactivation of mouse thyroid selenoproteins using a genetic loss-of-function approach was not associated with severe morphological or functional alterations. This suggests that selenoproteins are not essential for thyroid cell integrity, perhaps because other antioxidant enzymes can compensate for their activity when they are absent or inactivated (207).
ROS mediate a variety of signal transduction pathways (reviewed in Ref. 211). In accordance with this, a recent report demonstrated that ROS levels corresponding to the physiological oxidative load of the cell are required to safeguard the function and integrity of the thyroid (198). In goitrous thyrocytes, the oxidative load is higher than under basal conditions, resulting in OS, likely because of increased H2O2 production or impaired consumption when iodide is lacking and/or when TPO is blocked (212). However, OS under these conditions is probably neutralized by antioxidant defenses and is thus harmless. Likewise, OS is required for cell division because goiter formation is significantly hampered when OS is blocked by N-acetylcysteine, a potent antioxidant (212). These results are not entirely surprising, because they are in accordance with the growth-promoting effects of ROS (213). For instance, ROS, including H2O2, stimulate the proliferation of many cell types through various mechanisms that affect growth receptor tyrosine kinase pathways, the MAPK pathway, the phosphoinositide-3-kinase pathway (PI3K/Akt), several cyclin-dependent kinases, and the nuclear factor κB pathway, making them important mediators of thyroid carcinogenesis (213–221). These observations support a role for H2O2 in the high frequency of thyroid tumors and microcarcinomas, particularly under conditions of antioxidant deficiency (201–203). They are also in agreement with a study that showed that the high rate of mutations in the thyroid is due to constitutive elevated H2O2 levels (202) and with other studies showing that large amounts of H2O2 cause RET/PTC1 rearrangement and DNA double-strand and single-strand breaks in thyroid cells (199, 200). In addition, imbalances in intracellular redox systems have been linked to the molecular mechanisms of tumorigenesis in patients with thyroid cancers (222). Although the link between OS and thyroid carcinogenesis still needs to be worked out, some explanations are now put forward to explain why H2O2 becomes a carcinogen when not adequately contained. Thus, DUOX protein expression is generally normal or slightly increased in thyroid cancers, whereas NIS, TPO, and pendrin protein expression is decreased, mislocalized, or even sometimes absent, which implies less H2O2 utilization and therefore longer exposure to H2O2 (54, 223–225). This longer exposure, even though mild, may have long-term consequences considering the mean 8.5-yr life span of thyroid cells (226). In addition, it is not unusual to observe aberrant localization of the H2O2-DUOX-generating system, for instance in the cytoplasm, where antioxidant defenses are again inadequate to deal with increased OS (223, 225). Under such uncontrolled conditions, moderately high levels of H2O2 that are not enough to kill the cell (as seen in the aforementioned model of Cav-1-knockout mice and in PDS patients) might, however, diffuse freely into the nucleus and provoke DNA damage. Thyroid cancer tissues are also infiltrated by inflammatory cells that are sources of ROS, which may promote tumor growth by stimulating the major oncogene-initiated signaling pathways (227–229). TSH-stimulated H2O2 production could be an additional explanation for the carcinogenic properties of H2O2, because cancerous thyroid cells still express the TSH receptor (224). This may explain why high TSH plasma levels are associated with an increased risk of malignancy in thyroid nodules (230, 231).
OS is also highly toxic in experimental models of iodine-induced thyroid involution, Th1 cytokine-induced cytotoxicity, and autoimmune thyroiditis (232–236). Iodine treatment of hyperplastic thyrocytes results in their impairment, as shown by the increase in necrosis/apoptosis, the presence of many cellular fragments, and the massive interstitial inflammatory reaction (234, 237–242). In these cases, iodine-induced cytotoxicity is due to a more aggressive attack associated with a massive release of very toxic free radicals (OH·, O2·−, ONOO−, and iodocompounds) in addition to H2O2, most of which are also produced by infiltrating inflammatory cells (196, 234). Nevertheless, cell destruction is transient, and the thyroid always returns to its original aspect. This is not the case in individuals genetically susceptible to developing autoimmune diseases such as Hashimoto's thyroiditis. In experimental models of nonobese diabetic mice for instance, the acute inflammation that occurs after iodine administration nearly always evolves toward long-term destructive autoimmune thyroiditis (236, 240, 243–246). A clinical connection can be easily made with these experimental data, because human thyroid autoimmune diseases have been shown to be associated with increased OS (247–250). Taken together, the data indicate that, although thyroid antioxidant systems are ideally adapted for the detoxification of H2O2 and other ROS produced under normal conditions or even in excess in goiters, it is likely that they are overwhelmed under conditions of high free radical release, which would lead to neoplastic and autoimmune diseases.
IV. The Angiofollicular Unit: From Concept to Demonstration
A. The role of the local microvasculature in the maintenance of endocrine function
The complex functional and morphological multistep changes that occur during goitrogenesis have been described in great detail in the past (196, 201, 240, 243, 251–257). It is, of course, widely accepted that TSH stimulates thyrocytes, as shown by the increases in hypertrophy, hyperplasia, and expression of NIS, TPO, and Tg (251–253, 255, 258–260). It has also been known for decades that epithelial changes accompany increased thyroid blood flow and vascular expansion (253–255, 261–267). The microvascular expansion is even essential for goitrogenesis, as evidenced by the inhibition of goiter formation when three important angiogenic growth factor signaling pathways [vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and angiopoietins] are blocked (268).
Each follicle is enclosed by an independent basket-like plexus of anastomotic fenestrated capillaries that covers 20–50% of its surface (269). After ID or goitrogen treatment, each vascular nest increases in size as a result of cell proliferation and capillary fusion to cover up to 70–80% of the follicle surface area (269). This vascular expansion can be rather impressive; for example, a 250% increase in thyroid blood flow was observed in rats rendered iodine deficient for 2 wk (270), whereas in humans, a 44% decrease in iodine intake resulted in a 49% increase in thyroid blood flow (271). By contrast, thyroid blood flow falls promptly in cases of massive iodine supply (potentially triggered by endothelins), which is necessary to prevent excessive TH synthesis and to protect the gland against free radicals (196, 239, 253, 272, 273). Considering the emerging role of the microvasculature in thyroid homeostasis, it is becoming clear that the morphological and functional unit of the thyroid is not restricted solely to the follicular cells of the follicle but involve also the surrounding capillaries that, along with thyrocytes, form the AFU (258–260, 274) (Fig. 4).
Figure 4.

The concept of AFUs in the thyroid gland. The thyroid gland is composed of many AFUs. They are individualized entities with their own genotypic and phenotypic assets under the local control of a host of autocrine and paracrine factors, as well as exerted by TSH. A and B, Each unit is composed of thyrocytes gathered in a follicle that is surrounded by capillaries made of endothelial cells and pericytes independent from their neighbors. In unstimulated conditions (i.e. in conditions of adequate iodine supply), the vascular bed covers about 20–50% of the follicle surface (this AFU corresponds to the so-called active follicle in Fig. 2). C and D, As soon as the intracellular iodide (I−) content drops, the thyrocytes react immediately by secreting VEGF, which triggers an early and modest TSH-independent microvascular reaction. If the increased local clearance of I− associated with this compensatory mechanism is not enough to preserve TH synthesis, TSH plasma levels start rising. Upon TSH stimulation, a more robust but always tightly controlled microvascular reaction occurs. E and F, By the end of the process, the area occupied by the expanded vascular bed may cover up to 80% of the follicle surface made of hyperplastic and hypertrophic thyrocytes. Scale bar, 10 μm. Arrows indicate microvessels.
Such interconnected changes between epithelial and endothelial compartments are not unique to the thyroid. In the pancreatic islets of Langerhans, for example, dynamic interactions occur between blood vessels and ß-cells to improve insulin secretion and ß-cell proliferation (275, 276). Islet blood flow is for instance regulated by specific metabolic factors such as glucose that almost doubles it (277, 278), but also by less specific factors such as ATP signals through A1 adenosine receptors as well as neuronal and vasoactive factors (279, 280). In all cases, these changes occur independently of blood flow in the rest of the pancreas. As conceptualized in the case of the thyroid where capillaries close to thyrocytes behave differently from one AFU to another and from those irrigating the adjacent parathyroids, pancreatic islets are also arranged so that regions containing highly vascularized ß-cells are distinct from those containing other cell types (281). The pituitary gland is another interesting example where the microvasculature is involved in growth, development, and function (282), and there is evidence that abnormal microvascular development or altered angiogenesis is associated with some forms of central diabetes insipidus and pituitary adenomas (283, 284). Also bearing in mind the peculiar vascular architecture in the ovaries (285–287) and in the adrenals (288, 289), it appears that the involvement of the microvasculature is part of a more global system in endocrine glands where the local vasculature cooperates with endocrine cells to preserve their function and maintain homeostasis.
B. ID: the trigger of an immediate TSH-independent microvascular response
The question of how epithelial and endothelial growth is coordinated in the thyroid has been addressed extensively in the past. The involvement of TSH, although obvious at first sight (290, 291), was challenged quickly (270, 292, 293), and additional hypotheses, including the neuronal hypothesis and the growth and vasoactive factor hypothesis were proposed (274, 294). According to the neuronal hypothesis, thyroid blood flow and TH secretion are influenced by adrenergic, cholinergic, and peptidergic stimuli (270, 292, 293, 295–301), whereas the growth and vasoactive factor hypothesis implies the involvement of paracrine/autocrine factors of epithelial, neuronal, interstitial, and endothelial origin (258–260, 268, 272–274, 302–306). Additionally, the vascular status of the thyroid gland may also be influenced by estrogens (307). Again, this kind of complex regulation is not restricted solely to the thyroid because similar complex interactions between vasodilators and vasoconstrictors, gastrointestinal peptides, and the autonomic nervous system were reported to control islet blood flow in the pancreas (308).
In addition to not being the sole participant in this complex interplay, it is known that TSH acts somewhat late in response to ID, once intrathyroidal reserves of iodine become insufficient to maintain steady TH production (more than a week in animal models of ID-induced goiter) (251–255, 258, 267, 273, 302). Thus, to cope with daily variations in iodine supply, thyrocytes use various TSH-independent intra- and extrathyroidal self-regulatory mechanisms including the stimulation of I− trapping by NIS, the preferential synthesis of T3 over T4, the recycling of intracellular I− by DEHAL1, the peripheral conversion of T4 into T3 (309–311), and the rapid inflation of its own microcirculation (251–255, 258–260, 267, 269–271, 273, 292, 293, 299, 302).
These changes in the endothelial compartment are the earliest morphological events observed in response to ID and occur a few days before changes are visible in epithelial cells (252, 254, 267). The vascular reaction occurs in two waves. The delayed phase is TSH dependent, starts at least a week after goiter onset, and is accompanied by the hypertrophy and hyperplasia of follicular cells (253–255, 261–267). By contrast, the early phase is TSH independent and, besides endothelial cells, also involves pericytes, as evidenced by the increased expression of the proteoglycan neuron-glial antigen 2, a marker of pericyte activation (263, 267). Because this early phase takes place as an immediate response to the drop in intrathyroid I− content, the intracellular I− shortage has been suggested to initiate the changes that affect microvessels, even though a role for the organified form of I− or for other iodinated molecules cannot be ruled out (267). A link between I− shortage and the quick release of VEGF from thyroid epithelial cells was established recently in vitro (49) and in experiments where bevacizumab (an antibody against VEGF) was administered to iodine-deficient animals (267). In contrast to animals receiving control IgGs, this antibody prevented an increase in thyroid blood flow and blocked an increase in the relative volumes of vessels.
C. ID-induced microvascular response: which molecular mechanism?
The molecular links between ID and TSH-independent VEGF-driven vascular reactions were dissected recently in an in vitro model of thyrocytes acutely deprived of iodine (49). When the intracellular 127I content drops sharply in thyrocytes incubated without I− for 4 h or treated with perchlorate, an immediate increase in hypoxia-inducible factor-1α (HIF-1α) and VEGF expression occurs along with a concomitant surge in intracytoplasmic ROS. Thus, when I− is no longer available or when its transport is blocked, its intracellular stocks drop rapidly, which causes an increase in ROS and the stabilization of HIF-1α. In turn, HIF-1α heterodimerizes with HIF-1ß, which is constitutively present in the cell. After binding to the promoter region of the VEGF gene, the HIF-1α/HIF-1ß heterodimer initiates transcription. As a result, VEGF mRNA and protein expression increases. VEGF is then secreted and activates adjacent pericytes and endothelial cells, which leads to microvascular expansion and increased blood flow (Fig. 5).
Figure 5.

Proposed mechanism of early ID-induced TSH-independent microvascular reaction in thyroid AFUs. A, TSH-independent microvascular activation. In unstimulated AFUs (left), capillaries close to thyrocytes are fenestrated and surrounded by pericytes. Endothelial cells and pericytes are separated only by fused basal membranes. If iodide (I−) supply drops, independently from any stimulation by TSH (right), VEGF expression increases in thyrocytes, which induces a microvascular activation, implying, as initial step, the detachment of chondroitin sulfate proteoglycan 4 (NG2)-expressing pericytes from endothelial cells. B, Molecular mechanisms. When I−supply is altered or when its transport is blocked, the thyrocyte intracellular content drops rapidly, thereby increasing ROS, which leads to the stabilization of HIF-1α. The exact nature of ROS and where they are produced still deserve further investigation. HIF-1α heterodimerizes with HIF-1β, which is constitutively present in the cell. The HIF-1α/HIF-1β heterodimer, after binding to the hypoxia response element (HRE) site of the promoter region of the VEGF gene, turns on its transcription. As a result, VEGF mRNA and protein expression increases. VEGF is then secreted and activates adjacent endothelial cells and pericytes, thereby leading to microvascular activation, increased blood flow, and proliferation of endothelial cells and, in turn, to increased local clearance of I−. When this and other protective measures become insufficient to safeguard TH synthesis, TSH enters into the action, in coordination with many other locally generated growth and vascular regulatory factors.
This pathway is transitory and is not associated with abnormal tissue growth, which implies tight control. By contrast, in malignant cells, the pattern of intracellular events in response to ID is somewhat different. ID-induced VEGF expression is stronger and is not controlled over time, which implies that the intracellular pathways are different from those activated in nonmalignant cells (50). Thus, although the angiogenic stimulus should be viewed as a physiological adaptation of the gland to optimize its functional yield in normal cells (49, 267), ID is associated with a less controlled angiogenic switch in malignant cells (50). This notion of microvascular control in normal tissues vs. malignant cells was also reinforced by the recent discovery of some components of the highly conserved cell signaling system Delta-Notch pathway in the thyroid (312). Beyond VEGF, angiogenic stimuli are monitored by many other controlling pathways, including the Delta-Notch pathway (313–316). This pathway is activated by VEGF to prevent excessive vessel proliferation. In humans, four Notch receptors (1–4), and five ligands [Delta-like ligand (DLL)-1, -3, and -4 and Jagged-1 and -2] were identified (316, 317). Notch1, -2, and -3 were reported in a study that suggested a role for the Notch pathway in thyroid cell differentiation (318), whereas Notch4 and DLL4 expression was reported to be lower in normal thyroids than in nonmalignant hyperplastic and malignant tissues (312). Only capillaries of Graves' disease samples were positive for DLL4, suggesting that DLL4 plays a role as a downstream controller of VEGF in hyperplastic nonmalignant tissues to keep the expanding microcirculation under control. Thus, in contrast to carcinomas where coherent and coordinated cross talk between cells of different tissue compartments is altered and leads to anarchic vascular growth, such cross talk is kept intact under nonmalignant conditions.
D. AFUs: a heterogeneous population of three-dimensional structures that protects the thyroid gland against functional failure
Although AFUs are perfectly adapted to respond to goitrogen stimuli in a synchronized manner, they do not work as a uniform population, but instead as heterogeneous clusters of distinct units characterized by particular growth and functional properties. This functional and morphological heterogeneity is a hallmark of the thyroid that helps the gland to adapt to changing physiological conditions and explains its propensity to form nodules.
In a model of elderly mice, characterized by marked morphological heterogeneity, thyroid capillaries are closely related to the functional status of the follicle upon which they depend. In fact, two types of follicles coexist in the thyroid of aged animals. Based on morphological and autoradiographic criteria, they are either active (hot) or inactive (cold) (258, 319–321). A correlation was drawn between the degree of activity of each follicle and the expansion of its respective microcirculation, which was significantly larger around active (hot) follicles than around inactive (cold) ones (258). When applied to humans, using tissue sections of multinodular goiters, the follicles were also sorted out into active or inactive, based on morphological criteria. In contrast to richly vascularized active follicles, inactive follicles, such as those in old mice, were surrounded by a hypotrophic microcirculation (259). Beyond epithelial function, vascular changes are also more marked around proliferative epithelial cells, as demonstrated in a model of transgenic hyperthyroid mice expressing the A2a adenosine receptor (Tg-A2aR) and in human Graves' disease (260) where the number of proliferating cell nuclear antigen-positive cells was highest in the most vascularized areas.
V. Angiofollicular Heterogeneity, Nodulogenesis, and Multinodular Goiter: Why and How?
The limitations of adaptive autoregulatory mechanisms in the thyroid gland in the face of a widely fluctuating iodine supply may lead to the development of goiters. Goiter formation should be considered as an adaptive process rather than pathological, as long as thyroid function is preserved and even though they may sometimes occur along with risks of local compression (35, 255). Throughout this process, not only TSH, but also many locally produced growth, vasoactive, and angiogenic factors [IGF-I, epidermal growth factor (EGF), FGF, endothelin-1, nitric oxide, VEGF, and others] are involved in the growth and replication of epithelial and endothelial cells (Fig. 2) (reviewed in Refs. 274, 294, 303, and 306). Of course, ID is not the unique etiological factor of goiters, because many individual genetic determinants and environmental factors (cigarette smoking, nutritional goitrogens, radiations, gender, age, and body mass index) are other well-known causative factors (256, 257, 322).
Starting from diffuse hyperplasia, nontoxic goiters always tend to become heterogeneous over time and are gradually transformed into multinodular goiters (MNGs) (201, 256, 257, 322). The tissue heterogeneity may be explained by two theories (perhaps complementary): the most studied and accepted polyclonal and mutation theory and the more recently proposed and less studied stem cell theory.
A. The polyclonal and mutation theory
The heterogeneity of MNG is evidenced in humans in 123I and 99technetium scans, which show a complex mixture of normal, hyperfunctioning (hot), and hypofunctioning (cold) nodules in variable proportions, the functional status (eu-, hyper-, or hypothyroidism) of a given individual being determined by the overall functional balance of these nodules (257). At the microscopic level, epithelial cells of each AFU also show heterogeneity in growth, endocytic responses, and Tg synthesis (319, 323–329). For instance, only a small fraction of thyrocytes reacts with TSH via the formation of endocytic colloid droplets (324, 325). In addition, the production of Tg varies considerably between cells; the colloid content of each follicle depends on the cells producing the largest amounts of Tg (72, 319, 323). The considerable tissue heterogeneity is therefore the result of the phenotypic diversity of thyroid cells. This phenotypic diversity may be explained by the polyclonality theory, which postulates that for a given stimulus, cells react differently from their neighbors depending on their intrinsic properties, which reflect their polyclonal origin (330–336). The principle of polyclonality can be understood at the cellular level. Thus, some cells, even within the same AFU, could for instance enter the cell cycle more quickly than others in response to growth stimuli. But the same principle can also be understood at the level of the organ as a whole. Accordingly, a rising nodule may result from the coordinated replication of a cluster of follicles characterized by the same growth properties, which may differ from those of their neighbors. When this trait is amplified, the process may eventually give rise to nodules having their own specific morphological and functional properties (201, 255–257, 260, 322, 337). This is supported by a study that reported that the thyroid epithelium exhibits large embryonal patch size. According to this paper, cellular monoclonal zones that react to growth-regulating signals would be relatively large instead of being restricted as initially thought (338). Thus, when it is applied to an organ made of many large monoclonal cell patches, but with different somatic genetic properties, the principle of polyclonality could account for organ heterogeneity and explain the development of macroscopic nodules distributed among otherwise morphologically and functionally quiescent areas. These resting zones are composed of large AFUs lined with flat epithelium and filled with dense colloid-containing globules of compact Tg (see Section III.B and Fig. 2) (157, 258, 259, 274). Their adjacent vasculature is hypotrophic. In experimental models, they are considered to act as a functional reserve that works as a rescue system when needed. Of note, proteins of the apical pole (TPO, pendrin, and DUOX) are absent in these hypofunctioning follicles, as is TgI. Upon TSH stimulation, the progressive dilution of dense Tg globules into soluble Tg is accompanied by the reexpression of proteins involved in TH synthesis (TPO, DUOX, and pendrin) and the proliferation of endothelial cells, as demonstrated by the increased number of [3H]thymidine-labeled endothelial nuclei (157, 258, 259). This morphological and functional awakening is necessarily followed by a stabilization phase mediated by locally generated regulatory factors such as TGF-ß that, on the one hand, limit cell growth and function but, on the other hand, facilitate the deposition of connective tissue, which may aggravate tissue heterogeneity (208, 303, 333, 339, 340). Over time, nodules of different size and aspect develop. Sometimes they degenerate and a cyst is formed with traces of hemorrhage and calcium deposits, and it is not unusual to also observe areas of focal lymphocytic infiltration. As in cold hypofunctioning nontransformed nodules, there is also a loss of TgI in thyroid cancers, but the causative defects differ. In cold hypofunctioning nontransformed nodules, NIS and TSH receptor expression persists, whereas the apical markers (pendrin, DUOX, and TPO) vanish (224). In cancers, NIS expression either disappears or increases in association with a mislocalization of the protein that is no longer retained in the basolateral membrane, which suggests that the decrease in I− uptake in thyroid carcinomas is due instead to alterations in NIS trafficking (reviewed in Ref. 54). The presence of apical markers depends on the type of tumor. For instance, pendrin and TPO are still observed in follicular carcinomas, in contrast to papillary carcinomas where they are undetectable. This suggests that the defect in Tg iodination has different causes in nontransformed and transformed tissues and in different types of carcinomas. Accordingly, the anti-TgI antibody (159) was shown to help the diagnosis of thyroid carcinomas, especially when the differential diagnosis with nontransformed follicular adenomas is difficult (224).
During tissue transformation, some nodules may acquire growth and functional advantages, as in autonomously functioning thyroid nodules. This is likely to be associated with an increased number of mutation events resulting from increased cell proliferation and functional activity (reviewed in Refs. 201, 256, 257, and 322). As already mentioned, during thyroid hyperplasia, DNA damage occurs because of increased H2O2 production, which results in a higher mutation load, and H2O2-associated mutagenesis is now a well-established fact (178, 199, 200, 202, 203, 213). Some of these mutations (ras mutations or those in the RAS/RAF/MEK/ERK/MAP cascade) confer a growth advantage, as is sometimes seen in cold thyroid nodules, whereas others (TSHr and Gsα mutations) rather favor both cell growth and function, as seen in autonomously functioning thyroid nodules, because they result in constitutive activation of the cAMP cascade (201, 256, 257, 341). These small clones with activating mutations will proliferate further irrespective of intra- and extrathyroid mechanisms (e.g. TSH) and even after cessation of the initial stimulatory stimulus (for example, ID). In addition, some defects in genes directly involved in thyroid physiology and TH synthesis (Tg, TPO, NIS, TSH receptor, PDS, and DEHAL1) will also predispose the thyroid to the development of goiter, especially in areas of borderline or insufficient iodine supply (reviewed in Ref. 201).
B. The stem cell theory
Currently, the most widely accepted theory of thyroid tissue heterogeneity, ranging from adenomas to MNGs and finally carcinomas, is based on the notion that this is caused by multiple molecular aberrations in well-differentiated thyrocytes, including epigenetic changes, profound alterations in chromosomes, and dysregulated signaling and regulatory pathways. An alternative, and perhaps complementary, explanation for thyroid heterogeneity was presented recently using the concept of adult stem cells (342–348). Adult stem cells are undifferentiated, quiescent, or slow cycling cells found in differentiated tissues (349). In addition to colon, skin, pancreas, liver, and brain, they have been reported in the human thyroid gland (342–345). Through asymmetric cell division, adult stem cells are capable of making identical copies of themselves, resulting in one self-copy and one precursor or progenitor cell that divides further, eventually giving rise to differentiated cells. Adult stem cells are characterized by their resistance to growth stimulation, which is explained by their close interactions with niche cells (reviewed in Refs. 350 and 351). These cells protect stem cells from uncontrolled differentiation stimuli, apoptotic stimuli, and excessive proliferation. Under conditions where strict niche cell-induced control is reduced, stem cells gradually escape growth control and start dividing. Some progenitor cells, under the influence of locally produced growth factors, may then proliferate faster than the surrounding differentiated thyrocytes to form nodules or hyperplastic lesions. If progenitor cells are not fully differentiated, they could serve as the starting point of cold nonfunctioning nodules (347). A paper by Lan et al. (344) in 2007 indicated that thyroid adult stem cells can be isolated directly from primary thyroid cultures as three-dimensional spheres (called thyrospheres) in a medium enriched with EGF and FGF, but only under serum-free medium conditions and in the absence of TSH (i.e. in conditions that reflect differentiated cell apoptosis). When isolated from these nonadherent, three-dimensional spheres, these stem cells can then differentiate into thyrocytes (with the ability to express PAX8, Tg, NIS, TSHr, and TPO mRNA and to show TSH-dependent 125I uptake) when they are incubated into serum-enriched medium containing TSH, even though no TH secretion was found in this model (344) or in models of mouse embryonic stem cells (352, 353). Whether these stem cell-derived thyrocytes can reconstitute functional AFUs is currently unknown. Recently, thyroid cancerous stem cells derived from mutated adult stem cells or their progenitor cells were reported in papillary, follicular, and undifferentiated thyroid cancers (12, 347, 348, 354–357). Their identification is important because they could account for the aggressiveness and recurrence of undifferentiated carcinomas and for their resistance to radioiodine treatment and conventional chemo and radiotherapy (12, 347, 348). Further investigations are necessary to assemble into one coherent model current knowledge about the events affecting differentiated thyroid cells and stem cells.
VI. Concluding Remarks
The thyrocyte is a specialized cell that adapts itself continuously to fulfill its main function, that is, TH synthesis, a potentially hazardous biochemical reaction, while optimizing the uptake of an essential trace element, iodine. Oxidizing and coupling reactions occur at the interface with the colloid and are regulated in such a way that H2O2 and ROS production are tightly controlled. ROS are more than by-products of aerobic respiration; they are key players in thyroid function, and thyrocytes are well equipped to deal with ROS by virtue of their potent detoxifying systems. When the synthesis complex is altered, oxidation reactions start to occur in the cytoplasm with devastating consequences, such as morphological and functional breakdown, which engender disease processes, including those of an autoimmune or neoplastic nature.
A crucial function that thyrocytes must perform on a daily basis is the constant incorporation of iodine into Tg. To face daily variations in iodine supply, each thyrocyte uses various intra- and extrathyroidal TSH-independent self-regulatory mechanisms, including the regulation of its own microvasculature. In this context, the structural and functional unit of the thyroid includes the adjacent microvasculature in the three-dimensional structure called AFU. According to this concept, the vascular plasticity acts in conjunction with the multiple homeostatic processes that control TH synthesis. ID-associated vascular responses occur in two successive phases: the first early phase involves a TSH-independent ROS/HIF/VEGF pathway, whereas the second delayed phase starts when TSH plasma levels rise; however, the nature of the intracellular sensor that triggers the quick vascular remodeling in reaction to ID is still unknown. It is worth investigating the identity of this trigger, because AFU must tightly control the adjacent vascular response at all times. It is easy to understand how strategic this control must be for the thyroid to avoid anarchic and uncontrolled growth of the vascular compartment. Such strict control of the endothelial compartment by epithelial cells, irrespective of TSH, is an inherent property of healthy AFU and could explain why so many in situ microcancers are quiescent in the thyroid gland. Recent evidence suggests that ID regulates vascular changes in thyroid carcinomas through VEGF, but in a way different from that in nonmalignant tissues or at least using an alternative sequence of events. This suggests that altered autocrine-paracrine signals in unstructured AFUs facing ID may induce an angiogenic switch that leads to uncontrolled cellular proliferation.
When protective measures become insufficient for thyrocytes to sustain TH synthesis, TSH leaps into action along with many other locally generated growth and vascular regulatory factors in very dense and complex paracrine networks, resulting in the emergence of a goiter. Although goiter growth is controlled in time and space, it is highly heterogeneous from inception because of the polyclonal origin of the cells. Evidence suggests that the concept of polyclonality accounts for organ heterogeneity and the growth of nodules distributed among morphologically and functionally quiescent areas. These zones correspond to tissue reserves that become easily accessible upon TSH stimulation. This morphological and functional awakening is always followed by a stabilization phase attributable to locally generated regulatory factors, such as TGF-ß that, on one hand, limit cell growth and function but, on the other hand, facilitate the deposition of connective tissue, which may aggravate the heterogeneous character of the thyroid tissue.
To conclude, the thyroid gland has somehow succeeded during its phylogenic maturation in squaring the circle between iodine uptake, TH synthesis, and protection against ROS by setting up highly efficient structures, termed AFUs, which enable it to face its own functional exigencies. This morphological and functional integration warrants an adequate TH supply, at any time and whenever necessary.
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AFU
- Angiofollicular unit
- AHDS
- Allan-Herndon-Dudley syndrome
- Cav-1
- caveolin-1
- DEHAL1
- dehalogenase 1
- DIT
- di-iodotyrosine
- DLL
- Delta-like ligand
- DUOX1
- dual oxidase 1
- DUOXA2
- DUOX maturation factor 2
- EFP1
- EF-hand binding protein 1
- EGF
- epidermal growth factor
- ER
- endoplasmic reticulum
- FGF
- fibroblast growth factor
- HIF-1α
- hypoxia-inducible factor-1α
- ID
- iodine deficiency
- MCT8
- monocarboxylate transporter 8
- MIT
- mono-iodotyrosine
- MNG
- multinodular goiter
- NADPH
- reduced nicotinamide adenine dinucleotide phosphate
- NIS
- sodium-iodide symporter
- NOX
- adenine dinucleotide phosphate oxidase
- OS
- oxidative stress
- PDI
- protein disulfide isomerase
- PDS
- Pendred syndrome
- ROS
- reactive oxygen species
- SLC5A5
- soluble carrier 5A5
- Tg
- thyroglobulin
- Tgl
- antibody against iodinated Tg containing T4 at the N-terminal hormonogenic site
- TH
- thyroid hormone
- TPO
- thyroid peroxidase
- TR
- TH receptor
- VEGF
- vascular endothelial growth factor.
References
- 1. Boelaert K, Franklyn JA. 2005. Thyroid hormone in health and disease. J Endocrinol 187:1–15 [DOI] [PubMed] [Google Scholar]
- 2. Zimmermann MB. 2011. The role of iodine in human growth and development. Semin Cell Dev Biol 22:645–652 [DOI] [PubMed] [Google Scholar]
- 3. Zimmermann MB. 2009. Iodine deficiency. Endocr Rev 30:376–408 [DOI] [PubMed] [Google Scholar]
- 4. Courtois B. 1813. Découverte d'une substance nouvelle dans le Vareck. Ann Chim (Paris) 88:304–310 [Google Scholar]
- 5. Taurog A. 2004. Iodine. In: Martini L, ed. Encyclopedia of endocrine diseases. New York: Academic Press; 82–88 [Google Scholar]
- 6. Kendall E. 1914. The determination of iodine in connection with studies in thyroid activity. J Biol Chem 19:251–256 [Google Scholar]
- 7. Houston R. 1999. Iodine: physiology, dietary sources and requirements. In: Strain JJ, Caballero B, eds. Encyclopedia of human nutrition. London: Academic Press; 66–74 [Google Scholar]
- 8. Muramatsu Y, Wedepohl KH. 1998. The distribution of iodine in the earth's crust. Chem Geol 147:201–216 [Google Scholar]
- 9. Osterc A, Stibilj V, Raspor P. 2011. Iodine in the environment. In: Nriagu J, ed. Encyclopedia of environmental health. Amsterdam, London: Elsevier; 280–287 [Google Scholar]
- 10. Venturi S, Venturi M. 1999. Iodide, thyroid and stomach carcinogenesis: evolutionary story of a primitive antioxidant? Eur J Endocrinol 140:371–372 [DOI] [PubMed] [Google Scholar]
- 11. Küpper FC, Carpenter LJ, McFiggans GB, Palmer CJ, Waite TJ, Boneberg EM, Woitsch S, Weiller M, Abela R, Grolimund D, Potin P, Butler A, Luther GW, 3rd, Kroneck PM, Meyer-Klaucke W, Feiters MC. 2008. Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc Natl Acad Sci USA 105:6954–6958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin RY. 2011. Thyroid cancer stem cells. Nat Rev Endocrinol 7:609–616 [DOI] [PubMed] [Google Scholar]
- 13. Kluge B, Renault N, Rohr KB. 2005. Anatomical and molecular reinvestigation of lamprey endostyle development provides new insight into thyroid gland evolution. Dev Genes Evol 215:32–40 [DOI] [PubMed] [Google Scholar]
- 14. Müller W. 1873. Über die Hypobranchialrinne der Tunicaten und deren Vorhandenstein bei Amphioxus und den Cyclostomen. Jena Z Med Naturw 7:327–332 [Google Scholar]
- 15. Suzuki S, Kondo Y. 1973. Thyroidal morphogenesis and biosynthesis of thyroglobulin before and after metamorphasis in the lamprey, Lampetra reissneri. Gen Comp Endocrinol 21:451–460 [DOI] [PubMed] [Google Scholar]
- 16. Fujita H, Honma Y. 1969. Iodine metabolism of the endostyle of larval lampreys, Ammocoetes of Lampetra japonica. Electron microscopic autoradiography of 125-I. Z Zellforsch Mikrosk Anat 98:525–537 [DOI] [PubMed] [Google Scholar]
- 17. Wright GM, Filosa MF, Youson JH. 1978. Immunocytochemical localization of thyroglobulin in the endostyle of the anadromous sea lamprey, Petromyzon marinus L. Am J Anat 152:263–268 [DOI] [PubMed] [Google Scholar]
- 18. Wright GM, Filosa MF, Youson JH. 1978. Light and electron microscopic immunocytochemical localization of thyroglobulin in the thyroid gland of the anadromous sea lamprey, Petromyzon marinus L., during its upstream migration. Cell Tissue Res 187:473–478 [DOI] [PubMed] [Google Scholar]
- 19. Wright GM, Filosa MF, Youson JH. 1980. Immunocytochemical localization of thyroglobulin in the transforming endostyle of anadromous sea lampreys, Petromyzon marinus L., during metamorphosis. Gen Comp Endocrinol 42:187–194 [DOI] [PubMed] [Google Scholar]
- 20. Dunn AD. 1980. Properties of an iodinating enzyme in the ascidian endostyle. Gen Comp Endocrinol 40:484–493 [DOI] [PubMed] [Google Scholar]
- 21. Ogasawara M, Di Lauro R, Satoh N. 1999. Ascidian homologs of mammalian thyroid peroxidase genes are expressed in the thyroid-equivalent region of the endostyle. J Exp Zool 285:158–169 [DOI] [PubMed] [Google Scholar]
- 22. Ogasawara M. 2000. Overlapping expression of amphioxus homologs of the thyroid transcription factor-1 gene and thyroid peroxidase gene in the endostyle: insight into evolution of the thyroid gland. Dev Genes Evol 210:231–242 [DOI] [PubMed] [Google Scholar]
- 23. Ogasawara M, Shigetani Y, Suzuki S, Kuratani S, Satoh N. 2001. Expression of thyroid transcription factor-1 (TTF-1) gene in the ventral forebrain and endostyle of the agnathan vertebrate, Lampetra japonica. Genesis 30:51–58 [DOI] [PubMed] [Google Scholar]
- 24. McCauley DW, Bronner-Fraser M. 2002. Conservation of Pax gene expression in ectodermal placodes of the lamprey. Gene 287:129–139 [DOI] [PubMed] [Google Scholar]
- 25. Venturi S, Donati FM, Venturi A, Venturi M. 2000. Environmental iodine deficiency: a challenge to the evolution of terrestrial life? Thyroid 10:727–729 [DOI] [PubMed] [Google Scholar]
- 26. De Felice M, Di Lauro R. 2004. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev 25:722–746 [DOI] [PubMed] [Google Scholar]
- 27. De Felice M, Di Lauro R. 2011. Intrinsic and extrinsic factors in thyroid gland development: an update. Endocrinology 152:2948–2956 [DOI] [PubMed] [Google Scholar]
- 28. Dumont JE, Opitz R, Christophe D, Vassart G, Roger PP, Maenhaut C. 2008. The phylogeny, ontogeny, anatomy and regulation of the iodine metabolizing thyroid. In: DeGroot LJ, ed. Thyroid disease manager. South Dartmouth, MA: Endocrine Education, Inc.; 1–109 [Google Scholar]
- 29. Cocchi M, Venturi S. 2000. Iodide, antioxidant function and omega-6 and omega-3 fatty acids: a new hypothesis of a biochemical cooperation? Prog Nutr 2:15–19 [Google Scholar]
- 30. Venturi S. 2001. Is there a role for iodine in breast diseases? Breast 10:379–382 [DOI] [PubMed] [Google Scholar]
- 31. Smyth PP. 2003. Role of iodine in antioxidant defence in thyroid and breast disease. Biofactors 19:121–130 [DOI] [PubMed] [Google Scholar]
- 32. Burek CL, Talor MV. 2009. Environmental triggers of autoimmune thyroiditis. J Autoimmun 33:183–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zimmermann MB, Jooste PL, Pandav CS. 2008. Iodine-deficiency disorders. Lancet 372:1251–1262 [DOI] [PubMed] [Google Scholar]
- 34. Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. 2009. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol 296:C654–C662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunn JT. 1998. What's happening to our iodine? J Clin Endocrinol Metab 83:3398–3400 [DOI] [PubMed] [Google Scholar]
- 36. Dai G, Levy O, Carrasco N. 1996. Cloning and characterization of the thyroid iodide transporter. Nature 379:458–460 [DOI] [PubMed] [Google Scholar]
- 37. Smanik PA, Liu Q, Furminger TL, Ryu K, Xing S, Mazzaferri EL, Jhiang SM. 1996. Cloning of the human sodium lodide symporter. Biochem Biophys Res Commun 226:339–345 [DOI] [PubMed] [Google Scholar]
- 38. Bizhanova A, Kopp P. 2009. The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology 150:1084–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eskandari S, Loo DD, Dai G, Levy O, Wright EM, Carrasco N. 1997. Thyroid Na+/I− symporter. Mechanism, stoichiometry, and specificity. J Biol Chem 272:27230–27238 [DOI] [PubMed] [Google Scholar]
- 40. Dohán O, Portulano C, Basquin C, Reyna-Neyra A, Amzel LM, Carrasco N. 2007. The Na+/I symporter (NIS) mediates electroneutral active transport of the environmental pollutant perchlorate. Proc Natl Acad Sci USA 104:20250–20255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paroder-Belenitsky M, Maestas MJ, Dohán O, Nicola JP, Reyna-Neyra A, Follenzi A, Dadachova E, Eskandari S, Amzel LM, Carrasco N. 2011. Mechanism of anion selectivity and stoichiometry of the Na+/I− symporter (NIS). Proc Natl Acad Sci USA 108:17933–17938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Riedel C, Levy O, Carrasco N. 2001. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem 276:21458–21463 [DOI] [PubMed] [Google Scholar]
- 43. Uyttersprot N, Pelgrims N, Carrasco N, Gervy C, Maenhaut C, Dumont JE, Miot F. 1997. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I− symporter mRNAs in dog thyroid. Mol Cell Endocrinol 131:195–203 [DOI] [PubMed] [Google Scholar]
- 44. Eng PH, Cardona GR, Fang SL, Previti M, Alex S, Carrasco N, Chin WW, Braverman LE. 1999. Escape from the acute Wolff-Chaikoff effect is associated with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid and protein. Endocrinology 140:3404–3410 [DOI] [PubMed] [Google Scholar]
- 45. Eng PH, Cardona GR, Previti MC, Chin WW, Braverman LE. 2001. Regulation of the sodium iodide symporter by iodide in FRTL-5 cells. Eur J Endocrinol 144:139–144 [DOI] [PubMed] [Google Scholar]
- 46. Serrano-Nascimento C, Calil-Silveira J, Goulart-Silva F, Nunes MT. 2012. New insights about the posttranscriptional mechanisms triggered by iodide excess on sodium/iodide symporter (NIS) expression in PCCl3 cells. Mol Cell Endocrinol 349:154–161 [DOI] [PubMed] [Google Scholar]
- 47. Wolff J, Chaikoff IL. 1948. Plasma inorganic iodide as a homeostatic regulator of thyroid function. J Biol Chem 174:555–564 [PubMed] [Google Scholar]
- 48. Bernier-Valentin F, Trouttet-Masson S, Rabilloud R, Selmi-Ruby S, Rousset B. 2006. Three-dimensional organization of thyroid cells into follicle structures is a pivotal factor in the control of sodium/iodide symporter expression. Endocrinology 147:2035–2042 [DOI] [PubMed] [Google Scholar]
- 49. 2009. Iodide deficiency-induced angiogenic stimulus in the thyroid occurs via HIF- and ROS-dependent VEGF-A secretion from thyrocytes. Am J Physiol Endocrinol Metab 296:E1414–E1422 [DOI] [PubMed] [Google Scholar]
- 50. Gérard AC, Humblet K, Wilvers C, Poncin S, Derradji H, de Ville de Goyet C, Abou-el-Ardat K, Baatout S, Sonveaux P, Denef JF, Colin IM. 2012. Iodine-deficiency-induced long lasting angiogenic reaction in thyroid cancers occurs via a vascular endothelial growth factor-hypoxia inducible factor-1-dependent, but not a reactive oxygen species-dependent, pathway. Thyroid 22:699–708 [DOI] [PubMed] [Google Scholar]
- 51. Roepke TK, King EC, Reyna-Neyra A, Paroder M, Purtell K, Koba W, Fine E, Lerner DJ, Carrasco N, Abbott GW. 2009. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med 15:1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Purtell K, Paroder-Belenitsky M, Reyna-Neyra A, Nicola JP, Koba W, Fine E, Carrasco N, Abbott GW. 2012. The KCNQ1-KCNE2 K+ channel is required for adequate thyroid I- uptake. FASEB J 26:3252–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De La Vieja A, Dohan O, Levy O, Carrasco N. 2000. Molecular analysis of the sodium/iodide symporter: impact on thyroid and extrathyroid pathophysiology. Physiol Rev 80:1083–1105 [DOI] [PubMed] [Google Scholar]
- 54. Dohán O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, Ginter CS, Carrasco N. 2003. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 24:48–77 [DOI] [PubMed] [Google Scholar]
- 55. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. 1997. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17:411–422 [DOI] [PubMed] [Google Scholar]
- 56. Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. 2000. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141:839–845 [DOI] [PubMed] [Google Scholar]
- 57. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. 1999. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21:440–443 [DOI] [PubMed] [Google Scholar]
- 58. Pesce L, Bizhanova A, Caraballo JC, Westphal W, Butti ML, Comellas A, Kopp P. 2012. TSH regulates Pendrin membrane abundance and enhances iodide efflux in thyroid cells. Endocrinology 153:512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kopp P, Bizhanova A. 2011. Clinical and molecular characteristics of Pendred syndrome. Ann Endocrinol (Paris) 72:88–94 [DOI] [PubMed] [Google Scholar]
- 60. van den Hove MF, Croizet-Berger K, Jouret F, Guggino SE, Guggino WB, Devuyst O, Courtoy PJ. 2006. The loss of the chloride channel, ClC-5, delays apical iodide efflux and induces a euthyroid goiter in the mouse thyroid gland. Endocrinology 147:1287–1296 [DOI] [PubMed] [Google Scholar]
- 61. Bizhanova A, Kopp P. 2011. Controversies concerning the role of pendrin as an apical iodide transporter in thyroid follicular cells. Cell Physiol Biochem 28:485–490 [DOI] [PubMed] [Google Scholar]
- 62. Twyffels L, Massart C, Golstein PE, Raspe E, Van Sande J, Dumont JE, Beauwens R, Kruys V. 2011. Pendrin: the thyrocyte apical membrane iodide transporter? Cell Physiol Biochem 28:491–496 [DOI] [PubMed] [Google Scholar]
- 63. Fong P. 2011. Thyroid iodide efflux: a team effort? J Physiol 589:5929–5939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Coady MJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, Markowitz SD, Romero MF, Lapointe JY. 2004. The human tumour suppressor gene SLC5A8 expresses a Na+-monocarboxylate cotransporter. J Physiol 557:719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miyauchi S, Gopal E, Fei YJ, Ganapathy V. 2004. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem 279:13293–13296 [DOI] [PubMed] [Google Scholar]
- 66. Paroder V, Spencer SR, Paroder M, Arango D, Schwartz S, Jr, Mariadason JM, Augenlicht LH, Eskandari S, Carrasco N. 2006. Na+/monocarboxylate transport (SMCT) protein expression correlates with survival in colon cancer: molecular characterization of SMCT. Proc Natl Acad Sci USA 103:7270–7275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rodriguez AM, Perron B, Lacroix L, Caillou B, Leblanc G, Schlumberger M, Bidart JM, Pourcher T. 2002. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. J Clin Endocrinol Metab 87:3500–3503 [DOI] [PubMed] [Google Scholar]
- 68. Rivolta CM, Targovnik HM. 2006. Molecular advances in thyroglobulin disorders. Clin Chim Acta 374:8–24 [DOI] [PubMed] [Google Scholar]
- 69. Malthiéry Y, Lissitzky S. 1987. Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur J Biochem 165:491–498 [DOI] [PubMed] [Google Scholar]
- 70. Lee J, Wang X, Di Jeso B, Arvan P. 2009. The cholinesterase-like domain, essential in thyroglobulin trafficking for thyroid hormone synthesis, is required for protein dimerization. J Biol Chem 284:12752–12761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vono-Toniolo J, Rivolta CM, Targovnik HM, Medeiros-Neto G, Kopp P. 2005. Naturally occurring mutations in the thyroglobulin gene. Thyroid 15:1021–1033 [DOI] [PubMed] [Google Scholar]
- 72. Suzuki K, Kawashima A, Yoshihara A, Akama T, Sue M, Yoshida A, Kimura HJ. 2011. Role of thyroglobulin on negative feedback autoregulation of thyroid follicular function and growth. J Endocrinol 209:169–174 [DOI] [PubMed] [Google Scholar]
- 73. Kimura S, Hong YS, Kotani T, Ohtaki S, Kikkawa F. 1989. Structure of the human thyroid peroxidase gene: comparison and relationship to the human myeloperoxidase gene. Biochemistry 28:4481–4489 [DOI] [PubMed] [Google Scholar]
- 74. Arnljots K, Olsson I. 1987. Myeloperoxidase precursors incorporate heme. J Biol Chem 262:10430–10433 [PubMed] [Google Scholar]
- 75. Fayadat L, Niccoli-Sire P, Lanet J, Franc JL. 1999. Role of heme in intracellular trafficking of thyroperoxidase and involvement of H2O2 generated at the apical surface of thyroid cells in autocatalytic covalent heme binding. J Biol Chem 274:10533–10538 [DOI] [PubMed] [Google Scholar]
- 76. Niccoli P, Fayadat L, Panneels V, Lanet J, Franc JL. 1997. Human thyroperoxidase in its alternatively spliced form (TPO2) is enzymatically inactive and exhibits changes in intracellular processing and trafficking. J Biol Chem 272:29487–29492 [DOI] [PubMed] [Google Scholar]
- 77. Taurog A, Wall M. 1998. Proximal and distal histidines in thyroid peroxidase: relation to the alternatively spliced form, TPO-2. Thyroid 8:185–191 [DOI] [PubMed] [Google Scholar]
- 78. Kuliawat R, Ramos-Castañeda J, Liu Y, Arvan P. 2005. Intracellular trafficking of thyroid peroxidase to the cell surface. J Biol Chem 280:27713–27718 [DOI] [PubMed] [Google Scholar]
- 79. Ahn CS, Rosenberg IN. 1971. Oxidation of 14C-formate in thyroid slices: effects of TSH, dibutyric cyclic 30,50-AMP (dbc-AMP) and prostaglandin E1 (PGE1). In: Hofer K, Fellinger R, eds. Vienna: Verlag Der Wiener Medizinischen Akademie; 825–837 [Google Scholar]
- 80. Bénard B, Brault J. 1971. [Production of peroxide in the thyroid]. Union Med Can 100:701–705 (French) [PubMed] [Google Scholar]
- 81. Björkman U, Ekholm R, Denef JF. 1981. Cytochemical localization of hydrogen peroxide in isolated thyroid follicles. J Ultrastruct Res 74:105–115 [DOI] [PubMed] [Google Scholar]
- 82. Björkman U, Ekholm R. 1984. Generation of H2O2 in isolated porcine thyroid follicles. Endocrinology 115:392–398 [DOI] [PubMed] [Google Scholar]
- 83. Dupuy C, Ohayon R, Valent A, Noël-Hudson MS, Dème D, Virion A. 1999. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J Biol Chem 274:37265–37269 [DOI] [PubMed] [Google Scholar]
- 84. De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, Miot F. 2000. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 275:23227–23233 [DOI] [PubMed] [Google Scholar]
- 85. Lambeth JD. 2002. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol 9:11–17 [DOI] [PubMed] [Google Scholar]
- 86. Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. 2003. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 17:1502–1504 [DOI] [PubMed] [Google Scholar]
- 87. Geiszt M, Leto TL. 2004. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem 279:51715–51718 [DOI] [PubMed] [Google Scholar]
- 88. Meitzler JL, Ortiz de Montellano PR. 2009. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) “peroxidase” domains: insights into heme binding and catalytic activity. J Biol Chem 284:18634–18643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Meitzler JL, Ortiz de Montellano PR. 2011. Structural stability and heme binding potential of the truncated human dual oxidase 2 (DUOX2) peroxidase domain. Arch Biochem Biophys 512:197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dupuy C, Kaniewski J, Dème D, Pommier J, Virion A. 1989. NADPH-dependent H2O2 generation catalyzed by thyroid plasma membranes. Studies with electron scavengers. Eur J Biochem 185:597–603 [DOI] [PubMed] [Google Scholar]
- 91. De Deken X, Wang D, Dumont JE, Miot F. 2002. Characterization of ThOX proteins as components of the thyroid H2O2-generating system. Exp Cell Res 273:187–196 [DOI] [PubMed] [Google Scholar]
- 92. Morand S, Chaaraoui M, Kaniewski J, Dème D, Ohayon R, Noel-Hudson MS, Virion A, Dupuy C. 2003. Effect of iodide on nicotinamide adenine dinucleotide phosphate oxidase activity and Duox2 protein expression in isolated porcine thyroid follicles. Endocrinology 144:1241–1248 [DOI] [PubMed] [Google Scholar]
- 93. Dupuy C, Virion A, Ohayon R, Kaniewski J, Dème D, Pommier J. 1991. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. J Biol Chem 266:3739–3743 [PubMed] [Google Scholar]
- 94. Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R, Noël-Hudson MS, Francon J, Lalaoui K, Virion A, Dupuy C. 2005. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J Biol Chem 280:30046–30054 [DOI] [PubMed] [Google Scholar]
- 95. Forteza R, Salathe M, Miot F, Forteza R, Conner GE. 2005. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol 32:462–469 [DOI] [PubMed] [Google Scholar]
- 96. Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, De Deken X. 2009. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol Chem 284:6725–6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Milenkovic M, De Deken X, Jin L, De Felice M, Di Lauro R, Dumont JE, Corvilain B, Miot F. 2007. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. J Endocrinol 192:615–626 [DOI] [PubMed] [Google Scholar]
- 98. Grasberger H, Refetoff S. 2006. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 281:18269–18272 [DOI] [PubMed] [Google Scholar]
- 99. Hoste C, Dumont JE, Miot F, De Deken X. 2012. The type of DUOX-dependent ROS production is dictated by defined sequences in DUOXA. Exp Cell Res 318:2353–2364 [DOI] [PubMed] [Google Scholar]
- 100. Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T, Ris-Stalpers C. 2002. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 347:95–102 [DOI] [PubMed] [Google Scholar]
- 101. Maruo Y, Takahashi H, Soeda I, Nishikura N, Matsui K, Ota Y, Mimura Y, Mori A, Sato H, Takeuchi Y. 2008. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab 93:4261–4267 [DOI] [PubMed] [Google Scholar]
- 102. Hoste C, Rigutto S, Van Vliet G, Miot F, De Deken X. 2010. Compound heterozygosity for a novel hemizygous missense mutation and a partial deletion affecting the catalytic core of the H2O2-generating enzyme DUOX2 associated with transient congenital hypothyroidism. Hum Mutat 31:E1304–E1319 [DOI] [PubMed] [Google Scholar]
- 103. Corvilain B, Collyn L, Van Sande J, Dumont JE. 2000. Stimulation by iodide of H2O2 generation in thyroid slices from several species. Am J Physiol Endocrinol Metab 278:E692–E699 [DOI] [PubMed] [Google Scholar]
- 104. Panneels V, Van den Bergen H, Jacoby C, Braekman JC, Van Sande J, Dumont JE, Boeynaems JM. 1994. Inhibition of H2O2 production by iodoaldehydes in cultured dog thyroid cells. Mol Cell Endocrinol 102:167–176 [DOI] [PubMed] [Google Scholar]
- 105. Corvilain B, Van Sande J, Dumont JE. 1988. Inhibition by iodide of iodide binding to proteins: the “Wolff-Chaikoff” effect is caused by inhibition of H2O2 generation. Biochem Biophys Res Commun 154:1287–1292 [DOI] [PubMed] [Google Scholar]
- 106. Thomasz L, Oglio R, Rivandeira DT, Dagrosa MA, Jahn G, Pignataro OP, Piganataro OP, Sartorio G, Pisarev MA, Juvenal GJ. 2010. Inhibition of goiter growth and of cyclic AMP formation in rat thyroid by 2-iodohexadecanal. Mol Cell Endocrinol 317:141–147 [DOI] [PubMed] [Google Scholar]
- 107. Thomasz L, Oglio R, Dagrosa MA, Krawiec L, Pisarev MA, Juvenal GJ. 2010. 6 Iodo-delta-lactone reproduces many but not all the effects of iodide. Mol Cell Endocrinol 323:161–166 [DOI] [PubMed] [Google Scholar]
- 108. Panneels V, Van Sande J, Van den Bergen H, Jacoby C, Braekman JC, Dumont JE, Boeynaems JM. 1994. Inhibition of human thyroid adenylyl cyclase by 2-iodoaldehydes. Mol Cell Endocrinol 106:41–50 [DOI] [PubMed] [Google Scholar]
- 109. Pisarev MA, Gartner R. 2000. Autoregulatory action of iodine. In: Braverman LE, Utigers RD, eds. The thyroid. 9th ed Philadelphia: Lippincott; 85–90 [Google Scholar]
- 110. Panneels V, Juvenal G, Boeynaems JM, Dumont JE, Van Sande J. 2009. Iodide effects on the thyroid. In: Preedy VR, Burrow GN, Watson R, eds. Comprehensive handbook of iodine: nutritional, endocrine, and pathological aspects. Oxford, UK: Academic Press; 99305–99316 [Google Scholar]
- 111. Nunez J, Pommier J. 1982. Formation of thyroid hormones. Vitam Horm 39:175–229 [DOI] [PubMed] [Google Scholar]
- 112. Corvilain B, van Sande J, Laurent E, Dumont JE. 1991. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology 128:779–785 [DOI] [PubMed] [Google Scholar]
- 113. Taurog A, Dorris ML, Doerge DR. 1996. Mechanism of simultaneous iodination and coupling catalyzed by thyroid peroxidase. Arch Biochem Biophys 330:24–32 [DOI] [PubMed] [Google Scholar]
- 114. Lamas L, Anderson PC, Fox JW, Dunn JT. 1989. Consensus sequences for early iodination and hormonogenesis in human thyroglobulin. J Biol Chem 264:13541–13545 [PubMed] [Google Scholar]
- 115. Moreno JC. 2003. Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res 60(Suppl 3):96–102 [DOI] [PubMed] [Google Scholar]
- 116. Gnidehou S, Caillou B, Talbot M, Ohayon R, Kaniewski J, Noël-Hudson MS, Morand S, Agnangji D, Sezan A, Courtin F, Virion A, Dupuy C. 2004. Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J 18:1574–1576 [DOI] [PubMed] [Google Scholar]
- 117. Moreno JC, Klootwijk W, van Toor H, Pinto G, D'Alessandro M, Lèger A, Goudie D, Polak M, Grüters A, Visser TJ. 2008. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med 358:1811–1818 [DOI] [PubMed] [Google Scholar]
- 118. Moreno JC, Visser TJ. 2010. Genetics and phenomics of hypothyroidism and goiter due to iodotyrosine deiodinase (DEHAL1) gene mutations. Mol Cell Endocrinol 322:91–98 [DOI] [PubMed] [Google Scholar]
- 119. Gnidehou S, Lacroix L, Sezan A, Ohayon R, Noël-Hudson MS, Morand S, Francon J, Courtin F, Virion A, Dupuy C. 2006. Cloning and characterization of a novel isoform of iodotyrosine dehalogenase 1 (DEHAL1) DEHAL1C from human thyroid: comparisons with DEHAL1 and DEHAL1B. Thyroid 16:715–724 [DOI] [PubMed] [Google Scholar]
- 120. Bjorkman U, Ekholm R. 1990. Biochemistry of thyroid hormone formation and secretion. In: Greer M, ed. The thyroid gland. New-York: Raven Press; 83–125 [Google Scholar]
- 121. Pommier J, Deme D, Nunez J. 1973. Effect of iodide concentration on thyroxine synthesis catalysed by thyroid peroxidase. Eur J Biochem 37:406–414 [DOI] [PubMed] [Google Scholar]
- 122. Morris DR, Hager LP. 1966. Mechanism of the inhibition of enzymatic halogenation by antithyroid agents. J Biol Chem 241:3582–3589 [PubMed] [Google Scholar]
- 123. Ohtaki S, Nakagawa H, Kimura S, Yamazaki I. 1981. Analyses of catalytic intermediates of hog thyroid peroxidase during its iodinating reaction. J Biol Chem 256:805–810 [PubMed] [Google Scholar]
- 124. Nakamura M, Yamazaki I, Nakagawa H, Ohtaki S. 1983. Steady state kinetics and regulation of thyroid peroxidase-catalyzed iodination. J Biol Chem 258:3837–3842 [PubMed] [Google Scholar]
- 125. Ohtaki S, Nakagawa H, Nakamura M, Kotani T. 1996. Thyroid peroxidase: experimental and clinical integration. Endocr J 43:1–14 [DOI] [PubMed] [Google Scholar]
- 126. Morrison M, Schonbaum GR. 1976. Peroxidase-catalyzed halogenation. Annu Rev Biochem 45:861–888 [DOI] [PubMed] [Google Scholar]
- 127. Magnusson RP, Taurog A, Dorris ML. 1984. Mechanisms of thyroid peroxidase- and lactoperoxidase-catalyzed reactions involving iodide. J Biol Chem 259:13783–13790 [PubMed] [Google Scholar]
- 128. Magnusson RP, Taurog A, Dorris ML. 1984. Mechanism of iodide-dependent catalatic activity of thyroid peroxidase and lactoperoxidase. J Biol Chem 259:197–205 [PubMed] [Google Scholar]
- 129. Dawson JH. 1988. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science 240:433–439 [DOI] [PubMed] [Google Scholar]
- 130. Taurog A, Dorris M, Doerge DR. 1994. Evidence for a radical mechanism in peroxidase-catalyzed coupling. I. Steady-state experiments with various peroxidases. Arch Biochem Biophys 315:82–89 [DOI] [PubMed] [Google Scholar]
- 131. Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH. 2006. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 20:2761–2772 [DOI] [PubMed] [Google Scholar]
- 132. Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S. 2010. Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest 120:3377–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Visser WE, Friesema EC, Visser TJ. 2011. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol 25:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. 2004. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364:1435–1437 [DOI] [PubMed] [Google Scholar]
- 135. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. 2004. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. 2005. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Friesema EC, Visser WE, Visser TJ. 2010. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol 322:107–113 [DOI] [PubMed] [Google Scholar]
- 138. Wirth EK, Sheu SY, Chiu-Ugalde J, Sapin R, Klein MO, Mossbrugger I, Quintanilla-Martinez L, de Angelis MH, Krude H, Riebel T, Rothe K, Köhrle J, Schmid KW, Schweizer U, Grüters A. 2011. Monocarboxylate transporter 8 deficiency: altered thyroid morphology and persistent high triiodothyronine/thyroxine ratio after thyroidectomy. Eur J Endocrinol 165:555–561 [DOI] [PubMed] [Google Scholar]
- 139. Zung A, Visser TJ, Uitterlinden AG, Rivadeneira F, Friesema EC. 2011. A child with a deletion in the monocarboxylate transporter 8 gene: 7-year follow-up and effects of thyroid hormone treatment. Eur J Endocrinol 165:823–830 [DOI] [PubMed] [Google Scholar]
- 140. Liao XH, Di Cosmo C, Dumitrescu AM, Hernandez A, Van Sande J, St Germain DL, Weiss RE, Galton VA, Refetoff S. 2011. Distinct roles of deiodinases on the phenotype of Mct8 defect: a comparison of eight different mouse genotypes. Endocrinology 152:1180–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bartalena L, Robbins J. 1993. Thyroid hormone transport proteins. Clin Lab Med 13:583–598 [PubMed] [Google Scholar]
- 142. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. 2003. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278:40128–40135 [DOI] [PubMed] [Google Scholar]
- 143. Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. 2005. Integrin αVβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 146:2864–2871 [DOI] [PubMed] [Google Scholar]
- 144. Scarlett A, Parsons MP, Hanson PL, Sidhu KK, Milligan TP, Burrin JM. 2008. Thyroid hormone stimulation of extracellular signal-regulated kinase and cell proliferation in human osteoblast-like cells is initiated at integrin αVβ3. J Endocrinol 196:509–517 [DOI] [PubMed] [Google Scholar]
- 145. Cao X, Kambe F, Moeller LC, Refetoff S, Seo H. 2005. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol 19:102–112 [DOI] [PubMed] [Google Scholar]
- 146. Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. 2006. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci USA 103:14104–14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kenessey A, Ojamaa K. 2006. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem 281:20666–20672 [DOI] [PubMed] [Google Scholar]
- 148. Storey NM, Gentile S, Ullah H, Russo A, Muessel M, Erxleben C, Armstrong DL. 2006. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc Natl Acad Sci USA 103:5197–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Verga Falzacappa C, Panacchia L, Bucci B, Stigliano A, Cavallo MG, Brunetti E, Toscano V, Misiti S. 2006. 3,5,3′-Triiodothyronine (T3) is a survival factor for pancreatic β-cells undergoing apoptosis. J Cell Physiol 206:309–321 [DOI] [PubMed] [Google Scholar]
- 150. Verga Falzacappa C, Petrucci E, Patriarca V, Michienzi S, Stigliano A, Brunetti E, Toscano V, Misiti S. 2007. Thyroid hormone receptor TRβ1 mediates Akt activation by T3 in pancreatic β-cells. J Mol Endocrinol 38:221–233 [DOI] [PubMed] [Google Scholar]
- 151. Leonard JL. 2008. Non-genomic actions of thyroid hormone in brain development. Steroids 73:1008–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Cheng SY, Leonard JL, Davis PJ. 2010. Molecular aspects of thyroid hormone actions. Endocr Rev 31:139–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Schneider PB. 1964. Thyroidal iodine heterogeneity: “last come, first served” system of thyroid turnover. Endocrinology 74:973–980 [DOI] [PubMed] [Google Scholar]
- 154. Studer H, Gerber H. 1991. Intrathyroidal iodine heterogeneity of iodocompounds and kinetic compartmentalization. Trends Endocrinol Metab 2:29–35 [DOI] [PubMed] [Google Scholar]
- 155. Herzog V, Berndorfer U, Saber Y. 1992. Isolation of insoluble secretory product from bovine thyroid: extracellular storage of thyroglobulin in covalently cross-linked form. J Cell Biol 118:1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Berndorfer U, Wilms H, Herzog V. 1996. Multimerization of thyroglobulin (TG) during extracellular storage: isolation of highly cross-linked TG from human thyroids. J Clin Endocrinol Metab 81:1918–1926 [DOI] [PubMed] [Google Scholar]
- 157. Gérard AC, Denef JF, Colin IM, van den Hove MF. 2004. Evidence for processing of compact insoluble thyroglobulin globules in relation with follicular cell functional activity in the human and the mouse thyroid. Eur J Endocrinol 150:73–80 [DOI] [PubMed] [Google Scholar]
- 158. Baudry N, Lejeune PJ, Delom F, Vinet L, Carayon P, Mallet B. 1998. Role of multimerized porcine thyroglobulin in iodine storage. Biochem Biophys Res Commun 242:292–296 [DOI] [PubMed] [Google Scholar]
- 159. Den Hartog MT, De Boer M, Veenboer GJ, De Vijlder JJ. 1990. Generation and characterization of monoclonal antibodies directed against noniodinated and iodinated thyroglobulin, among which are antibodies against hormonogenic sites. Endocrinology 127:3160–3165 [DOI] [PubMed] [Google Scholar]
- 160. Brix K, Lemansky P, Herzog V. 1996. Evidence for extracellularly acting cathepsins mediating thyroid hormone liberation in thyroid epithelial cells. Endocrinology 137:1963–1974 [DOI] [PubMed] [Google Scholar]
- 161. Brix K, Linke M, Tepel C, Herzog V. 2001. Cysteine proteinases mediate extracellular prohormone processing in the thyroid. Biol Chem 382:717–725 [DOI] [PubMed] [Google Scholar]
- 162. Linke M, Herzog V, Brix K. 2002. Trafficking of lysosomal cathepsin B-green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J Cell Sci 115:4877–4889 [DOI] [PubMed] [Google Scholar]
- 163. Friedrichs B, Tepel C, Reinheckel T, Deussing J, von Figura K, Herzog V, Peters C, Saftig P, Brix K. 2003. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest 111:1733–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dauth S, Arampatzidou M, Rehders M, Yu D, Führer D, Brix K. 2011. Thyroid cathepsin K: roles in physiology and thyroid desease. Clin Rev Bone Miner Metab 9:94–106 [Google Scholar]
- 165. Jordans S, Jenko-Kokalj S, Kühl NM, Tedelind S, Sendt W, Brömme D, Turk D, Brix K. 2009. Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC Biochem 10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Tedelind S, Jordans S, Resemann H, Blum G, Bogyo M, Fuhrer D, Brix K. 2011. Cathepsin B trafficking in thyroid carcinoma cells. Thyroid Res 4(Suppl 1):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sloane BF, Moin K, Krepela E, Rozhin J. 1990. Cathepsin B and its endogenous inhibitors: the role in tumor malignancy. Cancer Metastasis Rev 9:333–352 [DOI] [PubMed] [Google Scholar]
- 168. Marinò M, McCluskey RT. 2000. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. Am J Physiol Cell Physiol 279:C1295–C1306 [DOI] [PubMed] [Google Scholar]
- 169. van den Hove MF, Couvreur M, de Visscher M, Salvatore G. 1982. A new mechanism for the reabsorption of thyroid iodoproteins: selective fluid pinocytosis. Eur J Biochem 122:415–422 [DOI] [PubMed] [Google Scholar]
- 170. Dunn JT. 1996. Thyroglobulin retrieval and the endocytic pathway. In: Braverman LE, Utigers RD, eds. The thyroid. A fundamental and clinical text. Philadelphia: Lippincott-Raven; 81–84 [Google Scholar]
- 171. Marinò M, Friedlander JA, McCluskey RT, Andrews D. 1999. Identification of a heparin-binding region of rat thyroglobulin involved in megalin binding. J Biol Chem 274:30377–30386 [DOI] [PubMed] [Google Scholar]
- 172. Lisi S, Pinchera A, McCluskey RT, Willnow TE, Refetoff S, Marcocci C, Vitti P, Menconi F, Grasso L, Luchetti F, Collins AB, Marino M. 2003. Preferential megalin-mediated transcytosis of low-hormonogenic thyroglobulin: a control mechanism for thyroid hormone release. Proc Natl Acad Sci USA 100:14858–14863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Miquelis R, Courageot J, Jacq A, Blanck O, Perrin C, Bastiani P. 1993. Intracellular routing of GLcNAc-bearing molecules in thyrocytes: selective recycling through the Golgi apparatus. J Cell Biol 123:1695–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kostrouch Z, Bernier-Valentin F, Munari-Silem Y, Rajas F, Rabilloud R, Rousset B. 1993. Thyroglobulin molecules internalized by thyrocytes are sorted in early endosomes and partially recycled back to the follicular lumen. Endocrinology 132:2645–2653 [DOI] [PubMed] [Google Scholar]
- 175. Mezghrani A, Courageot J, Mani JC, Pugniere M, Bastiani P, Miquelis R. 2000. Protein-disulfide isomerase (PDI) in FRTL5 cells. pH-dependent thyroglobulin/PDI interactions determine a novel PDI function in the post-endoplasmic reticulum of thyrocytes. J Biol Chem 275:1920–1929 [DOI] [PubMed] [Google Scholar]
- 176. Botta R, Lisi S, Pinchera A, Giorgi F, Marcocci C, Taddei AR, Fausto AM, Bernardini N, Ippolito C, Mattii L, Persani L, de Filippis T, Calebiro D, Madsen P, Petersen CM, Marinò M. 2009. Sortilin is a putative postendocytic receptor of thyroglobulin. Endocrinology 150:509–518 [DOI] [PubMed] [Google Scholar]
- 177. Wang D, De Deken X, Milenkovic M, Song Y, Pirson I, Dumont JE, Miot F. 2005. Identification of a novel partner of duox: EFP1, a thioredoxin-related protein. J Biol Chem 280:3096–3103 [DOI] [PubMed] [Google Scholar]
- 178. Song Y, Driessens N, Costa M, De Deken X, Detours V, Corvilain B, Maenhaut C, Miot F, Van Sande J, Many MC, Dumont JE. 2007. Roles of hydrogen peroxide in thyroid physiology and disease. J Clin Endocrinol Metab 92:3764–3773 [DOI] [PubMed] [Google Scholar]
- 179. Schweizer U, Chiu J, Köhrle J. 2008. Peroxides and peroxide-degrading enzymes in the thyroid. Antioxid Redox Signal 10:1577–1592 [DOI] [PubMed] [Google Scholar]
- 180. Ohye H, Sugawara M. 2010. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood) 235:424–433 [DOI] [PubMed] [Google Scholar]
- 181. Song Y, Ruf J, Lothaire P, Dequanter D, Andry G, Willemse E, Dumont JE, Van Sande J, De Deken X. 2010. Association of duoxes with thyroid peroxidase and its regulation in thyrocytes. J Clin Endocrinol Metab 95:375–382 [DOI] [PubMed] [Google Scholar]
- 182. Fortunato RS, Lima de Souza EC, Ameziane-el Hassani R, Boufraqech M, Weyemi U, Talbot M, Lagente-Chevallier O, de Carvalho DP, Bidart JM, Schlumberger M, Dupuy C. 2010. Functional consequences of dual oxidase-thyroperoxidase interaction at the plasma membrane. J Clin Endocrinol Metab 95:5403–5411 [DOI] [PubMed] [Google Scholar]
- 183. Senou M, Costa MJ, Massart C, Thimmesch M, Khalifa C, Poncin S, Boucquey M, Gérard AC, Audinot JN, Dessy C, Ruf J, Feron O, Devuyst O, Guiot Y, Dumont JE, Van Sande J, Many MC. 2009. Role of caveolin-1 in thyroid phenotype, cell homeostasis, and hormone synthesis: in vivo study of caveolin-1 knockout mice. Am J Physiol Endocrinol Metab 297:E438–E451 [DOI] [PubMed] [Google Scholar]
- 184. Senou M, Khalifa C, Thimmesch M, Jouret F, Devuyst O, Col V, Audinot JN, Lipnik P, Moreno JC, Van Sande J, Dumont JE, Many MC, Colin IM, Gérard AC. 2010. A coherent organization of differentiation proteins is required to maintain an appropriate thyroid function in the Pendred thyroid. J Clin Endocrinol Metab 95:4021–4030 [DOI] [PubMed] [Google Scholar]
- 185. Morand S, Ueyama T, Tsujibe S, Saito N, Korzeniowska A, Leto TL. 2009. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J 23:1205–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Johnson KR, Marden CC, Ward-Bailey P, Gagnon LH, Bronson RT, Donahue LR. 2007. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol Endocrinol 21:1593–1602 [DOI] [PubMed] [Google Scholar]
- 187. Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S, Persani L, Weber G. 2008. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab 93:605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Grasberger H. 2010. Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol Cell Endocrinol 322:99–106 [DOI] [PubMed] [Google Scholar]
- 189. Grasberger H, De Deken X, Mayo OB, Raad H, Weiss M, Liao XH, Refetoff S. 2012. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol Endocrinol 26:481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. 1992. Caveolin, a protein component of caveolae membrane coats. Cell 68:673–682 [DOI] [PubMed] [Google Scholar]
- 191. Pilch PF, Liu L. 2011. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol Metab 22:318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Costa MJ, Senou M, Van Rode F, Ruf J, Capello M, Dequanter D, Lothaire P, Dessy C, Dumont JE, Many MC, Van Sande J. 2007. Reciprocal negative regulation between thyrotropin/3′,5′-cyclic adenosine monophosphate-mediated proliferation and caveolin-1 expression in human and murine thyrocytes. Mol Endocrinol 21:921–932 [DOI] [PubMed] [Google Scholar]
- 193. Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. 1997. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 272:6525–6533 [DOI] [PubMed] [Google Scholar]
- 194. Corvilain B, Laurent E, Lecomte M, Vansande J, Dumont JE. 1994. Role of the cyclic adenosine 3′,5′-monophosphate and the phosphatidylinositol-Ca2+ cascades in mediating the effects of thyrotropin and iodide on hormone synthesis and secretion in human thyroid slices. J Clin Endocrinol Metab 79:152–159 [DOI] [PubMed] [Google Scholar]
- 195. Mishina NM, Tyurin-Kuzmin PA, Markvicheva KN, Vorotnikov AV, Tkachuk VA, Laketa V, Schultz C, Lukyanov S, Belousov VV. 2011. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal 14:1–7 [DOI] [PubMed] [Google Scholar]
- 196. Denef JF, Many MC, van den Hove MF. 1996. Iodine-induced thyroid inhibition and cell necrosis: two consequences of the same free-radical mediated mechanism? Mol Cell Endocrinol 121:101–103 [DOI] [PubMed] [Google Scholar]
- 197. Ehrenshaft M, Mason RP. 2006. Protein radical formation on thyroid peroxidase during turnover as detected by immuno-spin trapping. Free Radic Biol Med 41:422–430 [DOI] [PubMed] [Google Scholar]
- 198. Poncin S, Colin IM, Gérard AC. 2009. Minimal oxidative load: a prerequisite for thyroid cell function. J Endocrinol 201:161–167 [DOI] [PubMed] [Google Scholar]
- 199. Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De Deken X, Van Sande J, Dumont JE, Miot F, Corvilain B. 2009. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr Relat Cancer 16:845–856 [DOI] [PubMed] [Google Scholar]
- 200. Ameziane-El-Hassani R, Boufraqech M, Lagente-Chevallier O, Weyemi U, Talbot M, Métivier D, Courtin F, Bidart JM, El Mzibri M, Schlumberger M, Dupuy C. 2010. Role of H2O2 in RET/PTC1 chromosomal rearrangement produced by ionizing radiation in human thyroid cells. Cancer Res 70:4123–4132 [DOI] [PubMed] [Google Scholar]
- 201. Paschke R. 2011. Molecular pathogenesis of nodular goiter. Langenbecks Arch Surg 396:1127–1136 [DOI] [PubMed] [Google Scholar]
- 202. Maier J, van Steeg H, van Oostrom C, Karger S, Paschke R, Krohn K. 2006. Deoxyribonucleic acid damage and spontaneous mutagenesis in the thyroid gland of rats and mice. Endocrinology 147:3391–3397 [DOI] [PubMed] [Google Scholar]
- 203. Krohn K, Maier J, Paschke R. 2007. Mechanisms of disease: hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat Clin Pract Endocrinol Metab 3:713–720 [DOI] [PubMed] [Google Scholar]
- 204. Björkman U, Ekholm R. 1995. Hydrogen peroxide degradation and glutathione peroxidase activity in cultures of thyroid cells. Mol Cell Endocrinol 111:99–107 [DOI] [PubMed] [Google Scholar]
- 205. Kim H, Lee TH, Park ES, Suh JM, Park SJ, Chung HK, Kwon OY, Kim YK, Ro HK, Shong M. 2000. Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cells. J Biol Chem 275:18266–18270 [DOI] [PubMed] [Google Scholar]
- 206. Gérard AC, Many MC, Daumerie Ch, Knoops B, Colin IM. 2005. Peroxiredoxin 5 expression in the human thyroid gland. Thyroid 15:205–209 [DOI] [PubMed] [Google Scholar]
- 207. Chiu-Ugalde J, Wirth EK, Klein MO, Sapin R, Fradejas-Villar N, Renko K, Schomburg L, Köhrle J, Schweizer U. 2012. Thyroid function is maintained despite increased oxidative stress in mice lacking selenoprotein biosynthesis in thyroid epithelial cells. Antioxid Redox Signal 17:902–913 [DOI] [PubMed] [Google Scholar]
- 208. Contempre B, Le Moine O, Dumont JE, Denef JF, Many MC. 1996. Selenium deficiency and thyroid fibrosis. A key role for macrophages and transforming growth factor β (TGF-β). Mol Cell Endocrinol 124:7–15 [DOI] [PubMed] [Google Scholar]
- 209. Köhrle J, Jakob F, Contempré B, Dumont JE. 2005. Selenium, the thyroid, and the endocrine system. Endocr Rev 26:944–984 [DOI] [PubMed] [Google Scholar]
- 210. Schomburg L. 2012. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol 8:160–171 [DOI] [PubMed] [Google Scholar]
- 211. Dröge W. 2002. Free radicals in the physiological control of cell function. Physiol Rev 82:47–95 [DOI] [PubMed] [Google Scholar]
- 212. Poncin S, Van Eeckoudt S, Humblet K, Colin IM, Gérard AC. 2010. Oxidative stress: a required condition for thyroid cell proliferation. Am J Pathol 176:1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Xing M. 2012. Oxidative stress: a new risk factor for thyroid cancer. Endocr Relat Cancer 19:C7–C11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Xing M. 2010. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid 20:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Liu D, Xing M. 2008. Potent inhibition of thyroid cancer cells by the MEK inhibitor PD0325901 and its potentiation by suppression of the PI3K and NF-κB pathways. Thyroid 18:853–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Pacifico F, Leonardi A. 2010. Role of NF-κB in thyroid cancer. Mol Cell Endocrinol 321:29–35 [DOI] [PubMed] [Google Scholar]
- 217. Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R, Parthasarathy S, Petros JA, Lambeth JD. 2001. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci USA 98:5550–5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Polytarchou C, Hatziapostolou M, Papadimitriou E. 2005. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J Biol Chem 280:40428–40435 [DOI] [PubMed] [Google Scholar]
- 219. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. 2005. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120:649–661 [DOI] [PubMed] [Google Scholar]
- 220. Tonks NK. 2005. Redox redux: revisiting PTPs and the control of cell signaling. Cell 121:667–670 [DOI] [PubMed] [Google Scholar]
- 221. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. 2003. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J 22:5501–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wang D, Feng JF, Zeng P, Yang YH, Luo J, Yang YW. 2011. Total oxidant/antioxidant status in sera of patients with thyroid cancers. Endocr Relat Cancer 18:773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Lacroix L, Nocera M, Mian C, Caillou B, Virion A, Dupuy C, Filetti S, Bidart JM, Schlumberger M. 2001. Expression of nicotinamide adenine dinucleotide phosphate oxidase flavoprotein DUOX genes and proteins in human papillary and follicular thyroid carcinomas. Thyroid 11:1017–1023 [DOI] [PubMed] [Google Scholar]
- 224. Gérard AC, Daumerie C, Mestdagh C, Gohy S, De Burbure C, Costagliola S, Miot F, Nollevaux MC, Denef JF, Rahier J, Franc B, De Vijlder JJ, Colin IM, Many MC. 2003. Correlation between the loss of thyroglobulin iodination and the expression of thyroid-specific proteins involved in iodine metabolism in thyroid carcinomas. J Clin Endocrinol Metab 88:4977–4983 [DOI] [PubMed] [Google Scholar]
- 225. Faggiano A, Caillou B, Lacroix L, Talbot M, Filetti S, Bidart JM, Schlumberger M. 2007. Functional characterization of human thyroid tissue with immunohistochemistry. Thyroid 17:203–211 [DOI] [PubMed] [Google Scholar]
- 226. Coclet J, Foureau F, Ketelbant P, Galand P, Dumont JE. 1989. Cell population kinetics in dog and human adult thyroid. Clin Endocrinol (Oxf) 31:655–665 [DOI] [PubMed] [Google Scholar]
- 227. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. 2004. Oxidative stress and cell signalling. Curr Med Chem 11:1163–1182 [DOI] [PubMed] [Google Scholar]
- 228. Lu H, Ouyang W, Huang C. 2006. Inflammation, a key event in cancer development. Mol Cancer Res 4:221–233 [DOI] [PubMed] [Google Scholar]
- 229. Nucera C, Lawler J, Parangi S. 2011. BRAF(V600E) and microenvironment in thyroid cancer: a functional link to drive cancer progression. Cancer Res 71:2417–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Boelaert K, Horacek J, Holder RL, Watkinson JC, Sheppard MC, Franklyn JA. 2006. Serum thyrotropin concentration as a novel predictor of malignancy in thyroid nodules investigated by fine-needle aspiration. J Clin Endocrinol Metab 91:4295–4301 [DOI] [PubMed] [Google Scholar]
- 231. Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H. 2009. Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 71:434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. van den Hove MF, Stoenoiu MS, Croizet K, Couvreur M, Courtoy PJ, Devuyst O, Colin IM. 2002. Nitric oxide is involved in interleukin-1α-induced cytotoxicity in polarised human thyrocytes. J Endocrinol 173:177–185 [DOI] [PubMed] [Google Scholar]
- 233. Gérard AC, Boucquey M, van den Hove MF, Colin IM. 2006. Expression of TPO and ThOXs in human thyrocytes is downregulated by IL-1α/IFN-γ, an effect partially mediated by nitric oxide. Am J Physiol Endocrinol Metab 291:E242–E253 [DOI] [PubMed] [Google Scholar]
- 234. Poncin S, Gérard AC, Boucquey M, Senou M, Calderon PB, Knoops B, Lengelé B, Many MC, Colin IM. 2008. Oxidative stress in the thyroid gland: from harmlessness to hazard depending on the iodine content. Endocrinology 149:424–433 [DOI] [PubMed] [Google Scholar]
- 235. Poncin S, Lengelé B, Colin IM, Gérard AC. 2008. Differential interactions between Th1/Th2, Th1/Th3, and Th2/Th3 cytokines in the regulation of thyroperoxidase and dual oxidase expression, and of thyroglobulin secretion in thyrocytes in vitro. Endocrinology 149:1534–1542 [DOI] [PubMed] [Google Scholar]
- 236. Poncin S, Colin IM, Decallonne B, Clinckspooor I, Many MC, Denef JF, Gérard AC. 2010. N-acetylcysteine and 15 deoxy-Δ12,14-prostaglandin J2 exert a protective effect against autoimmune thyroid destruction in vivo but not against interleukin-1α/interferon γ-induced inhibitory effects in thyrocytes in vitro. Am J Pathol 177:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Many MC, Denef JF, Haumont S, van den Hove-Vandenbroucke MF, Cornette C, Beckers C. 1985. Morphological and functional changes during thyroid hyperplasia and involution in C3H mice: effects of iodine and 3,5,3′-triiodothyronine during involution. Endocrinology 116:798–806 [DOI] [PubMed] [Google Scholar]
- 238. Many MC, Denef JF, Hamudi S, Cornette C, Haumont S, Beckers C. 1986. Effects of iodide and thyroxine on iodine-deficient mouse thyroid: a morphological and functional study. J Endocrinol 110:203–210 [DOI] [PubMed] [Google Scholar]
- 239. Mahmoud I, Colin I, Many MC, Denef JF. 1986. Direct toxic effect of iodide in excess on iodine-deficient thyroid glands: epithelial necrosis and inflammation associated with lipofuscin accumulation. Exp Mol Pathol 44:259–271 [DOI] [PubMed] [Google Scholar]
- 240. Many MC, Denef JF. 1992. Iodine and goiter involution. Thyroidology 4:23–26 [PubMed] [Google Scholar]
- 241. Many MC, Mestdagh C, van den Hove MF, Denef JF. 1992. In vitro study of acute toxic effects of high iodide doses in human thyroid follicles. Endocrinology 131:621–630 [DOI] [PubMed] [Google Scholar]
- 242. Mutaku JF, Poma JF, Many MC, Denef JF, van Den Hove MF. 2002. Cell necrosis and apoptosis are differentially regulated during goitre development and iodine-induced involution. J Endocrinol 172:375–386 [DOI] [PubMed] [Google Scholar]
- 243. Toussaint-Demylle D, Many MC, Theisen H, Kraal G, Denef JF. 1990. Effects of iodide on class II-MHC antigen expression in iodine deficient hyperplastic thyroid glands. Autoimmunity 7:51–62 [DOI] [PubMed] [Google Scholar]
- 244. Nagataki S, Eguchi K. 1992. Cytokines and immune regulation in thyroid autoimmunity. Autoimmunity 13:27–34 [DOI] [PubMed] [Google Scholar]
- 245. Many MC, Maniratunga S, Varis I, Dardenne M, Drexhage HA, Denef JF. 1995. Two-step development of Hashimoto-like thyroiditis in genetically autoimmune prone non-obese diabetic mice: effects of iodine-induced cell necrosis. J Endocrinol 147:311–320 [DOI] [PubMed] [Google Scholar]
- 246. Many MC, Maniratunga S, Denef JF. 1996. The non-obese diabetic (NOD) mouse: an animal model for autoimmune thyroiditis. Exp Clin Endocrinol Diabetes 104(Suppl 3):17–20 [DOI] [PubMed] [Google Scholar]
- 247. Andryskowski G, Owczarek T. 2007. [The evaluation of selected oxidative stress parameters in patients with hyperthyroidism]. Pol Arch Med Wewn 117:285–289 (Polish) [PubMed] [Google Scholar]
- 248. Erdamar H, Demirci H, Yaman H, Erbil MK, Yakar T, Sancak B, Elbeg S, Biberoðlu G, Yetkin I. 2008. The effect of hypothyroidism, hyperthyroidism, and their treatment on parameters of oxidative stress and antioxidant status. Clin Chem Lab Med 46:1004–1010 [DOI] [PubMed] [Google Scholar]
- 249. Torun AN, Kulaksizoglu S, Kulaksizoglu M, Pamuk BO, Isbilen E, Tutuncu NB. 2009. Serum total antioxidant status and lipid peroxidation marker malondialdehyde levels in overt and subclinical hypothyroidism. Clin Endocrinol (Oxf) 70:469–474 [DOI] [PubMed] [Google Scholar]
- 250. Aslan M, Cosar N, Celik H, Aksoy N, Dulger AC, Begenik H, Soyoral YU, Kucukoglu ME, Selek S. 2011. Evaluation of oxidative status in patients with hyperthyroidism. Endocrine 40:285–289 [DOI] [PubMed] [Google Scholar]
- 251. Denef JF, Haumont S, Beckers C. 1980. Morphological changes in mice thyroid induced by iodine deficiency. Virchows Arch B Cell Pathol Incl Mol Pathol 32:191–199 [DOI] [PubMed] [Google Scholar]
- 252. Denef JF, Haumont S, Cornette C, Beckers C. 1981. Correlated functional and morphometric study of thyroid hyperplasia induced by iodine deficiency. Endocrinology 108:2352–2358 [DOI] [PubMed] [Google Scholar]
- 253. Many MC, Denef JF, Gathy P, Haumont S. 1983. Morphological and functional changes during thyroid hyperplasia and involution in C3H Mice: evidence for folliculoneogenesis during involution. Endocrinology 112:1292–1302 [DOI] [PubMed] [Google Scholar]
- 254. Many MC, Denef JF, Haumont S. 1984. Precocity of the endothelial proliferation during a course of rapid goitrogenesis. Acta Endocrinol (Copenh) 105:487–491 [DOI] [PubMed] [Google Scholar]
- 255. Denef JF, Ovaert C, Many MC. 1989. [Experimental goitrogenesis]. Ann Endocrinol (Paris) 50:1–15 (French) [PubMed] [Google Scholar]
- 256. Krohn K, Führer D, Bayer Y, Eszlinger M, Brauer V, Neumann S, Paschke R. 2005. Molecular pathogenesis of euthyroid and toxic multinodular goiter. Endocr Rev 26:504–524 [DOI] [PubMed] [Google Scholar]
- 257. Paschke R. 2011. Nodulogenesis and goitrogenesis. Ann Endocrinol (Paris) 72:117–119 [DOI] [PubMed] [Google Scholar]
- 258. Gérard AC, Xhenseval V, Colin IM, Many MC, Denef JF. 2000. Evidence for co-ordinated changes between vascular endothelial growth factor and nitric oxide synthase III immunoreactivity, the functional status of the thyroid follicles, and the microvascular bed during chronic stimulation by low iodine and propylthiouracyl in old mice. Eur J Endocrinol 142:651–660 [DOI] [PubMed] [Google Scholar]
- 259. Gérard AC, Many MC, Daumerie C, Costagliola S, Miot F, DeVijlder JJ, Colin IM, Denef JF. 2002. Structural changes in the angiofollicular units between active and hypofunctioning follicles align with differences in the epithelial expression of newly discovered proteins involved in iodine transport and organification. J Clin Endocrinol Metab 87:1291–1299 [DOI] [PubMed] [Google Scholar]
- 260. Gérard AC, Denef JF, Many MC, Gathy P, de Burbure C, van den Hove MF, Coppée F, Ledent C, Colin IM. 2003. Relationships between cell division, expression of growth factors and microcirculation in the thyroids of Tg-A2aR transgenic mice and patients with Graves' disease. J Endocrinol 177:269–277 [DOI] [PubMed] [Google Scholar]
- 261. Wollman SH, Herveg JP, Zeligs JD, Ericson LE. 1978. Blood capillary enlargement during the development of thyroid hyperplasia in the rat. Endocrinology 103:2306–2314 [DOI] [PubMed] [Google Scholar]
- 262. Ericson LE, Wollman SH. 1980. Ultrastructural aspects of capillary fusion during the development of thyroid hyperplasia. J Ultrastruct Res 72:300–315 [DOI] [PubMed] [Google Scholar]
- 263. Ericson LW, Wollman SH. 1980. Increase in the rough endoplasmic reticulum in capillary endothelial cells and pericytes in hyperplastic rat thyroid glands. Endocrinology 107:732–737 [DOI] [PubMed] [Google Scholar]
- 264. Zeligs JD, Wollman SH. 1981. Ultrastructure of cytokinesis in blood capillary endothelial cells in thyroid gland in vivo. J Ultrastruct Res 75:291–299 [DOI] [PubMed] [Google Scholar]
- 265. Smeds S, Wollman SH. 1983. 3H-thymidine labeling of endothelial cells in thyroid arteries, veins, and lymphatics during thyroid stimulation. Lab Invest 48:285–291 [PubMed] [Google Scholar]
- 266. Wollman SH, Herveg JP, Tachiwaki O. 1990. Histologic changes in tissue components of the hyperplastic thyroid gland during its involution in the rat. Am J Anat 189:35–44 [DOI] [PubMed] [Google Scholar]
- 267. Gérard AC, Poncin S, Caetano B, Sonveaux P, Audinot JN, Feron O, Colin IM, Soncin F. 2008. Iodine deficiency induces a thyroid stimulating hormone-independent early phase of microvascular reshaping in the thyroid. Am J Pathol 172:748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Ramsden JD, Buchanan MA, Egginton S, Watkinson JC, Mautner V, Eggo MC. 2005. Complete inhibition of goiter in mice requires combined gene therapy modification of angiopoietin, vascular endothelial growth factor, and fibroblast growth factor signaling. Endocrinology 146:2895–2902 [DOI] [PubMed] [Google Scholar]
- 269. Imada M, Kurosumi M, Fujita H. 1986. Three-dimensional aspects of blood vessels in thyroids from normal, low iodine diet-treated, TSH-treated, and PTU-treated rats. Cell Tissue Res 245:291–296 [DOI] [PubMed] [Google Scholar]
- 270. Michalkiewicz M, Huffman LJ, Connors JM, Hedge GA. 1989. Alterations in thyroid blood flow induced by varying levels of iodine intake in the rat. Endocrinology 125:54–60 [DOI] [PubMed] [Google Scholar]
- 271. Arntzenius AB, Smit LJ, Schipper J, van der Heide D, Meinders AE. 1991. Inverse relation between iodine intake and thyroid blood flow: color Doppler flow imaging in euthyroid humans. J Clin Endocrinol Metab 73:1051–1055 [DOI] [PubMed] [Google Scholar]
- 272. Colin I, Berbinschi A, Denef JF, Ketelslegers JM. 1992. Detection and identification of endothelin-1 immunoreactivity in rat and porcine thyroid follicular cells. Endocrinology 130:544–546 [DOI] [PubMed] [Google Scholar]
- 273. Colin IM, Selvais PL, Rebai T, Maiter DM, Adam E, vandenHove MF, Ketelslegers JM, Denef JF. 1994. Expression of the endothelin-1 gene in the rat thyroid gland and changes in its peptide and mRNA levels in goiter formation and iodide-induced involution. J Endocrinol 143:65–74 [DOI] [PubMed] [Google Scholar]
- 274. Colin IM, Gerard AC. 2010. The thyroid angiofollicular units, a biological model of functional and morphological integration. Bull Mémoires Acad Royale Belgique 165:218–230 [PubMed] [Google Scholar]
- 275. Richards OC, Raines SM, Attie AD. 2010. The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr Rev 31:343–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Eberhard D, Kragl M, Lammert E. 2010. ‘Giving and taking’: endothelial and β-cells in the islets of Langerhans. Trends Endocrinol Metab 21:457–463 [DOI] [PubMed] [Google Scholar]
- 277. Jansson L, Hellerström C. 1983. Stimulation by glucose of the blood flow to the pancreatic islets of the rat. Diabetologia 25:45–50 [DOI] [PubMed] [Google Scholar]
- 278. Nyman LR, Ford E, Powers AC, Piston DW. 2010. Glucose-dependent blood flow dynamics in murine pancreatic islets in vivo. Am J Physiol Endocrinol Metab 298:E807–E814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Carlsson PO, Olsson R, Källskog O, Bodin B, Andersson A, Jansson L. 2002. Glucose-induced islet blood flow increase in rats: interaction between nervous and metabolic mediators. Am J Physiol Endocrinol Metab 283:E457–E464 [DOI] [PubMed] [Google Scholar]
- 280. Ballian N, Brunicardi FC. 2007. Islet vasculature as a regulator of endocrine pancreas function. World J Surg 31:705–714 [DOI] [PubMed] [Google Scholar]
- 281. Orci L, Unger RH. 1975. Functional subdivision of islets of Langerhans and possible role of D cells. Lancet 2:1243–1244 [DOI] [PubMed] [Google Scholar]
- 282. Bergland RM, Page RB. 1979. Pituitary-brain vascular relations: a new paradigm. Science 204:18–24 [DOI] [PubMed] [Google Scholar]
- 283. Maghnie M, Altobelli M, Di Iorgi N, Genovese E, Meloni G, Manca-Bitti ML, Cohen A, Bernasconi S. 2004. Idiopathic central diabetes insipidus is associated with abnormal blood supply to the posterior pituitary gland caused by vascular impairment of the inferior hypophyseal artery system. J Clin Endocrinol Metab 89:1891–1896 [DOI] [PubMed] [Google Scholar]
- 284. de la Torre NG, Turner HE, Wass JA. 2005. Angiogenesis in prolactinomas: regulation and relationship with tumour behaviour. Pituitary 8:17–23 [DOI] [PubMed] [Google Scholar]
- 285. Ferrara N, Houck K, Jakeman L, Leung DW. 1992. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev 13:18–32 [DOI] [PubMed] [Google Scholar]
- 286. Macchiarelli G, Nottola SA, Palmerini MG, Bianchi S, Maione M, Lorenzo C, Stifano G, Di Marco E, Correr S. 2010. Morphological expression of angiogenesis in the mammalian ovary as seen by SEM of corrosion casts. Ital J Anat Embryol 115:109–114 [PubMed] [Google Scholar]
- 287. Araujo VR, Duarte AB, Bruno JB, Pinho Lopes CA, de Figueiredo JR. 2011. Importance of vascular endothelial growth factor (VEGF) in ovarian physiology of mammals. Zygote 1–10 [DOI] [PubMed] [Google Scholar]
- 288. Hinson JP, Vinson GP, Whitehouse BJ. 1986. The relationship between perfusion medium flow rate and steroid secretion in the isolated perfused rat adrenal gland in situ. J Endocrinol 111:391–396 [DOI] [PubMed] [Google Scholar]
- 289. Feige JJ. 2009. Angiogenesis in adrenocortical physiology and tumor development. Ann Endocrinol (Paris) 70:153–155 [DOI] [PubMed] [Google Scholar]
- 290. Kapitola J, Schullerova M, Schreiberova O. 1971. Blood flow and radioiodine uptake in the thyroid gland of rats after administration and discontinuation of carbimazole and perchlorate. Acta Endocrinol (Copenh) 68:817–825 [DOI] [PubMed] [Google Scholar]
- 291. Kapitola J, Schullerova M, Schreiberova O, Vilimovska D, Josifko M. 1974. Relation of TSH concentration in blood to the radioactive rubidium 86Rb uptake in the thyroid gland of rats: evidence of TSH regulatory effect on thyroid gland blood flow. Acta Endocrinol (Copenh) 77:266–275 [PubMed] [Google Scholar]
- 292. Connors JM, Huffman LJ, Hedge GA. 1988. Effects of thyrotropin on the vascular conductance of the thyroid gland. Endocrinology 122:921–929 [DOI] [PubMed] [Google Scholar]
- 293. Michalkiewicz M, Connors JM, Huffman LJ, Pietrzyk Z, Hedge GA. 1991. Compensatory changes in thyroid blood flow are only partially mediated by thyrotropin. Am J Physiol 260:E608–E612 [DOI] [PubMed] [Google Scholar]
- 294. Eggo MC, Quiney VM, Campbell S. 2003. Local factors regulating growth and function of human thyroid cells in vitro and in vivo. Mol Cell Endocrinol 213:47–58 [DOI] [PubMed] [Google Scholar]
- 295. Melander A, Ericson LE, Sundler F, Ingbar SH. 1974. Sympathetic innervation of the mouse thyroid and its significance in thyroid hormone secretion. Endocrinology 94:959–966 [DOI] [PubMed] [Google Scholar]
- 296. Melander A, Sundler F. 1979. Presence and influence of cholinergic nerves in the mouse thyroid. Endocrinology 105:7–9 [DOI] [PubMed] [Google Scholar]
- 297. Huffman L, Hedge GA. 1986. Effects of vasoactive intestinal peptide on thyroid blood flow and circulating thyroid hormone levels in the rat. Endocrinology 118:550–557 [DOI] [PubMed] [Google Scholar]
- 298. Ahrén B. 1986. Thyroid neuroendocrinology: neural regulation of thyroid hormone secretion. Endocr Rev 7:149–155 [DOI] [PubMed] [Google Scholar]
- 299. Michalkiewicz M, Connors JM, Huffman LJ, Hedge GA. 1989. Increases in thyroid gland blood flow after hemithyroidectomy in the rat. Endocrinology 124:1118–1123 [DOI] [PubMed] [Google Scholar]
- 300. Ahren B. 1991. Regulatory peptides in the thyroid gland: a review on their localization and function. Acta Endocrinol (Copenh) 124:225–232 [PubMed] [Google Scholar]
- 301. Young JB, Bürgi-Saville ME, Bürgi U, Landsberg L. 2005. Sympathetic nervous system activity in rat thyroid: potential role in goitrogenesis. Am J Physiol Endocrinol Metab 288:E861–E867 [DOI] [PubMed] [Google Scholar]
- 302. Colin IM, Nava E, Toussaint D, Maiter DM, vanDenhove MF, Lüscher TF, Ketelslegers JM, Denef JF, Jameson JL. 1995. Expression of nitric oxide synthase isoforms in the thyroid gland: evidence for a role of nitric oxide in vascular control during goiter formation. Endocrinology 136:5283–5290 [DOI] [PubMed] [Google Scholar]
- 303. Bidey SP, Hill DJ, Eggo MC. 1999. Growth factors and goitrogenesis. J Endocrinol 160:321–332 [DOI] [PubMed] [Google Scholar]
- 304. Ramsden JD. 2000. Angiogenesis in the thyroid gland. J Endocrinol 166:475–480 [DOI] [PubMed] [Google Scholar]
- 305. Feige JJ, Baird A. 1995. Crinopexy: extracellular regulation of growth factor action. Kidney Int Suppl 49:S15–S18 [PubMed] [Google Scholar]
- 306. Eggo MC. 2010. Molecular regulation of thyroid gland function. Curr Opin Endocrinol Diabetes Obes 17:396–401 [DOI] [PubMed] [Google Scholar]
- 307. de Araujo LF, Grozovsky R, dos Santos Pereira MJ, de Carvalho JJ, Vaisman M, Carvalho DP. 2010. Expressions of vascular endothelial growth factor and nitric oxide synthase III in the thyroid gland of ovariectomized rats are upregulated by estrogen and selective estrogen receptor modulators. Thyroid 20:85–92 [DOI] [PubMed] [Google Scholar]
- 308. Jansson L, Andersson A, Bodin B, Kallskog O. 2007. Pancreatic islet blood flow during euglycaemic, hyperinsulinaemic clamp in anaesthetized rats. Acta Physiol (Oxf) 189:319–324 [DOI] [PubMed] [Google Scholar]
- 309. Pedraza PE, Obregon MJ, Escobar-Morreale HF, del Rey FE, de Escobar GM. 2006. Mechanisms of adaptation to iodine deficiency in rats: thyroid status is tissue specific. Its relevance for man. Endocrinology 147:2098–2108 [DOI] [PubMed] [Google Scholar]
- 310. Visser TJ. 2006. The elemental importance of sufficient iodine intake: a trace is not enough. Endocrinology 147:2095–2097 [DOI] [PubMed] [Google Scholar]
- 311. Kopp PA. 2008. Reduce, recycle, reuse: iodotyrosine deiodinase in thyroid iodide metabolism. N Engl J Med 358:1856–1859 [DOI] [PubMed] [Google Scholar]
- 312. Geers C, Colin IM, Gérard AC. 2011. Delta-like 4/Notch pathway is differentially regulated in benign and malignant thyroid tissues. Thyroid 21:1323–1330 [DOI] [PubMed] [Google Scholar]
- 313. Rehman AO, Wang CY. 2006. Notch signaling in the regulation of tumor angiogenesis. Trends Cell Biol 16:293–300 [DOI] [PubMed] [Google Scholar]
- 314. Sainson RC, Harris AL. 2007. Anti-Dll4 therapy: can we block tumour growth by increasing angiogenesis? Trends Mol Med 13:389–395 [DOI] [PubMed] [Google Scholar]
- 315. Roy M, Pear WS, Aster JC. 2007. The multifaceted role of Notch in cancer. Curr Opin Genet Dev 17:52–59 [DOI] [PubMed] [Google Scholar]
- 316. Dufraine J, Funahashi Y, Kitajewski J. 2008. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene 27:5132–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Bray SJ. 2006. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7:678–689 [DOI] [PubMed] [Google Scholar]
- 318. Ferretti E, Tosi E, Po A, Scipioni A, Morisi R, Espinola MS, Russo D, Durante C, Schlumberger M, Screpanti I, Filetti S, Gulino A. 2008. Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J Clin Endocrinol Metab 93:4080–4087 [DOI] [PubMed] [Google Scholar]
- 319. Studer H, Forster R, Conti A, Kohler H, Haeberli A, Engler H. 1978. Transformation of normal follicles into thyrotropin-refractory “cold” follicles in the aging mouse thyroid gland. Endocrinology 102:1576–1586 [DOI] [PubMed] [Google Scholar]
- 320. Tamura S, Fujita H. 1981. Fine structural aspects on the cold follicles in the aged mouse thyroid. Arch Histol Jpn 44:177–188 [DOI] [PubMed] [Google Scholar]
- 321. Mestdagh C, Many MC, Halpern S, Briançon C, Fragu P, Denef JF. 1990. Correlated autoradiographic and ion-microscopic study of the role of iodine in the formation of “cold” follicles in young and old mice. Cell Tissue Res 260:449–457 [DOI] [PubMed] [Google Scholar]
- 322. Meier CA. 2000. Thyroid nodules: pathogenesis, diagnosis and treatment. Baillieres Best Pract Res Clin Endocrinol Metab 14:559–575 [DOI] [PubMed] [Google Scholar]
- 323. Peter HJ, Gerber H, Studer H, Smeds S. 1985. Pathogenesis of heterogeneity in human multinodular goiter. A study on growth and function of thyroid tissue transplanted onto nude mice. J Clin Invest 76:1992–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Gerber H, Peter HJ, Studer H. 1987. Age-related failure of endocytosis may be the pathogenetic mechanism responsible for “cold” follicle formation in the aging mouse thyroid. Endocrinology 120:1758–1764 [DOI] [PubMed] [Google Scholar]
- 325. Gerber H, Peter HJ, Bachmeier C, Kaempf J, Studer H. 1987. Progressive recruitment of follicular cells with graded secretory responsiveness during stimulation of the thyroid gland by thyrotropin. Endocrinology 120:91–96 [DOI] [PubMed] [Google Scholar]
- 326. Smeds S, Peter HJ, Jörtsö E, Gerber H, Studer H. 1987. Naturally occurring clones of cells with high intrinsic proliferation potential within the follicular epithelium of mouse thyroids. Cancer Res 47:1646–1651 [PubMed] [Google Scholar]
- 327. Huber G, Derwahl M, Kaempf J, Peter HJ, Gerber H, Studer H. 1990. Generation of intercellular heterogeneity of growth and function in cloned rat thyroid cells (FRTL-5). Endocrinology 126:1639–1645 [DOI] [PubMed] [Google Scholar]
- 328. Derwahl M, Studer H, Huber G, Gerber H, Peter HJ. 1990. Intercellular propagation of individually programmed growth bursts in FRTL-5 cells. Implications for interpreting growth factor actions. Endocrinology 127:2104–2110 [DOI] [PubMed] [Google Scholar]
- 329. Roger PP, Baptist M, Dumont JE. 1992. A mechanism generating heterogeneity in thyroid epithelial cells: suppression of the thyrotropin/cAMP-dependent mitogenic pathway after cell division induced by cAMP-independent factors. J Cell Biol 117:383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Studer H, Peter HJ, Gerber H. 1989. Natural heterogeneity of thyroid cells: the basis for understanding thyroid function and nodular goiter growth. Endocr Rev 10:125–135 [DOI] [PubMed] [Google Scholar]
- 331. Kopp P, Kimura ET, Aeschimann S, Oestreicher M, Tobler A, Fey MF, Studer H. 1994. Polyclonal and monoclonal thyroid nodules coexist within human multinodular goiters. J Clin Endocrinol Metab 79:134–139 [DOI] [PubMed] [Google Scholar]
- 332. Studer H, Derwahl M. 1995. Mechanisms of nonneoplastic endocrine hyperplasia: a changing concept: a review focused on the thyroid gland. Endocr Rev 16:411–426 [DOI] [PubMed] [Google Scholar]
- 333. Kimura ET, Kopp P, Zbaeren J, Asmis LM, Ruchti C, Maciel RM, Studer H. 1999. Expression of transforming growth factor β1, β2, and β3 in multinodular goiters and differentiated thyroid carcinomas: a comparative study. Thyroid 9:119–125 [DOI] [PubMed] [Google Scholar]
- 334. Derwahl M, Studer H. 2000. Multinodular goitre: ‘much more to it than simply iodine deficiency.’ Baillieres Best Pract Res Clin Endocrinol Metab 14:577–600 [DOI] [PubMed] [Google Scholar]
- 335. Derwahl M, Studer H. 2001. Nodular goiter and goiter nodules: Where iodine deficiency falls short of explaining the facts. Exp Clin Endocrinol Diabetes 109:250–260 [DOI] [PubMed] [Google Scholar]
- 336. Derwahl M, Studer H. 2002. Hyperplasia versus adenoma in endocrine tissues: are they different? Trends Endocrinol Metab 13:23–28 [DOI] [PubMed] [Google Scholar]
- 337. Many MC, Denef JF, Hamudi S, Haumont S. 1986. Increased follicular heterogeneity in experimental colloid goiter produced by refeeding iodine excess after thyroid hyperplasia. Endocrinology 118:637–644 [DOI] [PubMed] [Google Scholar]
- 338. Jovanovic L, Delahunt B, McIver B, Eberhardt NL, Grebe SK. 2003. Thyroid gland clonality revisited: the embryonal patch size of the normal human thyroid gland is very large, suggesting X-chromosome inactivation tumor clonality studies of thyroid tumors have to be interpreted with caution. J Clin Endocrinol Metab 88:3284–3291 [DOI] [PubMed] [Google Scholar]
- 339. Chen K, Wei Y, Sharp GC, Braley-Mullen H. 2000. Characterization of thyroid fibrosis in a murine model of granulomatous experimental autoimmune thyroiditis. J Leukoc Biol 68:828–835 [PubMed] [Google Scholar]
- 340. Cyniak-Magierska A, Januszkiewicz-Caulier J, Brzeziañska E, Lewiñski A. 2010. Analysis of correlation between the process of thyroid fibrosis and TGFB1 gene expression level in fine-needle aspiration biopsy (FNAB) thyroid specimens collected from patients with Hashimoto's thyroiditis and non-toxic goitre. Exp Clin Endocrinol Diabetes 118:420–426 [DOI] [PubMed] [Google Scholar]
- 341. Krohn K, Wohlgemuth S, Gerber H, Paschke R. 2000. Hot microscopic areas of iodine-deficient euthyroid goitres contain constitutively activating TSH receptor mutations. J Pathol 192:37–42 [DOI] [PubMed] [Google Scholar]
- 342. Thomas T, Nowka K, Lan L, Derwahl M. 2006. Expression of endoderm stem cell markers: evidence for the presence of adult stem cells in human thyroid glands. Thyroid 16:537–544 [DOI] [PubMed] [Google Scholar]
- 343. Lin RY, Davies TF. 2006. Derivation and characterization of thyrocyte-like cells from embryonic stem cells in vitro. Methods Mol Biol 330:249–261 [DOI] [PubMed] [Google Scholar]
- 344. Lan L, Cui D, Nowka K, Derwahl M. 2007. Stem cells derived from goiters in adults form spheres in response to intense growth stimulation and require thyrotropin for differentiation into thyrocytes. J Clin Endocrinol Metab 92:3681–3688 [DOI] [PubMed] [Google Scholar]
- 345. Hoshi N, Kusakabe T, Taylor BJ, Kimura S. 2007. Side population cells in the mouse thyroid exhibit stem/progenitor cell-like characteristics. Endocrinology 148:4251–4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Fierabracci A, Puglisi MA, Giuliani L, Mattarocci S, Gallinella-Muzi M. 2008. Identification of an adult stem/progenitor cell-like population in the human thyroid. J Endocrinol 198:471–487 [DOI] [PubMed] [Google Scholar]
- 347. Derwahl M. 2011. Linking stem cells to thyroid cancer. J Clin Endocrinol Metab 96:610–613 [DOI] [PubMed] [Google Scholar]
- 348. Davies TF, Latif R, Minsky NC, Ma R. 2011. Clinical review: The emerging cell biology of thyroid stem cells. J Clin Endocrinol Metab 96:2692–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414:105–111 [DOI] [PubMed] [Google Scholar]
- 350. Moore KA, Lemischka IR. 2006. Stem cells and their niches. Science 311:1880–1885 [DOI] [PubMed] [Google Scholar]
- 351. Visvader JE, Lindeman GJ. 2008. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8:755–768 [DOI] [PubMed] [Google Scholar]
- 352. Arufe MC, Lu M, Kubo A, Keller G, Davies TF, Lin RY. 2006. Directed differentiation of mouse embryonic stem cells into thyroid follicular cells. Endocrinology 147:3007–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Lin RY, Kubo A, Keller GM, Davies TF. 2003. Committing embryonic stem cells to differentiate into thyrocyte-like cells in vitro. Endocrinology 144:2644–2649 [DOI] [PubMed] [Google Scholar]
- 354. Mitsutake N, Iwao A, Nagai K, Namba H, Ohtsuru A, Saenko V, Yamashita S. 2007. Characterization of side population in thyroid cancer cell lines: cancer stem-like cells are enriched partly but not exclusively. Endocrinology 148:1797–1803 [DOI] [PubMed] [Google Scholar]
- 355. Zito G, Richiusa P, Bommarito A, Carissimi E, Russo L, Coppola A, Zerilli M, Rodolico V, Criscimanna A, Amato M, Pizzolanti G, Galluzzo A, Giordano C. 2008. In vitro identification and characterization of CD133(pos) cancer stem-like cells in anaplastic thyroid carcinoma cell lines. PLoS One 3:e3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, Gulotta G, Dieli F, Giordano S, De Maria R, Stassi G. 2010. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res 70:8874–8885 [DOI] [PubMed] [Google Scholar]
- 357. Malaguarnera R, Frasca F, Garozzo A, Gianì F, Pandini G, Vella V, Vigneri R, Belfiore A. 2011. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab 96:766–774 [DOI] [PubMed] [Google Scholar]