

Abstract
The safety of progestogens as a class has come under increased scrutiny after the publication of data from the Women's Health Initiative trial, particularly with respect to breast cancer and cardiovascular disease risk, despite the fact that only one progestogen, medroxyprogesterone acetate, was used in this study. Inconsistency in nomenclature has also caused confusion between synthetic progestogens, defined here by the term progestin, and natural progesterone. Although all progestogens by definition have progestational activity, they also have a divergent range of other properties that can translate to very different clinical effects. Endometrial protection is the primary reason for prescribing a progestogen concomitantly with postmenopausal estrogen therapy in women with a uterus, but several progestogens are known to have a range of other potentially beneficial effects, for example on the nervous and cardiovascular systems. Because women remain suspicious of the progestogen component of postmenopausal hormone therapy in the light of the Women's Health Initiative trial, practitioners should not ignore the potential benefits to their patients of some progestogens by considering them to be a single pharmacological class. There is a lack of understanding of the differences between progestins and progesterone and between individual progestins differing in their effects on the cardiovascular and nervous systems, the breast, and bone. This review elucidates the differences between the substantial number of individual progestogens employed in postmenopausal hormone therapy, including both progestins and progesterone. We conclude that these differences in chemical structure, metabolism, pharmacokinetics, affinity, potency, and efficacy via steroid receptors, intracellular action, and biological and clinical effects confirm the absence of a class effect of progestogens.
Introduction
Classification of Progestogens
-
Structure-Function Relationships of Progestogens
Progestogens structurally related to progesterone
Progestogens structurally related to testosterone
Metabolism of Progestogens
-
Pharmacokinetics of Progestogens
Progestogens administered orally
Progestogens administered parenterally
Drug interactions
-
Intracellular Mechanisms of Action of Progestogens
Steroid receptor structure, distribution and ligand binding
Potency, efficacy and biocharacter of progestogens via steroid receptors
Regulation of transcription by progestogens: genomic effects
Nongenomic effects of progestogens
-
Clinical Effects of Progestogens in Postmenopausal Hormone Therapy
Effects on the endometrium
Effects on the breast
Effects on the cardiovascular system
Effects on the brain
Effects on bone
Conclusions
I. Introduction
Progestogens, compounds that exhibit progestational activity, include the only natural progestogen, progesterone, and a variety of synthetic progestogens. In postmenopausal women, progestogens are used therapeutically for protecting the endometrium against hyperplasia during estrogen therapy. One of the most widely used progestogens for that purpose is medroxyprogesterone acetate (MPA), which has been used for a considerable number of years, either continuously, combined with an estrogen, or sequentially. However, the safety of MPA and that of all other progestogens has been questioned after the results of the estrogen-plus-progestogen and estrogen-alone arms of the Women's Health Initiative (WHI) trial were published. The data showed increased breast cancer risk with the estrogen/MPA formulation but decreased risk with estrogen alone (1, 2). Although MPA was the only progestogen used in the WHI trial, safety concerns have recently been directed toward progestogens as a general class.
The objective of this review is to determine whether there is any reliable evidence to support the view for a general, uniform effect (class effect) of progestogens. To this end, progestogens will be compared with respect to their chemical structure, structure-function relationships, metabolism, pharmacokinetic parameters, potency, and efficacy via steroid receptors, intracellular mechanism of action, affinity, and biological and clinical effects.
II. Classification of Progestogens
The definition of a progestogen as a compound with progestational activity refers to its action of inducing a secretory endometrium to support gestation. This function of the rising levels of endogenous progesterone after ovulation prepares the endometrium for implantation of a fertilized egg, as well as supporting the uterine lining during a pregnancy, when circulating progesterone reaches characteristically high levels. The term progestogen has been used synonymously with other terms, such as progestagen, gestogen, gestagen, and progestin (3). However, recently, the term progestin has often been used exclusively to describe synthetic progestogens such as MPA, norethindrone, and levonorgestrel, thus excluding the natural progestogen, progesterone. Use of the term progestogen is consistent with the nomenclature of other hormone groups, such as androgens and estrogens, which are defined as compounds having androgenic and estrogenic activity, respectively. To avoid confusion in light of current practices, the North American Menopause Society has recommended that the term progestogen should be used when referring to progesterone and synthetic progestogens collectively, whereas the name progestin is specific only to synthetic progestogens (4). The nomenclature recommended by North American Menopause Society will be used in the present article.
Progestogens can be divided into two types: natural and synthetic (Table 1) (5). As stated earlier, there is only one natural progestogen, progesterone, which has the chemical structure shown in Fig. 1A. In contrast, there are a variety of progestins that are available for therapeutic use, which vary widely in their chemical structures, as evident in Figs. 2–6. For convenience, these have been classified into two groups: 1) those structurally related to progesterone and 2) those structurally related to testosterone. The chemical structure of testosterone is shown in Fig. 1B. These structural similarities have nothing to do with the actual precursor used to synthesize the progestins, which are derived by multiple chemical reactions from a variety of starting compounds.
Table 1.
Classification of progestogens
| Classification | Progestogen |
|---|---|
| Natural | Progesterone |
| Synthetic | |
| Structurally related to progesterone | |
| Pregnane derivatives | |
| Acetylated | MPA, megestrol acetate, chlormadinone acetate, cyproterone acetate |
| Nonacetylated | Dydrogesterone, medrogestone |
| 19-Norpregnane derivatives | |
| Acetylated | Nomegestrol acetate, nesterone |
| Nonacetylated | Demegestone, promegestone, trimegestone |
| Structurally related to testosterone | |
| Ethinylated | |
| Estranes | Norethindrone, norethindrone acetate, ethynodiol diacetate, norethynodrel, lynestrenol, tibolone |
| 13-Ethylgonanes | Levonorgestrel, desogestrel, norgestimate, gestodene |
| Nonethinylated | Dienogest, drospirenone |
Figure 1.

A, Chemical structure of the natural progestogen, progesterone; B, chemical structure of testosterone.
Figure 2.

Progestins structurally related to progesterone: pregnane derivatives, acetylated and nonacetylated.
Figure 3.

Progestins structurally related to progesterone: 19-norpregnane derivatives, acetylated and nonacetylated.
Figure 4.

Progestins structurally related to testosterone: ethinylated derivatives, estranes.
Figure 5.

Progestins structurally related to testosterone: ethinylated derivatives, 13-ethylgonanes.
Figure 6.

Progestins structurally related to testosterone: nonethinylated derivatives.
Progestins structurally related to progesterone can be subdivided into those with and without a methyl group at carbon 10, i.e. pregnane and 19-norpregnane derivatives, respectively. These derivatives are further classified as those that are acetylated and those that are not. Progestins structurally related to testosterone can be subdivided into those that contain an ethinyl group at carbon 17 and those that are nonethinylated. The ethinylated derivatives are further classified as those that have an estrane structure and those that have an 13-ethylgonane structure.
III. Structure-Function Relationships of Progestogens
The biological activity of a progestogen changes considerably, depending on its chemical structure, particularly with respect to pharmacokinetics and potency (5). Structural aspects are discussed below and depicted in Figs. 2–6; differences in pharmacokinetics, potency, and efficacy will be addressed later in this review.
A. Progestogens structurally related to progesterone
1. Pregnane derivatives (Fig. 2)
Starting with progesterone, the addition of a hydroxyl group at carbon 17 renders it devoid of biological progestational activity, but acetylation of that hydroxyl group restores some progestational activity, and the molecule is somewhat active when administered orally. Taking the additional step of adding a methyl group at carbon 6, the resulting molecule, MPA, exhibits relatively high progestational activity and is highly active when given orally (6).
Three highly potent progestogens, megestrol acetate, chlormadinone acetate, and cyproterone acetate, are structurally related to the MPA molecule. Megestrol acetate differs from MPA only in the presence of a double bond between carbons 6 and 7. Chlormadinone acetate and cyproterone acetate have, in addition to a double bond between carbons 6 and 7, a chloral group substituted for the methyl group at carbon 6. Cyproterone acetate differs from chlormadinone acetate only in that it has a methylene group attached to carbons 1 and 2.
Dydrogesterone is one of a group of compounds called retroprogesterones, which have a methyl group at carbon 10 but are not acetylated. They are unique in that the methyl group at carbon 10 is in the α-orientation, instead of the β-orientation seen in progesterone and the pregnanes. Dydrogesterone also has a double bond between carbons 6 and 7, and unlike progesterone, apparently does not inhibit ovulation when given throughout the menstrual cycle and does not alter the basal body temperature (7). These dramatic differences in peripheral and central effects therefore seem to be a consequence of the change in spatial orientation of the methyl group at carbon 10.
Medrogestone, also nonacetylated and having a methyl group at carbon 10, differs from progesterone in that it contains a methyl group at carbons 6 and 17 and a double bond between carbons 6 and 7.
2. 19-Norpregnane derivatives (Fig. 3)
The norpregnane derivatives all lack a methyl group at carbon 10 and include nomegestrol acetate, nesterone, demegestone, promegestone, and trimegestone. Apart from the absence of the methyl group at carbon 10, the norpregnane derivative nomegestrol acetate is identical to that of the pregnane derivative megestrol acetate. Nesterone differs from nomegestrol acetate in the presence of a methylene group at carbon 16 and absence of the methyl group at carbon 6 and the double bond between carbons 6 and 7. Unlike nomegestrol acetate and nesterone, demegestone, promegestone, and trimegestone all have a double bond between carbons 9 and 10 and a methyl group substituted for the acetate group at carbon 17. Promegestone and trimegestone also have a methyl group on the two-carbon side chain at carbon 17; in trimegestone, the penultimate carbon is also hydroxylated.
B. Progestogens structurally related to testosterone
1. Ethinylated derivatives: estranes
Starting with the testosterone molecule instead of progesterone, biological activity can be dramatically altered by small changes in the molecular structure. Androgenicity is substantially reduced by the addition of an ethinyl group to form the new compound 17α-ethinyltestosterone, commonly known as ethisterone, which has some progestational and oral activity. These progestogenic and oral activities of ethisterone are further enhanced, and androgenicity almost eliminated, by removal of the methyl group at carbon 10 to form norethindrone (U.S. name), known as norethisterone in Europe and elsewhere.
The norethindrone family of progestogens is referred to as the estranes (Fig. 4) because they all have the same 18-carbon steroid nucleus as the parent steroid, estrane. This family of progestogens also includes norethindrone acetate, ethynodiol diacetate, norethynodrel, and lynestrenol. Norethindrone acetate and ethynodiol diacetate differ from norethindrone by having an acetate group at carbon 3 and at carbons 3 and 17, respectively. Norethynodrel has an identical structure to norethindrone except for a shift in the double bond from carbons 4, 5 of norethindrone to carbons 5, 10 in the norethynodrel molecule. Lynestrenol differs from norethindrone only in the absence of an oxygenated functional group at carbon 3. Tibolone is identical in structure to norethynodrel except it has a methyl group at carbon 7. It is metabolized rapidly and extensively into three metabolites. Two of its metabolites, 3α- and 3β-hydroxytibolone, bind to the estrogen receptor (ER), whereas the Δ4 metabolite binds to the progesterone receptor (PR). Tibolone itself binds with low affinity to the progesterone and androgen receptors.
2. Ethinylated derivatives: 13-ethyl gonanes
When the methyl group at carbon 13 of norethindrone is replaced by an ethyl group, a racemic mixture of d-(−)-norgestrel (levonorgestrel) and l-(+)-norgestrel (dextronorgestrel) results (8). Levonorgestrel is the biologically active form of norgestrel and has proved to be one of the most potent orally active progestogens (7).
The levonorgestrel family of progestogens is sometimes referred to as gonanes; however, this is not appropriate because all steroids by definition are gonanes because they contain the 4-ring carbon nucleus (gonane). A more appropriate name for these progestogens is 13-ethyl gonanes (Fig. 5) (9).
Other progestogens in the levonorgestrel family of 13-ethylgonanes include desogestrel, norgestimate, and gestodene. Having arrived on the scene more recently than levonorgestrel, norethindrone, and progestogens structurally related to norethindrone, these compounds are often referred to as the new progestogens. Desogestrel differs from levonorgestrel by having no oxygenated functional group at carbon 3 but a methylene group at carbon 11, whereas norgestimate has an oxime group at carbon 3 and an acetate group at carbon 17. Gestodene is closer in structure to the parent compound levonorgestrel, merely having an additional double bond between carbons 15 and 16.
3. Nonethinylated derivatives
The nonethinylated subgroup of progestogens consists of the compounds dienogest and drospirenone (Fig. 6). Dienogest is similar in structure to norethindrone except for a cyanomethyl group instead of an ethinyl group at carbon 17 and a double bond between carbons 9 and 10. Drospirenone is structurally related to spironolactone and contains the androstane skeleton to which are attached methylene groups at carbons 6 and 7, as well as carbons 15 and 16, and a carbolactone group at carbon 17.
IV. Metabolism of Progestogens
The metabolism of progestogens is poorly understood, largely because relatively few studies on the metabolism of the different progestogens have been carried out.
Progestogens administered orally undergo hepatic first-pass metabolism. The extent to which this occurs varies and depends on the chemical structure of the progestogen. After oral ingestion, progestogens are first subjected to incomplete metabolism by enzymes in intestinal bacteria and the intestinal mucosa. The enzymes include reductases and dehydrogenases, which can add hydrogens to double bonds and ketone groups on progestogen molecules, forming 5α- or 5β-dihydro, 3α- or 3β-hydroxy, and/or 20α- or 20β-hydroxy metabolites.
The metabolized and unmetabolized progestogens are absorbed and enter the portal vein blood at high concentrations. In the liver, they are subjected to a plethora of steroidogenic enzymes, including cytochrome P450 enzymes, which are capable of transforming the metabolized and unmetabolized progestogen molecule into numerous metabolites. Progestogens can also undergo enterohepatic recirculation, but the extent to which this occurs for the different progestogens is poorly understood.
After parenteral administration of a progestogen, the liver is still a major site of progestogen metabolism, even though there is no hepatic first-pass metabolism. The major difference between the metabolism of a drug given orally and one administered parenterally is that the liver is initially exposed to a highly concentrated bolus of unmetabolized and metabolized progestogen.
Of all the studies on metabolism of different progestogens, we know most about progesterone metabolism. Progesterone is highly vulnerable to enzymatic reduction by reductases and hydroxysteroid dehydrogenases during hepatic first-pass metabolism, because its structure contains two ketone groups and a double bond (10). Thus, the molecule is transformed to two isomers of dihydroprogesterone, four pregnanolone isomers, and eight isomers of pregnanediol. In addition, progesterone can undergo hydroxylation by cytochrome P450 enzymes. Subsequently, all progesterone metabolites with a hydroxyl group can be sulfated and glucuronidated, and these conjugated products are then excreted in urine and feces. In addition to undergoing extensive transformation during the hepatic first pass, progesterone is also poorly absorbed when administered in a crystalline form. However, when the crystals are broken down to fine particles by the process of micronization, its absorption is improved substantially. The micronization process gives rise to a greater surface area of the compound, allowing it to be dissolved more readily in the aqueous medium of the intestine.
Surprisingly, very little is known about the metabolism of the progestogen most widely used for postmenopausal hormone therapy (HT), i.e. MPA. It has been shown that MPA undergoes ring A reduction, hydroxylation at carbons 6 and 21, and conjugation (primarily glucuronidation) (5). Because ring A of MPA possesses the Δ4-3-ketone structure found in progesterone, one would expect that the two functional groups would be reduced in a similar manner as those in progesterone; i.e. ring A dihydro and tetrahydro MPA metabolites would be formed. However, unlike progesterone, the reduction of the ketone group at carbon 20 may be impaired due to possible steric hindrance by the acetate group at carbon 17 on the MPA molecule.
Little is also known about the other progestins related in chemical structure to progesterone. However, one would expect those progestins that have a Δ4-3-ketone structure and/or a ketone group at carbon 20 to undergo reduction in a similar manner as progesterone. Again, reduction may be impaired at carbon 20 in the presence of a functional group (acetate or methyl) at carbon 17 due to steric hindrance.
Relatively more is known about the metabolism of progestins structurally related to testosterone (8). It has been shown that norethindrone and levonorgestrel undergo extensive ring A reactions forming reduced and, to a lesser extent, hydroxylated metabolites. The parent compounds and their metabolites can be conjugated, forming sulfated and glucuronidated products, which are excreted primarily in urine and also in feces. It has also been shown that significant amounts of ethinylestradiol are formed after administration of norethindrone orally to postmenopausal women (11, 12). In fact, it was estimated that oral administration of a 0.5- to 1.0-mg dose of norethindrone combined with ethinylestradiol may add as much as 2–10 μg ethinylestradiol to the existing dose (11).
What is the biological significance of progestogen metabolites? First, some progestogens are prodrugs and require biochemical transformation to active metabolites. The norethindrone derivatives, which include norethindrone acetate, ethynodiol diacetate, norethynodrel, and lynestrenol, have no progestational activity. However, after their oral administration, they are rapidly converted to the progestationally active compound, norethindrone. Desogestrel and norgestimate are also prodrugs. The former compound is converted to the active progestogen etonogestrel (previously called 3-ketodesogestrel), whereas norgestimate is converted to the progestationally active metabolites levonorgestrel and norelgestromin (previously called levonorgestrel-3-oxime). Second, conjugated progestogen metabolites, such as sulfates of norethindrone and levonorgestrel, which are inactive, may form circulating reservoirs from which the active progestogens may be obtained by sulfatase activity. Third, the steroidal milieu consisting of numerous metabolites obtained after administration of a progestogen is unique for each progestogen. Different biological effects may be produced by administered progestogens, due to the specific influence of each progestogen and its metabolites on the conformation of the progestogen receptor and its subsequent activation of transcription in target cells.
V. Pharmacokinetics of Progestogens
Pharmacokinetics (absorption, distribution, and excretion) determine how much of the progestogen administered is available to tissues, primarily by measuring its blood level, and the amount that enters the cells is determined by the extent to which it is bound to carrier proteins that cannot cross the cell membranes. After a progestogen enters the systemic circulation, it is distributed between blood and tissues by passive diffusion. The pattern of distribution of the progestogen is mainly regulated by its binding to transport proteins and tissue receptors. In the blood compartment, all progestogens are bound with low affinity and high capacity to albumin. In addition, some of the progestogens that are structurally related to testosterone also bind with high affinity but low capacity to SHBG; they include norethindrone, levonorgestrel, etonogestrel, and gestodene (13, 14) (Table 2). A relatively smaller amount of progesterone is also bound with high affinity and low capacity, but not to SHBG; instead, it is bound to corticosteroid-binding globulin (15) (Table 2). The binding of progestogens to transport proteins is reversible, so that a change in the concentration of a binding protein in one compartment is followed by a reequilibration of these compounds in that compartment. Alterations in binding protein concentrations may contribute to the kinetic variability of a progestogen.
Table 2.
Distribution of progestogens bound to SHBG or CBG in blood
| Progestogen (Ref.) | SHBG-bound (%) | CBG-bound (%) | Albumin-bound (%) | Free (%) |
|---|---|---|---|---|
| Norethindrone (13) | 35.5 | ND | 60.8 | 3.7 |
| Levonorgestrel (13) | 47.5 | ND | 50.0 | 2.5 |
| Etonogestrel (14) | 31.6 | ND | 65.9 | 2.5 |
| Gestodene (14) | 75.3 | ND | 24.1 | 0.6 |
| Progesterone (15) | 0.6 | 17.7 | 79.3 | 2.4 |
CBG, Corticosteroid-binding globulin; ND, not detected.
It is well recognized that the non-protein-bound (unbound or free) fraction of a steroid is available for metabolism in steroid-metabolizing cells or binding to a receptor in target cells. However, because the binding of steroids to albumin is relatively weak, albumin-bound steroids are also generally considered to be available for metabolism or binding to receptors. There is a paucity of data on free and bioavailable (albumin-bound plus free) fractions of progestogens.
A. Progestogens administered orally
The most common route of progestogen administration for postmenopausal HT and steroidal contraception is oral, yet there is a paucity of information on the pharmacokinetics of progestogens by this route. Progestogens given orally generally reach a maximum concentration within 1–3 h; the maximum concentration and area under the curve are dose dependent. Information on bioavailability and half-life has been derived from frequent blood sampling during 24 h after oral dosing. Bioavailability represents the amount of the progestogen that is found in the circulation after undergoing hepatic first-pass metabolism, estimated by plotting the blood level of the drug against time after administering a given dose both orally and iv and then comparing the areas under the curve; the resulting fraction is multiplied by 100%. Half-life is the time (in hours) over which a drug's blood level drops to one half of its highest value after dosing. Approximate values taken from the literature (16–31) for bioavailabilities and half-lives of progestogens are summarized in Table 3.
Table 3.
Average bioavailabilities and half-lives of progestogens
| Progestogen | Dose (mg) | Bioavailability (%) | Half-life (h) | Ref. |
|---|---|---|---|---|
| Progesterone | 100, 200, 300 | <5 | 16.2–18.3 | 16 |
| MPA | 10 | >90 | 24 | 17 |
| Megestrol acetate | 160 | NA | 22.3 | 18 |
| Cyproterone acetate | 2 | NA | 54.0–78.6 | 19 |
| Chlormadinone acetate | 2 | ∼100 | 80.1 | 20 |
| Medrogestone | 5 | NA | 34.9 | 21 |
| Dydrogesterone | 10 | 28 | 14–17 | —a |
| Nomegestrol acetate | 2.5 | 60 | 50 | 22 |
| Trimegestone | 0.5 | ∼100 | 15 | —a |
| Norethindrone | 1 | 64 | 8 | 23 |
| Levonorgestrel | 0.15–0.25 | 89/99b | 9.9/13.2b | 24 |
| Desogestrel | 0.15 | 62/76b | 11.9/23.8b | 25, 26 |
| Gestodene | 0.075 | 87/99b | 12–14 | 27, 28 |
| Dienogest | 4 | 96.2 | 10.8/11.6b | 29 |
| Drospirenone | 3 | 66 | 31.1–32.5 | 30, 31 |
NA, No data available.
The data were obtained from a package insert.
Multiple bioavailability or half-life values are shown.
Among progesterone and progestogens structurally related to progesterone, the highest bioavailabilities (>90%) are obtained with MPA, chlormadinone acetate, and trimegestone. In contrast, the bioavailability of progesterone is only less than 5%, and that of dydrogesterone and nomegestrol acetate is 28 and 60%, respectively. Chlormadinone acetate, cyproterone acetate, and nomegestrol acetate have the longest half-lives (80.1, 54.0–78.6, and 50 h, respectively), whereas that of medrogestone is substantially lower (34.9 h). Progesterone and other progestogens related to progesterone (including MPA, megestrol acetate, dydrogesterone, and trimegestone) have even shorter half-lives, ranging from 15–24 h.
Among progestogens structurally related to testosterone, the highest bioavailabilities are achieved with levonorgestrel, gestodene, and dienogest, reaching more than 90%, whereas norethindrone, desogestrel, and drospirenone have bioavailabilities in the range of 62–76%. The longest half-life occurs with drospirenone (31.1–32.5 h), whereas norethindrone has the shortest (8 h); intermediate half-lives between these two extremes are observed for levonorgestrel, desogestrel, gestodene, and dienogest.
Circulating levels and pharmacokinetic parameters of a progestogen given orally can vary considerably, by up to 5-fold or more, among women. Bioavailability can be significantly affected by age because of decreased hepatic cytochrome P450 content with aging, which reduces the extent of hepatic first-pass metabolism resulting in increased oral bioavailability. Elderly women may also have reduced renal clearance of circulating drug as well as a volume of distribution that is enhanced for lipid-soluble drugs and diminished for water-soluble drugs. To a lesser extent, pharmacokinetics can also vary within the same individual under different conditions.
B. Progestogens administered parenterally
In an attempt to avoid the hepatic first-pass metabolism of progestogens, a variety of parenteral routes of administration have been used, which include im, vaginal, percutaneous, intranasal, sublingual, and rectal. The limited data that exist concerning the pharmacokinetics of those that are more commonly used are discussed below.
1. Intramuscular route
In one study, four doses (10, 25, 50, or 100 mg) of progesterone in oil were injected im in six postmenopausal women (32). A typical depot effect was seen, with elevated progesterone levels persisting for 24–48 h. Circulating progesterone levels similar to those seen in a normal menstrual cycle luteal phase could be achieved with a single 25-mg im injection of progesterone in oil.
2. Intravaginal route
A comparative study of vaginal and im administration of micronized progesterone found the intravaginal dosing to be an effective, acceptable, and convenient alternative to im injections (33). In this study, 15 women received 200 mg micronized progesterone intravaginally every 6 h, whereas another group of 15 women were given two im injections of 50 mg progesterone in oil, during a 24-h period. In the im group, serum progesterone levels rose rapidly, plateauing at about 16 ng/ml after 4 h of treatment. In the vaginal administration group, however, serum progesterone levels rose more slowly and reached a peak of about 7 ng/ml after 4 h. Endometrial progesterone concentrations in biopsies taken after 7 d of treatment were considerably higher after intravaginal than im dosing, despite the higher serum progesterone levels after im injection. This study highlights the potential importance of the vaginal route in menopausal HT, because the endometrium is the most important target of progesterone action in this application.
3. Percutaneous route
The use of progesterone in the form of transdermal delivery via topical creams or gels has been a subject of some concern because of speculation that the low serum progesterone levels achieved with these agents indicate an insufficient secretory effect on the endometrium (34). However, despite such low serum levels below 4 ng/ml, antiproliferative effects on the endometrium have been demonstrated with progesterone creams (35), and in addition, salivary progesterone levels are found to be very high (36), indicating that progesterone levels in serum do not necessarily reflect those in tissues. The effects of topical progesterone creams on the endometrium should therefore be based on histological examination of the endometrium rather than on serum levels.
An important caveat with progesterone cream products that are readily available over the counter is that some of these products do not contain progesterone but instead contain wild yam extract in which the precursor for the synthesis of progesterone, diosgenin, is present. However, the chemical reactions required to convert the diosgenin in wild yam extract to progesterone can be carried out only in a laboratory and do not occur in the body.
Two different progestins, levonorgestrel and norethindrone acetate, are used in different transdermal systems, each in combination with estradiol. Both systems are adhesive-based matrix transdermal patches designed to release estradiol and levonogestrel or norethindrone acetate continuously for 7 or 3.5 d, respectively. The levonorgestrel/estradiol-containing system (Climara Pro) provides a levonorgestrel nominal delivery rate of 0.015 mg/d (37). After its application, in one study, levonogestrel concentrations were maximal after approximately 2.5 d, and average serum steady-state concentrations were 166 pg/ml (38). The norethindrone acetate/estradiol-containing system (CombiPatch) is available in two different doses of the progestin, with nominal delivery rates of 0.14 and 0.25 mg/d. In one study, norethindrone steady-state concentrations were attained within 24 h of application and the subsequent average serum steady-state concentrations were 489 and 840 pg/ml for the respective doses (39).
C. Drug interactions
The potential interaction of progestogens with other drugs has been the subject of numerous reports since the early 1970s. Some interactions are well documented and therapeutically relevant; however, many remain unproven or are the subject of continuing controversy. Strong evidence indicates that griseofulvin (an antifungal drug), rifampin (an antituberculosis drug), and certain anticonvulsants (phenobarbital and phenytoin) induce hepatic enzymes and decrease oral contraceptive (OC) effectiveness. An unproven, but widely accepted, drug interaction involves the effect of antibiotics on OC efficacy. Despite a number of reports implicating penicillins, tetracyclines, and other antibiotics in causing OC failure, no firm evidence links antibiotic administration with altered circulating levels of progestogens.
VI. Intracellular Mechanisms of Action of Progestogens
A. Steroid receptor structure, distribution, and ligand binding
The intracellular actions of progestogens are mediated predominantly via the PR, a ligand-activated transcription factor and member of the steroid receptor and nuclear receptor families of receptors (40). Progestins are designed to be potent, high-affinity PR agonists that mimic the actions of progesterone but with better bioavailability. However, many progestins bind to other members of the steroid receptor family, which includes the androgen receptor (AR), glucocorticoid receptor (GR), and mineralocorticoid receptor (MR), and exhibit off-target effects via these receptors (41, 42). Progestogens do not bind to the ER, the other member of the steroid receptor family. Moreover, current progestogens exhibit considerable variation in their binding affinities via the AR, MR, and GR.
It is not surprising that progestogens cross-react with several members of the steroid receptor family, because the PR, AR, GR, MR, and ER share significant amino acid homology in certain regions, while exhibiting a highly conserved overall domain structure. These domains include an unconserved amino-terminal domain of variable length, a highly variable transcriptional activation function-1 (TAF-1) domain situated near the N terminus, a highly conserved DNA-binding domain (DBD), as well as a moderately conserved C-terminal ligand-binding domain (LBD). The TAF-1 domain has been reported to be ligand independent and required for optimal transcriptional activity via protein-protein interactions with general transcription factors as well as cofactors (43). The DBD, the most conserved domain of the steroid receptors, contains two zinc finger motifs and is responsible for sequence-specific and high-affinity DNA binding, as well as playing a role in receptor dimerization, interaction with cofactors (44), and nuclear localization (45). The LBD, toward the C terminus, determines ligand specificity and affinity, as well as playing a role in dimerization, nuclear localization, and interaction with chaperone proteins and cofactors (45–47). A highly conserved TAF-2 domain is present within the LBD, which contains at least one cofactor interaction motif important for ligand-dependent transcriptional activity (46, 48). Despite the approximately 50–60% amino acid sequence homology between the LBDs of the PR, AR, GR, and MR, these steroid receptors exhibit subtle differences in their dimerization and cofactor binding sites due to differences in secondary structure, whereas the ER is even less conserved (49–54).
Progestogen action via steroid receptors is further complicated by the presence of several receptor isoforms for each receptor. The PR exists as two isoforms, PR-A and PR-B, transcribed from two promoters of a single gene (55). The longer PR-B isoform is more transcriptionally active and contains a third transactivation function domain that is absent from PR-A, allowing binding of coactivators to PR-B that do not bind to PR-A (56–58). Similarly, other steroid receptors exist in several isoforms that exhibit differential expression profiles and functions (40, 59, 60). The PR, ER, AR, and MR have a relatively selective distribution. The PR is expressed in the female reproductive tract, mammary gland, brain, and pituitary gland as well as some immune-function cells (61, 62). Ratios of the individual PR isoforms vary in the ovary, breast, and uterus (63), where they have different physiological functions in various target cells (63, 64), most likely in part due to the distinct and promoter-specific transactivation effects of PR-A and PR-B (65). Changes in the ratio of PR-A to PR-B have been implicated in the development of breast cancers, most likely via a mechanism involving MAPK-dependent PR phosphorylation and isoform stability (66). Changes in PR isoform expression levels and/or activity have also been associated with functional progesterone withdrawal in the human pregnant uterus (67). The two main ER isoforms, ERα and ERβ, have distinct tissue expression patterns and roles in disease and normal physiology in breast, ovary, colon, endometrium, and bone cells in women (68). The AR is expressed in the mammary gland, muscle, prostate, skin, vagina, bone marrow, and testes (40). Thus, AR effects are likely to be responsible for differential progestogen actions in these tissues, particularly in the breast. In contrast, the GR is ubiquitously expressed, although its levels are regulated in a tissue- and cell-cycle-specific manner (40). Therefore, differential progestogen effects mediated by the GR are likely to occur in most tissues and in particular those where GR levels are high, such as in immune-function cells. Interestingly, GR levels have been shown to vary widely in different breast carcinoma subtypes (69), suggesting a particularly important role of varying GR levels in the determination of effects of progestogens such as MPA in breast cancer. Differential expression profiles and functions of GR isoforms, such as GRα and GRβ (59), would increase the possibilities for differential progestogen actions via the GR. The MR, although not as widely expressed as the GR, is also expressed in many tissues, including the kidney, colon, central nervous system, heart, adipocytes, and vascular cells (40, 70–72). Thus, physiological functions in these tissues are likely to be modulated selectively by progestogens via the MR.
To determine the affinity of a progestogen for a particular receptor, binding studies have been developed. These have been performed in a wide range of models including animal or human tissue, human cell lines expressing endogenous receptors, cell lines deficient in endogenous receptors but overexpressing exogenous human steroid receptors, or even in in vitro systems using recombinant purified human receptor. Binding assays are usually performed using a constant concentration of radiolabeled reference agonist incubated with varying concentrations of unlabeled competitor test ligand to obtain an IC50 for the competitor steroid. Affinities are usually expressed as relative binding affinity (RBA), which is calculated by dividing the IC50 of the test steroid by the IC50 of the reference steroid, multiplying by 100, and expressing the RBA as a percentage. The IC50 is the concentration of the unlabeled steroid that corresponds to 50% inhibition of the total binding of the radiolabeled reference agonist. RBAs are often only an approximate measure of relative affinity because IC50 can vary with receptor concentration, concentration of radiolabeled steroid, and whether or not equilibrium has been reached for both steroids. More accurate affinities can be obtained by determination of time to reach equilibrium for the steroids under investigation as well as by performing homologous and heterologous displacement assays with determination of equilibrium dissociation constants using the Cheng-Prusoff equation or by saturation binding analysis (41).
From Table 4, which summarizes some of the available data on RBAs of progestogens to different steroid receptors, it is immediately apparent that the data show a wide variability. One of the reasons for this is undoubtedly due to different methods used to determine affinity, as discussed above. Another source of variability is the use of different cell or tissue models, which vary in the relative concentrations of different steroid receptors. Off-target binding of the progestogen to receptors other than the one under investigation could effectively lower the apparent RBA, especially if the progestogen has a relatively high affinity for a competing receptor, because the concentration of unlabeled competitor progestogen available for binding to the target receptor will be effectively less than the added concentration. Thus, experiments that determine equilibrium dissociation constants and those using cell lines deficient in endogenous receptors and overexpressing exogenous human steroid receptors or even in vitro systems using recombinant purified human receptor are likely to yield more accurate results. Another source of variability is the species from which tissue is obtained as well as the variation in age and pretreatment of the animal or human donor. Note that direct comparisons between the values determined by competition binding using different reference radiolabeled agonists [e.g. progesterone vs. promegestone for the PR or dihydrotestosterone (DHT) vs. mibolerone for the AR] for a particular receptor and competitor ligand cannot be made. Nevertheless, despite these sources of error and variability in binding experiments, several valuable insights have been obtained.
Table 4.
RBAs and hormonal activities of progestogens via the PR, AR, GR, and MR
| Progestogen | PR RBA (%) | AR |
GR |
MR |
||||
|---|---|---|---|---|---|---|---|---|
| RBA (%) | Androgenic activity | Antiandrogenic activity | RBA (%) | Glucocorticoid activity | RBA (%) | Antimineralocorticoid activity | ||
| Progesterone | 50a | 0a | ? | (+) | 10a | ? | 100a | + |
| 100b | 3b | 11b | 100b | |||||
| 1c | 2c | 9c | ||||||
| Chlormadinone acetate | 67a | 5a | + | — | 8a | + | 0a | — |
| Cyproterone acetate | 90a | 6a | + | — | 6a | + | 8a | — |
| Dienogest | 5a | 10a | — | + | 1a | — | 0 | — |
| Drospirenone | 35a | 65a | — | + | 6a | — | 230a | + |
| 19b | 2b | 3b | 500b | |||||
| Gestodene | 90a | 85a | ? | — | 27a | ? | 290a | ? |
| 864b | 71b | 38b | 97b | |||||
| Levonorgestrel | 150a | 45a | + | — | 1a | — | 75a | ? |
| 323b | 58b | 7.5b | 17b | |||||
| MPA | 115a | 5a | ? | — | 29a | + | 160a | — |
| 298b | 36b | 58b | 3.1b | |||||
| 2c | 39c | 0.08c | ||||||
| Nestorone | 136a | 0a | — | — | 38a | — | ND | ND |
| Nomegestrol acetate | 125a | 42a | — | + | 6a | — | 0a | — |
| Norethindrone | 75a | 15a | + | — | 0a | — | 0a | — |
| 134b | 55b | 1.4b | 2.7b | |||||
| Norethindrone acetate | ND | 1.7c | + | — | 1.6c | — | 0.07c | — |
| Promegestone | 100a | 0a | — | — | 5a | + | 53a | — |
| Trimegesterone | 330a | 1a | — | ? | 9a | — | 120a | ? |
| 588b | 2.4b | 13b | 42b | |||||
RBAs were determined by competitive binding assays using a radiolabeled reference ligand and increasing concentrations of unlabeled competitor ligand and are based on IC50 values in most cases (a and b), whereas Ki (equilibrium dissociation constant for an unlabelled competitor or inhibitor ligand competing for binding of the radiolabeled reference ligand to the receptor) values were determined by homologous and heterologous displacement and using the Cheng-Prusoff equation (c) (73). There is no evidence of significant direct binding for any of these steroids to the ER (RBAs all 0 to <1% relative to estradiol) (7, 41). Hormonal activities are based on animal experiments and taken from Refs. 7 and 41. All the steroids are progestogenic, and all exhibit antiestrogenic activity in animal models via a mechanism independent of the progestin binding to ER. None of them, except norethindrone, exhibits estrogenic activity (7). Key to activity levels: +, effective; (+), weakly effective; —, not effective; ?, literature inconsistent. ND, Not determined.
Values were compiled by cross-comparisons from several competitive binding studies that used different methods and were taken from Ref. 7. Most of the data are from animal tissues or cell lines expressing several receptors, and hence, some are likely to be inaccurate. The reference radiolabeled ligands (100% RBA) were as follows: PR, promegestone; AR, metribolone or R1881; GR, dexamethasone; MR, aldosterone.
Values were determined using recombinant human receptor binding in vitro (74). The reference radiolabeled ligands (100% RBA) were as follows: PR, progesterone; AR, testosterone; GR, dexamethasone; MR, aldosterone.
RBAs were calculated from Ki (equilibrium dissociation constant for an unlabelled competitor or inhibitor ligand competing for binding of the radiolabeled reference ligand to the receptor) values, determined by expressing the human recombinant GR in the A549 cell line (73) or the human recombinant AR or MR (314) in the COS-1 cell line, both deficient in steroid receptors, using the methods outlined in Ref. 73. The reference ligands (100% RBA) were as follows: AR, mibolerone; GR, dexamethasone; MR, aldosterone. Note that for the AR, the RBA for DHT in this assay was 1.3%.
Although all progestogens bind with high relative affinity to the PR, most bind with a greater affinity than progesterone (Table 4). As the natural progestational agent of all mammals, progesterone was an obvious choice as the reference steroid for many binding assays and was used in conjunction with [3H]progesterone in competitive binding studies with PRs. More recently, the highly potent synthetic progestin, promegestone (R5020), has replaced progesterone as the reference compound because most progestins have greater progestational activity than progesterone itself. Human and animal tissues can show profound differences in RBAs for the PR. RBAs for norgestimate and its principal active metabolites for uterine PRs were determined in two studies (75, 76); in one (75), norgestimate was bound to the PR in rabbit uterine tissue with an RBA of 124%, whereas norelgestromin and levonorgestrel had RBAs of 94 and 541%, respectively. In the other study (76), which used human uterine tissue instead of rabbit, norgestimate showed very little binding to the PR (RBA, 0.8%) and the binding of norelgestromin was low (RBA, 8%), whereas the RBA of levonorgestrel was 250%. This illustrates the difficulties of extrapolating animal RBA data to human tissues.
Progestogens vary greatly in their reported affinities for the AR, with some of the older-generation progestins such as MPA, norethindrone, and levonorgestrel binding with high affinity relative to testosterone (77–86), although some researchers report similar affinities for progesterone, MPA, norethindrone acetate, and DHT for the AR (Table 4). In contrast, drospirenone, dienogest, and trimegestone exhibit low RBA (74, 87, 88), although reported relative values differ for several progestogens, whereas nesterone does not bind at all to the AR (89).
Progesterone, trimegestone, and drospirenone have a relatively high affinity for the MR (Table 4) (90–93). The latter two progestogens were developed for their antimineralocorticoid properties for contraceptive usage (94) and for their predicted beneficial effects on blood pressure and cardiovascular function (31, 90, 95, 96). However, other progestins such as MPA and norethindrone acetate bind weakly to the MR (41), whereas several progestins such as dienogest, nomegestrol acetate, and promegestone do not bind at all (87, 97).
In contrast to PR and AR binding, relatively few progestogens bind to the GR with affinities in the significant pharmacological range, with the notable exceptions of MPA, gestodene, and nestorone (Table 4). MPA has a high RBA for the GR (73, 77, 81, 98–100), and it has been shown that MPA displays significantly higher binding affinity toward the GR than cortisol, the endogenous glucocorticoid in humans (100). Gestodene binds with a relatively high affinity to the GR (101). However, progestins such as norethindrone, levonorgestrel, dienogest, and trimegestone, like progesterone, bind the GR with low relative affinity (31, 73, 74, 82, 87, 88, 99, 100).
In summary, a major determinant of differential intracellular progestogen actions is the variable affinity of progestogens for binding to the PR and to other members of the steroid receptor family. Although all progestogens bind with relatively high affinity to the PR, they do not bind to the ER, and their reported relative affinities for the AR, GR, and MR differ substantially. Affinities, together with concentrations of progestogens and competing endogenous ligands, determine receptor occupancy for a particular steroid receptor. Fractional occupancy is in turn a major determinant of the biological response. Although the equilibrium dissociation constants for a particular progestogen or endogenous ligand for a particular steroid receptor do not change (41), the fractional occupancy of a receptor changes depending on ligand concentration, which in turn varies according to its relative affinity for, and concentrations of, the different steroid receptors present. Although useful binding data are available, much of it may be inaccurate; additional experiments are required to more accurately determine equilibrium binding constants for most of the progestogens for different steroid receptors and their isoforms, in the absence of confounding factors such as the sources of the receptors, the methods of binding analysis, and the presence of off-target receptors. Given that the relative levels of different receptors and their isoforms vary greatly in different tissues, this is also likely to be a major determinant of differential actions via progestogens.
B. Potency, efficacy, and biocharacter of progestogens via steroid receptors
Progestogens exhibit considerable variation in their potencies and efficacies as well as the resulting extent of agonist, partial agonist, or antagonist responses, i.e. their biocharacter, via steroid receptors. Potency is defined in this context as the concentration of ligand required for half of the maximal biological response, whereas efficacy is the maximal induced response for that particular ligand (41). Agonists, partial agonists, and antagonists all bind to a particular receptor, with an agonist resulting in an efficacy similar to that of the natural ligand, whereas a partial agonist gives a similar response to that of the natural ligand but with a lower efficacy, and an antagonist inhibits the response of an agonist. Partial agonists and antagonists can exhibit varying degrees of antagonism depending on the relative concentrations of competing ligands and their affinities for a particular receptor as well as on receptor concentration.
Much of the data on potency, efficacy, and biocharacter via different steroid receptors has been obtained from animal experiments (41) (Table 4). These data do reflect to some extent the actions of a progestogen via a particular steroid receptor but also suffer from the same source of variability as the binding studies when it comes to off-target effects via other receptors, which would lead to inaccurate potency estimates. In addition, the animal data are also confounded by pharmacokinetic factors, metabolism, binding to serum proteins, and indirect actions of the progestogens via target proteins other than steroid receptors.
Bioassays have been developed that test the effects of progestogens on uterine glandular proliferation, pregnancy maintenance, delay of parturition, or inhibition of ovulation in rabbits or rats. The Clauberg test is based on initial observations made by Clauberg in the 1920s and is the most widely used bioassay for progestational agents. It was later developed into specific protocols by McPhail in 1934 (102). The principle of the test is to measure glandular proliferation in rabbit endometrium that has been primed with estrogen, in response to progestogens given orally or parenterally. McPhail used a standardized scale for grading the complex glandular proliferation of the rabbit endometrium in response to the different progestogens. This scale starts from 0, corresponding to no glandular development, with a highest possible value of +4, corresponding to maximal glandular development. In practice, progestogens are compared at a dose level that produces a value of +2 on the McPhail scale.
The Clauberg test is, however, subject to considerable variation in estimates of potency (103). Problems arise in interpretation of the test because dose-response curves for commonly employed test substances are not parallel. Other commonly used bioassays also have various limitations (103). For example, bioassays that measure pregnancy maintenance as a progestational effect cannot use estrogens, which will inhibit the active progestogens when given at sufficient doses; the bioassay for delay of parturition cannot distinguish between the various progestogens; and the ovulation inhibition bioassay in the laboratory gives different progestogen potencies when compared with those obtained in women. Despite these limitations, bioassays have led to significant insights into progestogen actions, although they frequently do not correlate with the steroid receptor-binding affinity data, in particular for the AR, GR, and MR.
Several general tends have emerged from both the animal bioassays and in vitro binding affinity studies. Although all progestogens bind to the PR (Table 4) and act as progesterone agonists, they exhibit differences in the potency of the progestogenic responses (Table 4) (104–108). On the other hand, progestogens exhibit a wide spectrum of activities via the AR, ranging from no effect to agonist, partial agonist, and antiandrogenic activity (Table 4). For example, some of the older-generation progestins such as MPA, norethindrone acetate, norethindrone, and levonorgestrel, which bind with relatively high affinity to the AR, have been reported to act as agonists or partial agonists in some contexts, unlike progesterone (Table 4) (77–86), although the androgenic biological activities reported for MPA and progesterone vary greatly in the literature. In contrast, drospirenone, dienogest, and trimegestone, which exhibit low RBA for the AR, exhibit no AR-mediated agonist activity but exhibit variable to potent antiandrogenic properties (Table 4) (74, 87, 88). Nestorone has no activity via the AR (Table 4) (89), whereas nomegestrol acetate, which binds the AR, has no agonist activity and displays partial antiandrogenic activity (Table 4) (22, 109, 110). Consistent with their binding activities, MPA has partial to full agonist activity via the GR in some contexts (41), whereas gestodene exhibits partial agonist activity in some contexts (41). However, progestogens such as norethindrone, levonorgestrel, dienogest, and trimegestone show no or very little glucocorticoid-like activity in most contexts, whereas the reported effects of progesterone via the GR vary (Table 4) (41). Certain progestogens like trimegestone and drospirenone with a relatively high affinity for the MR exhibit weak partial MR agonist activity. However, both progesterone and drospirenone exhibit potent antagonist activity toward aldosterone via the MR, whereas the reported antagonistic effects of trimegesterone are variable (Table 4) (90–93). Other progestins such as MPA and norethindrone acetate, which bind weakly to the MR, exhibit no antimineralocorticoid activity in rat models (Table 4) (41), whereas dienogest neither binds to nor displays agonist or antagonist activity for the MR (Table 4) (87, 97).
In addition to in vitro binding affinity tests and bioassays, clinical tests have been used to assess the relative biological effects of progestogens in women; they include those based on delay of menses, induction of secretory changes in the endometrium, inhibition of ovulation, and changes in vaginal cytology and cervical mucus. Traditionally, in these clinical tests, the term potency is often used to refer to a relative response obtained at a chosen progestogen dose, using equivalent mass doses, without dose-response analysis. Alternatively, some assays refer to potency as the comparative dose (in mass) required to give a particular level of response, usually not a maximal response. Hence, these are not true measures of potency or efficacy in terms of the definitions discussed above. Thus, the term potency reported from such clinical studies needs to be interpreted with these limitations in mind. Greenblatt and co-workers (111) were the first to describe the delay-of-menses test for progestogenic potency. In this test, the progestogen is administered beginning on the sixth or seventh day after ovulation and continuing for 3 wk or more. If the progestogen is effective, it will delay menstrual bleeding until 2–3 d after treatment is discontinued. The delay-of-menses test was further developed and standardized by Swyer and Little (112) for assessing comparative potency of progestogens and is consequently referred to as the Swyer-Greenblatt test.
A literature review published in 1985 assessed the relative potency of progestogens used in oral contraception in the United States on the basis of available human data showing the effect of progestogens on the delay of menses by the Swyer-Greenblatt test as well as effects on subnuclear vacuolization (as an indirect determination of glycogen deposition) and lipid and lipoprotein levels (113). The review concluded that norethindrone, norethindrone acetate, and ethynodiol diacetate are approximately equivalent in potency, whereas norgestrel and its biologically active enantiomer, levonorgestrel, are about 5–10 and 10–20 times as potent as a similar weight of norethindrone, respectively. However, there are limitations in the studies that were reviewed. Parallelism of dose-response curves was not demonstrated in the delay-of-menses test, and high doses of ethinyl estradiol (50 and 100 μg) were used in this test and in the subnuclear vacuolization test. Also, only relative effects were obtained in the lipid/lipoprotein tests because the results were not obtained from dose-response curves.
In another approach to determine progestogen potency from clinical data, a series of studies by King and co-workers (114–120) assessed progestogenic effects by analyzing biochemical and morphological features of endometria from estrogen-primed postmenopausal women. First, the postmenopausal women were treated daily with either 0.625 or 1.25 mg of conjugated equine estrogens, and then the effects of 6 d of sequential progestogen treatment during the last 6–12 d of the month were assessed. At least three different doses of each of 5 orally administered progestogens, specifically, norethindrone, levonorgestrel, MPA, dydrogesterone, and progesterone, were studied. The endometria were analyzed for biochemical parameters including nuclear estradiol receptor levels, DNA synthesis, and isocitric and estradiol dehydrogenase activities. King and Whitehead (121) reexamined the results of these studies to allow comparisons with corresponding premenopausal secretory-phase values and reported the potency of progestogens relative to a value of 1 for norethindrone. The analysis showed that the potency of levonorgestrel was 8-fold greater, whereas the potencies of MPA, dydrogesterone, and progesterone were 10, 50, and 500 times lower, respectively.
The recommended oral progestogen doses for endometrial protection (Table 5) are based on the potencies established by the analysis of King and Whitehead (121); they are 1, 0.15–0.5, 2.5–10, 20, and 100–300 mg for norethindrone (or its acetate), levonorgestrel, MPA, dydrogesterone, and progesterone, respectively. The specific dose recommended also depends on whether the progestogen is given sequentially for 12–14 d/month or continuously as well as on the type of estrogen administered concurrently.
Table 5.
Comparison of different progestogen potencies determined experimentally with corresponding therapeutic oral doses
| Progestin | Potency |
|
|---|---|---|
| Experimental | Based on dose | |
| Levonorgestrel | 8 | 2–6.7 (0.15–0.5 mg) |
| Norethindrone | 1 | 1 (1 mg) |
| MPA | 0.1 | 0.1–0.4 (2.5–10 mg) |
| Dydrogesterone | 0.02 | 0.05 (20 mg) |
| Progesterone | 0.002 | 0.01–0.0033 (100–300 mg) |
Potency values are relative to a value of 1 for norethindrone.
In contrast to animal experiments and clinical data, several researchers have done experiments in cell culture to investigate more directly the relative potency, efficacy, and biocharacter of progestogens via specific steroid receptors and on specific target genes. These strategies include the use of cell lines as models for cells in a particular target tissue relevant to HT side effects, cell lines deficient in other receptors with transient overexpression of the receptor under investigation, or the genetic engineering of cell lines to overexpress a particular receptor. However, very few studies have verified the specificity of the response by, for example, small interfering RNA knockdown of a particular receptor or using receptor-specific antagonists. Nevertheless, much valuable information has been obtained from these in vitro activity studies, including evidence for a lack of a class effect of progestogens.
C. Regulation of transcription by progestogens: genomic effects
1. Overview of mechanisms of ligand-dependent transcriptional regulation by steroid receptors
Ligand-activated steroid receptors directly regulate transcription of specific target genes by several genomic mechanisms that are conserved within the family of steroid receptors, although some mechanistic differences do occur. Regulation of transcription of mammalian genes generally involves dynamic, regulated steroid receptor-mediated recruitment of multiprotein complexes. These complexes include chromatin-remodeling proteins that shift nucleosomes, coactivators that acetylate histone proteins to open up chromatin, or corepressors that deacetylate histone proteins resulting in more compact chromatin. Also involved are several other proteins such as mediator complexes, the basal transcription machinery including RNA polymerase and associated factors, and enzymes that modify components of the complexes, including methylases and kinases (122, 123). Steroid receptors are key proteins in this process (99). In the absence of ligand binding, the PR and ER are located predominantly in the nucleus, whereas the AR, GR, and MR are located predominantly in the cytoplasm (44). There is also evidence that receptor isoforms display differential subcellular localization in the absence of ligand. For example, in endometrial cancer cells, the unliganded PR-A is predominantly located in the nucleus, whereas the unliganded PR-B is predominantly cytoplasmic (124), but both PRs are distributed in the nucleus and in the cytoplasm of several cell lines when overexpressed (125). The receptors are held in an inactive conformation by the presence of a protein complex of the heat-shock proteins (hsp) hsp90 and hsp70, immunophilins, and other proteins (126). The lipid-soluble steroid ligands diffuse passively across the plasma membrane and bind to the LBD of steroid receptors, inducing hyperphosphorylation, a conformational change in the receptor, changes in the composition of the protein complex, and nuclear translocation of the cytoplasmic receptors (44, 60, 127).
The genomic mechanisms whereby ligand-bound steroid receptors directly increase transcription of many target genes via direct DNA binding, or transactivation, involve binding of a receptor dimer to specific palindromic DNA sequences in promoters of target genes known as steroid-responsive elements (SREs). This results in formation of a multiprotein complex on the promoter via protein-protein interactions, including chromatin-remodeling proteins, coactivators, and components of the transcriptional machinery, in a dynamic, complex interplay of factors leading to an increase in transcription initiation (41, 44, 47, 128) (Fig. 7). Although each steroid receptor exhibits selectivity and a higher affinity for specific SRE sequences, the high degree of structural and functional conservation within the DBDs of steroid receptors allows most steroid receptors to bind, at least in vitro, to the same DNA response element (reviewed in Ref. 129). Thus, the progesterone response element (PRE) also binds the AR, GR, and MR (reviewed in Ref. 130).
Figure 7.
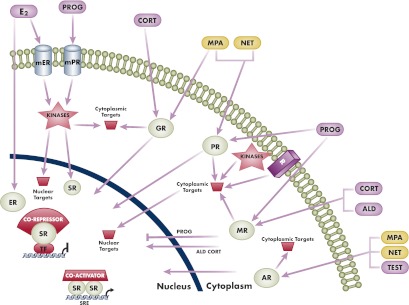
Schematic diagram to illustrate differential genomic (nuclear) and nongenomic (extranuclear, cytoplasmic) actions of progestogens and endogenous steroid hormones. The two progestins, MPA and norethindrone, were chosen to illustrate the concept of differential actions compared with each other and progesterone. In genomic actions, all progestogens bind to the PR and act as agonists. MPA is a partial to full agonist for the GR and AR but has no significant activity via the MR or ER. However, norethindrone is a partial to full agonist for the AR but has no significant activity via the GR, MR, or ER. Progesterone is a weak agonist for the GR and AR, has no significant activity via the ER, and is a full antagonist for the MR. The two best-characterized genomic mechanisms for steroid receptors are illustrated. The first is transactivation by steroid receptor dimers binding directly to SREs in the promoters of target genes, followed by recruitment of coactivators and increased transcription. The second is transrepression via tethering of a steroid receptor monomer to other positively acting transcription factors, followed by recruitment of a corepressor and inhibition of transcription. Other complexes and higher-order effects on chromatin structure as discussed in the text are not depicted for simplicity. The depicted nongenomic or cytoplasmic actions include activation of various cytoplasmic targets by the classical nuclear steroid receptors or by membrane steroid receptors. Progesterone is a full agonist for the mPR, whereas MPA and norethindrone have no significant activity via mPR. Note that cytoplasmic actions can also lead to genomic actions by targeting of nuclear proteins such as transcription factors, cofactors, and chromatin proteins or even steroid receptors. Also depicted is the cross talk between the classical PR and other plasma membrane receptors (R) such as the epidermal growth factor receptor, as discussed in the text. Note that actions of progesterone as a weak GR or AR agonist are not depicted. ALD, aldosterone; CORT, cortisol; E2, estradiol; mER, membrane ER; NET, norethindrone; PROG, progesterone; SR, steroid receptor; TEST, testosterone; TF, transcription factor.
Ligand-bound steroid receptors can also transrepress or directly and negatively regulate transcription via several genomic mechanisms, including direct DNA binding to negative SREs (131), or by protein-protein interaction and interference with other DNA-bound transcription factors such as nuclear factor-κB (NFκB) or activator protein-1 and CCAAT-enhancer-binding protein (41, 132–137) (Fig. 7). The latter mechanism is often referred to as a tethering mechanism, which can also result in an increase in transcription, depending on the transcription factors involved and promoter architecture (138). The details of these mechanisms are not well established for most members of the steroid receptor family but have been the focus of studies on GR actions due to their involvement in the antiinflammatory response (133, 135). All the members of the steroid receptor family have been shown to repress genes by antagonizing NFκB action (133, 135, 137, 139–142). In addition, the PR has been shown to increase transcription via tethering mechanisms involving interaction with specificity protein 1 and CCAAT/enhancer-binding protein (143–145).
Ligand-bound steroid receptors can thus lead to both increases or decreases in transcription and hence gene expression, via several direct genomic mechanisms where the outcome is cell and promoter dependent, depending on which cofactors are recruited by the receptor and the identity of the specific ligand. Several lines of evidence show that, in general, an agonist bound to a receptor induces a conformational change that facilitates binding of coactivators, resulting in transcriptional activation due to their intrinsic histone acetylase activity, which makes the chromatin more accessible for recruitment of the basal transcription machinery and other transcription factors (146). Antagonists, on the other hand, are generally accepted to promote either the recruitment of corepressors, resulting in a decrease of transcription initiation via their histone deacetylase activity, reducing accessibility of DNA-binding sites for transcription factors, or a failure to recruit coactivators (147, 148). However, this general description is likely to be an oversimplification, because the spatial architecture and three-dimensional packaging of chromatin inside the nucleus, as revealed by new chromatin immunoprecipitation-sequencing technology, may well play a major role in nuclear receptor action (149, 150). Furthermore, tissue-specific steroid responses are determined by tissue-specific expression profiles of cofactors that affect the differential recruitment of coactivators vs. corepressors (146). Thus, it is almost impossible to predict the transcriptional response for a particular steroid ligand on a particular gene in a specific cell type, and these responses need to be determined experimentally.
The epigenome is emerging as a major regulator of cell-type-specific responses, regulating cell-type-specific gene expression profiles induced by nuclear receptors in response to ligands. The epigenome is dynamic and is a function of many factors including DNA methylation, higher-order chromatin structure such as chromatin looping, posttranslational modification of histone tails, and localization of histone variants (123). Nuclear receptor binding sites are present in enhancer elements that are brought into proximity with promoters by chromatin looping mechanisms that are programmed during cell lineage commitment (123) and are important regulatory elements in cell-specific gene expression (151, 152). In addition to cell-specific responses being mediated by epigenetic preprogramming of enhancers, nuclear receptors can also reprogram the epigenome in response to ligands (123). Nuclear receptors associate with many of the enzymes that modify histones and chromatin structure, such as the histone lysine demethylase, LSD1, which has been shown to associate with the AR (153) and to be important for nuclear receptor-mediated gene expression (154).
2. Differential effects of progestogens on specific gene expression via steroid receptors
Despite the general trends discussed above, progestogens exhibit cell-type-specific and gene-specific effects in particular models relevant to disease and side effects, due to multiple factors as discussed in previous sections. Thus, it is useful to consider what is known about the effects of different progestogens via different steroid receptors on transcription of specific target genes. Unfortunately, very few such detailed mechanistic studies have been performed or designed to compare effects of different progestogens or establish the receptors involved. However, those that have been performed shed useful insights into differential intracellular progestogen actions.
a. Effects via PR.
Side effects associated with progestins in HT use include increased risk of breast cancer (155–157), cardiovascular complications such as strokes (1, 158, 159), effects on immune function (160–163), and neurological effects (164, 165). There is evidence that the dose and choice of progestin could determine risk outcome (166–169). However, surprisingly little is known about the molecular mechanisms, differential effects, and target genes of progestins acting via the PR in target tissues relevant to these side effects.
Much research has focused on the mechanism of action of progestogens in human breast cancer cell lines, where both pro- and antimitogenic effects have been ascribed to PR agonists (137). Some reports suggest that similar genomic effects occur with most progestins and progesterone via the PR on several target genes (104). For example, microarray analysis revealed that MPA and progesterone exhibit very similar qualitative expression profiles, with MPA being somewhat more efficacious, on endogenous PR-regulated genes in the human T47Dco breast cancer cell line expressing both the PR-A and PR-B isoforms (105). Interestingly, the same authors detected some cell-specific differences in breast cancer cell lines between the maximal responses and potencies of MPA compared with R5020 on a synthetic PRE-luciferase construct, most likely due to different relative concentrations of proteins other than the PR, although this was not established (105). However, consistent with its lower PR agonist potency (92), drospirenone has been shown to display weak effects compared with progesterone and other progestogens such as MPA, norethindrone acetate, levonorgestrel, and trimegestone on the transcriptional profile of PR-regulated gene expression in the PR-positive T47Dco breast cancer cell line (104). Some progestins such as norethindrone are implicated in increased proliferation and metastasis via an induction of vascular endothelial growth factor (VEGF) release into the media of cultured T47D breast cancer cells (106) by a mechanism involving transactivation via three functional PRE elements in the VEGF promoter (107). Some evidence also exists that progestogens play a role in the development of PR+ breast cancer by affecting the ability of cancer cells to invade the surrounding environment and interact with the extracellular environment. Progesterone, MPA, and drospirenone have also been implicated in PR-mediated increased breast cancer cell migration, with drospirenone being less potent than MPA (108), similar to the differential effects observed in the T47Dco cell line.
In contrast to results in breast cancer cell lines where most progestins appear to have similar qualitative effects on transcription of target genes compared with progesterone, results in endometrial cells suggest that some progestins may have opposite effects compared with progesterone. For example, MPA has been shown to repress expression of the chemokine regulated on activation, normal T cell expressed and secreted (RANTES) gene via the PR in cultured human endometrial stromal cells (170), whereas progesterone increased the expression of RANTES in primary endometrial T cells (171). As found in breast cancer cells, MPA appears to have similar genomic effects via the PR-A and PR-B in endometrial cells, as suggested by the finding that MPA increases VEGF synthetic promoter activity in Ishikawa endometrial adenocarcinoma cells (107) via both receptor isoforms.
Results in cell line models relevant to cardiovascular side effects also suggest different genomic effects of some progestins compared with progesterone. For example, unlike progesterone and dienogest, it was found that MPA, norethindrone acetate, and levonorgestrel increase expression of two markers of vascular inflammation, intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), in part via the PR (172). Consistent with this, studies on endothelial nitric oxide (NO) production, a marker for vasodilation (173), suggest differential actions of some progestins compared with progesterone. MPA was shown to have no effect on NO production in isolated human endothelial cells as well as in aortas from ovariectomized rats, unlike progesterone and drospirenone, which increased NO production, most likely via the PR (174, 175).
Similarly, studies in rat models suggest differential actions on brain mitochondrial function of MPA compared with other progestins and progesterone, which is of relevance to neurological health in premenopausal and postmenopausal women (169, 176). Unlike progesterone, MPA antagonizes estrogen up-regulation of brain mitochondrial function. Although the detailed mechanisms are unknown, these most likely involve differential steroid receptor-mediated changes in expression of key genes such as ATP synthase (169, 176).
The above evidence suggests that progestogens exhibit differential genomic effects in several cell models relevant to breast cancer and endometrial, cardiovascular, and brain function. However, the molecular mechanisms and occurrence of ligand-, cell-, isoform-, and promoter-specific effects of a range of progestogens remain to be further investigated in parallel in more physiologically relevant primary cell models. In particular, the contribution of off-target actions via steroid receptors other than the PR requires further investigation as a possible explanation for differential progestogen actions. In addition, some of the observed effects of progestogens on gene expression may occur by indirect genomic actions via the PR or other steroid receptors (42). A physiologically important example of indirect genomic effects on estrogenic activity of progestogens via the PR is the up-regulation by the PR due to its transactivation of the 17β-hydroxysteroid dehydrogenase type 2 gene, the product of which inactivates estradiol by converting it to estrone (7). In addition, progestogens exert indirect antiestrogenic effects in the endometrium by transrepression of the ER gene (7). These antiestrogenic actions of progestogens in the endometrium do not occur via binding of progestogens to the ER.
b. Effects via AR.
Several earlier-generation progestins possess androgenic activity but not antiandrogenic activity, whereas most of the newer progestins possess antiandrogenic activity but no androgenic activity in animal models. However, the relative advantages of androgenic or antiandrogenic actions of progestogens in HT, as well as the extent to which these are mediated via direct genomic AR actions, are unclear. Nevertheless, there is evidence that off-target effects of progestins via the AR are likely to be relevant to cardiovascular function and breast cancer in HT users. The rationale for using progestins with antiandrogenic activity in HT is to improve the poor lipid profile of postmenopausal women, attributable to decreased levels of estrogen and SHBG; the resulting increased levels of free biologically active androgen are associated with decreased levels of high-density lipoprotein (HDL) cholesterol and increased levels of low-density lipoprotein cholesterol (177). The antiandrogenic metabolic effects of several progestins are, however, not ascribed to binding to the AR, but rather to a competitive inhibition of 5α-reductase activity, thereby decreasing the conversion of testosterone to the more active DHT (177). Nevertheless, several studies have investigated the potency and efficacy of different progestogens for transactivation of the AR via androgen response elements in cell lines. For example, MPA, norethindrone, levonorgestrel, and gestodene, but not dienogest, exhibit strong to weak partial agonist activity for AR-mediated transactivation via androgen response elements (77, 105, 178–180), whereas dienogest, trimegestone, drospirenone, and progesterone, but not MPA or norethindrone, can antagonize DHT-mediated transactivation via the AR (97, 178, 179). A recent study showing differential effects of MPA, norethindrone, and progesterone on TNFα-induced RANTES mRNA levels in a human ectocervical cell line, with MPA being repressive via the AR, suggests that progestogens may have differential effects on markers of immune function via the AR in the ectocervix (181). In this study, an AR-specific antagonist was used to establish a role for the AR. Evidence from clinical studies showing that antiandrogenic progestogens such as cyproterone acetate and dienogest are associated with increased levels of HDL cholesterol, SHBG, and triglycerides (177) suggest that the effects of dienogest are likely to be AR mediated, although the mechanisms and target genes remain to be established.
The findings that some progestins like MPA, norethindrone, levonorgestrel, and gestodene bind to the AR with relatively high affinity and exhibit partial agonist activity via the AR in cell lines, and androgenic effects in rats, suggest that these progestins may result in AR-mediated androgenic genomic effects in women on HT. In particular, AR-mediated genomic effects by MPA or other AR partial agonists have been suggested to play a role in increasing the risk of breast cancer by disrupting some androgen signaling in the breast that may be protective for breast cancer (182).
c. Effects via GR.
Consistent with an apparent requirement for progestogens to lack GR activity, most progestins exhibit no transcriptional activity via the GR, with the exception of MPA, which has a high affinity for GR and exhibits potent GR agonist or partial agonist activity, and gestodene, which exhibits less potent GR partial agonist activity (22, 41). Strong evidence for GR-mediated agonist activity of MPA was obtained by chromatin immunoprecipitation analysis that showed the recruitment of the GR to a glucocorticoid response element (GRE)-containing endogenous promoter in response to MPA but not to norethindrone acetate or progesterone (183). Several lines of evidence suggest that MPA, which is widely used as an injectable contraceptive and in HT, and by implication to a lesser extent gestodene, have side effects on immune, cardiovascular, bone density, breast cancer, and neurological processes via direct GR-mediated effects on gene expression. Consistent with the immunosuppressive properties of glucocorticoid ligands acting genomically as agonists via the GR, MPA has been reported to inhibit proliferative responses to the T-cell mitogens, concanavalin A, and phytohemagglutinin (100); it also, together with estrogen, down-regulates release of the proinflammatory cytokines IL-2 and interferon-γ by phytohemagglutinin-stimulated peripheral blood mononuclear cells isolated from postmenopausal women using HT (163). This is further supported by the finding that MPA, but not progesterone, transrepresses IL-2 transcription in isolated human monocytes (77). Also, consistent with a role for GR-mediated transrepression by MPA in osteoporosis, MPA has been shown to display glucocorticoid-like negative effects on bone density, unlike other progestins such as norethindrone and levonorgestrel (184, 185).
Extensive work in cell lines has identified several target genes and provided evidence for direct GR-mediated effects of MPA in their transactivation and transrepression relevant to MPA's off-target side effects mentioned above. MPA has been shown to exhibit potent GR-mediated transcriptional activity on synthetic and endogenous GRE-, activator protein-1-, or NFκB-containing promoters, respectively, in several cell lines (73, 98, 99, 186–190). For example, of relevance to immune function, MPA has been shown to repress IL-6 and/or IL-8 mRNA and/or protein levels in the L929sA (98) and human thyroid cancer (KTC-2) cell lines (186) as well as RANTES mRNA levels in an ectocervical cell line (181). Given the central role of glucocorticoids and the GR in the immune and inflammatory response (191, 192), MPA and possibly gestodene are likely to exert side effects on other target genes involved in immune function. In contrast, dienogest, drospirenone, and trimegestone do not possess glucocorticoid activity via overexpressed GR and synthetic GRE-containing promoters (90, 95, 97). Other evidence suggests GR-mediated genomic effects of MPA via GRE-containing genes on kidney function, because MPA, but not progesterone, was shown to increase endogenous α-ENaC (α-subunit of epithelial Na channel) in serum and sgk1 (glucocorticoid-regulated kinase 1) mRNA levels in mouse cortical collecting duct cell lines (188). Similarly, GR-mediated transactivation is implicated in cardiovascular side effects for MPA and gestodene, which, unlike norethindrone and levonorgestrel, up-regulate proteolytically activatable thrombin receptor (PAR-1) mRNA in rat vascular smooth muscle primary cells to potentiate the vascular procoagulant effects of thrombin (193). Relevant to possible beneficial effects on cardiovascular function, MPA acting most likely via the GR down-regulates endothelial nitric oxide synthase (eNOS) mRNA expression and resulting nitric oxide levels in human umbilical vein endothelial cells, unlike levonorgestrel (190).
Strong evidence exists for an important role of the GR in determining the differential actions of progestogens in breast cancer (194) via genomic mechanisms (195). Progesterone and progestins demonstrate distinct but overlapping mRNA expression profiles in breast cancer cells (195, 196). Recent results suggest that MPA, acting via transactivation of the GR on target genes such as the fatty acid synthase gene, may promote tumorigenesis in normal cells and cancer progression in cancer cells, unlike progesterone (195). However, MPA acting via the GR has also been implicated in playing a positive role in breast cancer via increasing expression of nucleoside diphosphate kinase A (metastasis suppressor) protein expression in metastatic human breast carcinoma cells by transactivation via GREs (130). Unlike progestins such as norethindrone, MPA acting via the GR is likely to have an immunosuppressive effect via repression of cytokine gene expression in both systemic and local endocervical immune function (Hapgood, J. P., Y. Govender, R. M. Ray, C. Avenant, and M. Tomasicchio, unpublished results). Because progestins such as norethindrone, levonorgestrel, dienogest, drospirenone, and trimegestone do not act via the GR, they are likely to exhibit very different side-effect profiles on the above-mentioned targets compared with MPA and, to a lesser extent, gestodene.
d. Effects via MR.
Unlike most progestins, some such as drospirenone and trimegestone exhibit potent antimineralocorticoid properties in vitro via binding with relatively high affinity to the MR and acting as aldosterone antagonists (31, 90). Their development appears to be based on a strategy to mimic the antimineralocorticoid properties of progesterone and/or to prevent cardiovascular complications in postmenopausal women using estrogen/progestin treatment for HT (94–96). Estrogen can promote an increase in weight and blood pressure via its actions on the renin-angiotensin-aldosterone system, leading to sodium and water retention (197). Interestingly, no changes were observed in blood pressure with progesterone administration to normotensive postmenopausal women, although a slight reduction in blood pressure was observed in hypertensive women (198). However, the anti-MR effects of progesterone may be relevant only when endogenous progesterone concentrations are high, such as during the luteal phase of the menstrual cycle and pregnancy, because progesterone has a short half-life and is rapidly converted to metabolites without anti-MR activity (199). Given the established role of aldosterone in regulating blood pressure and cardiovascular function, as well as effects on renal inflammation and the central nervous system (70–72), it is likely that progestogens with anti-MR activity will exert biological effects on these processes, depending on their affinity for the MR and the concentration of progestogens and competing ligands. This antimineralocorticoid effect has been evident by a slight decrease in body weight and blood pressure in women using drospirenone, but not levonorgestrel, in combination with estrogen (200). This is consistent with a study in ovariectomized female rats treated with aldosterone and salt to induce renal injury, which showed that estradiol in combination with drospirenone did not increase sodium retention and blood pressure, unlike MPA (201). Very little is known about the target genes or precise MR-mediated genomic mechanisms of progestogens.
Consistent with their low affinity for the MR, progestogens like MPA and norethindrone do not display transactivation agonist activity via the expressed MR on a reporter gene in the COS-1 cell line, although both were able to weakly antagonize aldosterone-mediated transcription via the MR, albeit to a much lesser extent than progesterone (97). In vitro studies have also confirmed that drospirenone exhibits antagonist activity toward aldosterone and weak agonist activity in transactivation studies via the MR (90, 202). Drospirenone, like progesterone, has been shown to inhibit aldosterone-induced up-regulation of the adhesion molecule E-selectin, plasminogen activator inhibitor-1, and the chemokine monocyte chemoattractant protein-1 in human female aortic endothelial cells, consistent with MR-mediated antagonist activity toward transactivation (202). The effects of antagonism of MR-mediated transrepression by progestogens like drospirenone remain to be investigated. Because most progestogens do not act via the MR, they are likely to exhibit very different physiological effects compared with progestogens such as drospirenone with potent anti-MR activity in tissues containing MR.
e. Effects via ER.
Although most studies report no direct binding to, or genomic actions by, progestogens via the ER, some reports (203, 204) but not others (78, 81, 82) suggest that both MPA and norethindrone acetate or norethindrone do bind to the ER. The metabolites of norethindrone, gestodene, and levonorgestrel, have, however, been reported to activate the ER (180, 205, 206). Metabolites of norethindrone appear to discriminate between ER isoforms, its 5α-reduced metabolite (3β,5α-tetrahydro-norethindrone) having been shown to selectively transactivate ERα at low concentrations but being ERβ agonistic at high concentrations (206). Although the physiological significance of binding and genomic actions of some progestogens or their metabolites via the ER are unclear, the ER does play an important indirect genomic role in the actions of progestogens because the estrogen-activated ER regulates expression of the PR gene and hence the response to progestogens (7).
In summary, besides affinity, other major determinants of differential progestogen actions via a particular steroid receptor are their potency and efficacy for a particular biological response. However, the relationship between the affinity, potency, efficacy, and biocharacter of a ligand is not straightforward or predictable (41, 99) and appears to depend on which ligand, promoter, or cell is involved. For example, two ligands may bind to a particular receptor with a similar affinity, but one may be an agonist and another an antagonist, depending on the particular receptor conformation induced by that ligand (146). For a particular ligand and receptor, the potency and efficacy are also gene specific; i.e. a progestogen may be a partial agonist on one promoter but a full agonist on another in the same cell (99, 181). These promoter-specific differences are also dependent on chromatin structure and the particular promoter architecture and, hence, assembled multiprotein complexes that differ for each promoter. Relatively little work has been done to investigate the relative effects on gene expression via different progestogens in different cells and to investigate the mechanisms and receptors involved. Results to date do show, however, that many target genes relevant to disease and side effects are indeed differentially regulated by progestogens, most likely due to differential extents of involvement of different steroid receptors and their isoforms. However, much more research needs to be done, in particular to compare different progestogens in parallel as well as determine involvement of receptors and their isoforms and cofactors, e.g. by small interfering RNA knockdown experiments or by using receptor-specific antagonists. Cell-type-specific responses to progestogens are also most likely regulated by cell-type-specific epigenomic factors, as has been shown for other nuclear receptor ligands (123). It remains to be investigated whether differential cell-specific effects of progestins acting via the same nuclear receptor may be mediated by differential interaction of the liganded receptor with proteins involved in epigenetic preprogramming or reprogramming, such as histone methylases, demethylases, and chromatin-remodeling proteins.
D. Nongenomic effects of progestogens
Besides regulation of transcription via binding to intracellular steroid receptors by so-called nuclear or genomic mechanisms, progestogens have also been reported to result in a range of cytosolic effects such as activation of kinase pathways. These nongenomic effects have been reported to occur via the classical cytosolic and nuclear receptors as well as by plasma membrane-bound classical steroid receptors and via other novel membrane-bound receptor proteins (67, 207–210). Nongenomic signaling via a membrane-bound PR has been implicated in playing a role in brain signaling, oocyte maturation, and breast cancer (210). Progestogens acting via the PR have been reported to activate several kinase pathways such as Src, MAPK, phosphatidylinositol 3-kinase/protein kinase B, and human epidermal growth factor receptor 2 tyrosine kinase signaling cascades (211–214). For example, cytoplasmic nongenomic signaling has been reported to occur in breast cancer cells via the cytosolic PR-B, which activates c-src and MAPK as well as Wnt-1 and the epidermal growth factor receptor (144). MPA acting via the PR has been shown to activate the signal transducer and activator of transcription 3 pathway via a mechanism involving rapid, nongenomic tyrosine phosphorylation in breast cancer cells (214). Furthermore, the mechanism also involves nongenomic induction of human epidermal growth factor receptor 2 nuclear translocation (215) to stimulate late cell growth. In most cases, the effects of different progestogens on activation of kinases have not been investigated, with most researchers using either R5020 (144, 212, 215) or MPA (214) simply as a potent PR reference agonist. One study that compared the effects of MPA vs. progesterone on kinase activation found that whereas both MPA and progesterone activate the Erk MAPK, only progesterone resulted in Erk nuclear translocation (211), suggesting that more research needs to be done to investigate differential kinase activation by progestogens. Whether a membrane-associated PR is the same protein as the intracellular PR is controversial. Some studies have reported the possible involvement of a novel cell surface membrane PR (mPR), which is similar to G protein-coupled receptors and couples to a G protein (216). However, although this mPR was reported to bind progesterone, it did not bind to several progestins such as MPA and norethindrone (216–218).
Nongenomic actions of progestogens via the PR have been implicated in playing an important role in breast cancer. Work in human breast cancer cell lines shows that MPA induces cell proliferation by increasing cyclin D1 promoter activity via the PR-B isoform, but not PR-A (219). The mechanism appears to involve cytoplasmic activation by the PR-B of the phosphatidylinositol 3-kinase/protein kinase B/NFκB signaling cascade, resulting in activation of the cyclin D1 promoter, which does not contain PRE-related sequences (219).
The mechanisms and physiological significance of nongenomic signaling by different progestogens via the PR isoforms remain to be further investigated and could potentially be relevant to differential side effects. Off-target differential nongenomic signaling by progestogens via steroid receptors other than the PR may also be physiologically relevant. These may, for example, be involved in mediating differential actions of progestogens in brain mitochondrial function (169, 176). Interestingly nongenomic actions mediated via the GR and ER have been reported to be involved in neuroprotection (220, 221), whereas nongenomic actions via the AR are likely to be involved in spermatogenesis (222). For the MR, nongenomic actions have been implicated in several physiological processes including brain signaling, endothelial dysfunction, and inflammation (209). Given the differential binding affinities and genomic actions of most progestogens via the intracellular classical ER, AR, and GR, it is likely that progestogens also exhibit differential nongenomic actions via these receptors when membrane bound or possibly via other novel membrane-bound steroid receptors.
Clinical Effects of Progestogens in Postmenopausal Hormone Therapy
Estrogen therapy is effective in treating climacteric symptoms and in the prevention of menopausal osteoporosis. The addition of progestogen in combination or sequential regimens has risks and benefits with regard to the endometrium, breast, cardiovascular system, bone, and brain.
A. Effects on the endometrium
As stated earlier, a progestogen is defined as a substance that transforms an endometrium primed by estrogen into a secretory endometrium. Progestogens are therefore used in HT in women with uteri to prevent the endometrial hyperplasia and endometrial cancer that may result from the use of estrogen only, often referred to as unopposed estrogen. Progestogens exert their protective effects by decreasing nuclear mitotic activity induced by estrogens and by increasing 17β-hydroxysteroid dehydrogenase type 2 activity that converts estradiol to the biologically less potent estrone.
The development of endometrial hyperstimulation with estrogen use increases with higher doses and duration of unopposed estrogen (223–225). Most recently, the Million Women Study (226) estimated the number of endometrial cancers per 1000 women in 5 yr to be 3.0 (2.8–3.2) without HT, 4.9 (3.5–7.5) with unopposed estrogen therapy, and 2.0 (1.5–2.6) with combined estrogen and progestin therapy; expressed in terms of relative risk (RR), HT never-users had a RR of 1.0, unopposed estrogen users had a RR of 1.45 ]95% confidence interval (CI) = 1.02–2.06; P = 0.04] and combined continuous estrogen with progestogen users had the lowest RR of 0.71 (95% CI = 0.56–0.90; P = 0·005). Examination of the participants' endometrial histology in the Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial showed consistent endometrial protection across all treatment groups, comprising conjugated equine estrogens (CEEs) combined with MPA given either continuously (0.625 mg CEE/2.5 mg MPA) or sequentially (0.625 mg CEE/10 mg MPA), or 0.625 mg CEE plus 200 mg micronized progesterone, compared with placebo (the placebo group had one case of endometrial adenocarcinoma) (227). However, the European Prospective Investigation into Cancer and Nutrition (n = 115,474) noted an increased endometrial cancer risk in all hormone users including treatment with estrogen only [39 cases, hazard ratio (HR) = 2.52; 95% CI = 1.77–3.57], estrogen plus progestin (121 cases, HR = 1.41; 95% CI = 1.08–1.83), and estrogen plus micronized progesterone (26 cases, HR = 2.42; 95% CI = 1.53–3.83) (228). The authors stated that risks differed according to regimen and type of progestin used and that because the micronized progesterone findings are based on small numbers, they recommended further study. This study also found that continuous regimens may better reduce the risk of endometrial cancer; the group receiving continuous treatment had only three cases of endometrial cancer (HR = 0.24; 95% CI = 0.08–0.77), whereas the sequential group had 50 cases (HR = 1.52; 95% CI = 1.00–2.29) (228). In light of these data, continuous daily combined estrogen/progestogen treatment as well as daily estrogen with sequential progestin (for 12–14 d/month), has been advocated to decrease estrogen-stimulated risk of endometrial adenocarcinoma and its precursor hyperplasia. Typical daily dosages of progestogens used for endometrial protection in women using estrogen therapy are shown in Table 6.
Table 6.
Typical daily progestogen doses used for endometrial protection in hormone therapy
| Progestogen | Dose (mg) |
|---|---|
| Progesterone | 200–300 |
| MPA | 2.5–10.0 |
| Chlormadinone acetate | 10 |
| Cyproterone acetate | 1 |
| Dydrogesterone | 5–10 |
| Nomegestrol acetate | 5–10 |
| Promegestone | 0.25–0.5 |
| Trimegestone | 0.5 |
| Norethindrone acetate | 0.5–1.0 |
| Levonorgestrel | 0.075 |
| Desogestrel | 0.075 |
| Norgestimate | 0.09 |
| Gestodene | 0.05 |
| Dienogest | 3–4 |
| Drospirenone | 2 |
Values are based on Ref. 6.
Progestogens are available in various formulations that can be prescribed for endometrial protection. Progesterone vaginal gel (4%) used biweekly for 1 yr together with a transdermal estradiol patch releasing 50 μg/d estradiol resulted in atrophic endometrium being observed on biopsy in all subjects (229). Progesterone capsules (100 mg) administered orally every other day combined with a transdermal patch releasing 50 μg estradiol per day for a duration of 3 yr also resulted in endometrial biopsies showing atrophy in all women (230). Additionally, the levonorgestrel intrauterine system has been studied for endometrial protection when systemic estrogen is used. There was no endometrial hyperplasia in three studies using the levonorgestrel intrauterine system in postmenopausal women receiving 1.25 mg/d oral CEE (231), 1.5 mg/d estradiol gel (232), and either a 50-μg estradiol patch or 2 mg oral estradiol valerate (233), each for a duration of 5 yr.
B. Effects on the breast
Although in vitro experiments cannot replace clinical trials, they are useful to explore possible differences between substances tested in the same model, which can then be confirmed in clinical studies. There are numerous experimental data available on the effect of progestogens on proliferation of normal and cancerous breast epithelial cells, but only a limited number of experiments have been carried out testing multiple progestogens in the same cell model. Of particular interest is an in vitro study in which the effects of progesterone, MPA, chlormadinone acetate, norethindrone, levonorgestrel, gestodene, and dienogest on proliferation and apoptosis of normal breast epithelial cells were tested at various concentrations, and the ratio of apoptosis to proliferation was compared (234). MCF-10A, a human, nontumorigenic, estrogen- and PR-negative breast epithelial cell line, was used with a mixture of growth factors to stimulate the cells. In combination with growth factors, the apoptosis/proliferation ratio was reduced significantly by MPA and chlormadinone acetate, favoring a proliferative effect. MPA produced as much as a 4-fold reduction in the ratio. The other progestogens had no significant effect. The results suggest that the progestogens differ in their ability to induce proliferation or inhibit the growth of normal human breast epithelial cells. However, these results should be interpreted with caution because MCF-10A cells lack PRs and ERs and have a myoepithelial phenotype. This differs from the normal breast, which contains ER/PR-positive luminal cells that are likely the primary point of response to HT.
Information about the risk of a clinical breast cancer diagnosis associated with isolated progestogen use in postmenopausal women is theoretical in that there are few clinical indications other than HT, where it is used solely for endometrial protection in nonhysterectomized women. In premenopausal women using progestogens in hormonal contraception, clinical breast data on the progestogen-only effect is scant because the reproductive age group of women users has a low baseline risk of breast cancer. The result of adding a progestogen to estrogen in HT creates a more complex and confusing situation because there is no consensus on the background effect of estrogen itself as well as the effect of its dose, delivery system, treatment time, and the patient's background medical and lifestyle attributes. Publication of the WHI study in 2002 (1) and subsequent analyses of data have generated an intense interest in the issue of independent risk from the progestogen when added to estrogen therapy, specifically regarding breast cancer. As a result, epidemiological studies are moving away from investigating estrogen alone compared with estrogen combined with any progestogen and instead comparing estrogen alone with estrogen combined with specific unique progestogens.
The WHI CEE/MPA study arm was stopped prematurely at 5.2 yr because of increased risk of breast cancer with a HR of 1.26 (95% CI = 1.0–1.6) (1) after an average of 7.1 yr. Although there was no increased risk of breast cancer diagnosis of invasive breast cancer in the estrogen-only arm, CEE compared with placebo (HR = 0.80; 95% CI = 0.62–1.04; P = 0.09), there was little immediate follow-up on this different outcome in the two arms (235). The WHI CEE/MPA study arm remained the focus of analyses because the cancers were more advanced and more commonly node-positive (81cases, 23.7%, vs. 43 cases, 16.2%, respectively; HR = 1.96; 95% CI = 1.23–2.58). Breast cancer mortality appeared to be greater in the CEE/MPA group (25 deaths, 0.03% per year) compared with the placebo group (12 deaths, 0.01% per year) (236). It was noted that there was increased mammographic density that interfered with mammographic detection, resulting in diagnostic delay as well as diagnosis of breast cancers at a more advanced stage (237).
Mammographic density is a breast cancer risk factor and is associated with increased risks for hyperplasia, atypical hyperplasia, and ductal carcinoma in situ. The earlier PEPI trial found greater breast density over 12 months regardless of whether the regimen was continuous (0.625 mg CEE/2.5 mg MPA) or whether it was sequential (0.625 mg CEE/10 mg MPA or CEE/200 mg micronized progesterone) (238). When HT was discontinued, mammographic density quickly returned to normal levels. The impact of dose was then studied in the combination regimens. In a study evaluating ultra-low-dose HT (0.5 mg estradiol/0.25 mg norethindrone acetate or 0.5 mg estradiol/0.1 mg norethindrone acetate), no change was found in breast density after 6 months of use (239).
Besides the WHI results, additional epidemiological data on postmenopausal HT have consistently reported that the addition of any progestin to estrogen increases the risk of breast cancer diagnosis compared with estrogen alone (240–245). A case-control study (n = 33,000) evaluating breast cancer risk from 1996–2006 in an insurance database population of women aged 50–64 yr (246) reported an odds ratio of 0.96 (95% CI = 0.88–1.06) for developing breast cancer in women using estrogen-only therapy, whereas in those using estrogen/progestin HT, the odds ratio was 1.44 (95% CI = 1.31–1.58). Another study evaluated pooled data from six mammographic registries (n = 373,265); HT users with and without hysterectomy were recruited and followed prospectively, noting the intervals between mammograms (247). Use of estrogen and progestin for greater than 5 yr was associated with a greater risk of breast cancer diagnosis (RR = 1.49; 95% CI = 1.36–1.63) compared with nonusers. The increased risk was not observed either in estrogen plus progestin users who had used HT for less than 5 yr (RR = 0.85; 95% CI = 0.73–0.98) or in estrogen-only users irrespective of duration of use whether less than 5 yr (RR = 0.86; 95% CI = 0.71–1.03) or more than 5 yr (RR = 0.92; 95% CI = 0.84–1.00) (247).
A 2006 meta-analysis of studies to assess evidence for a link between postmenopausal HT and risk of breast cancer diagnosis (248) found an average RR of 0.79 (95% CI = 0.61–1.02) for invasive breast cancer diagnosis with estrogen use and of 1.24 (95% CI = 1.02–1.50) with estrogen-progestin use in four randomized trials, whereas epidemiological studies reported a RR of 1.18 (95% CI = 1.01–1.38) with estrogen alone and 1.70 (95% CI = 1.36–2.17) with estrogen plus progestin (248). Hormone use and breast cancer histology was also evaluated in a prospective cohort (n = 67,754) (249); in the estrogen and progestin group, both ductal cancer (RR = 1.75; 95% CI = 1.59–2.01) and lobular cancer (RR = 2.12; 95% CI = 1.62–2.77) diagnoses were increased compared with the estrogen-alone group, in which there was no increase in the diagnosis of ductal cancer (RR = 0.99; 95% CI = 0.84–1.17) or lobular cancer (RR = 1.13; 95% CI = 0.94–1.78) (249).
A recent WHI analysis (250) has reviewed the risk of breast cancer diagnosis in the CEE-alone study arm. After a median follow-up of 11.8 (9.1–12.9) yr with estrogen use for 5.9 (2.5–7.3) yr, there was a lower incidence of breast cancer (151 cases, 0.27% per year) compared with placebo (199 cases, 0.35% per year; HR = 0.77; 95% CI = 0.62–0.95; P = 0.02). In the estrogen-only arm, mortality per year from breast cancer (96 deaths, 0.009%) was less than that seen in controls who did not use HT (16 deaths, 0.024%), and in fact, fewer women in the estrogen-alone group than the control group died from any cause.
A current review (251) summarizes the conceptual change since 2002, based on WHI CEE and CEE/MPA data, in light of the fact that epidemiological studies consistently show estrogen when combined with progestin carries a different breast cancer risk compared with estrogen alone when used for 5 yr. The review reiterates the conclusions noted in previous citations with regard to CEE/MPA risks and states that estrogen-alone use reduces breast cancer risk and does not substantially interfere with breast cancer detection by mammography.
Clarification of the differential effects of different progestogens combined with estrogen is limited because few epidemiological studies have had sufficient sample sizes and/or accurate information to assess breast cancer risk in relation to different types and routes of estrogen administration associated with different types of progestogens. However, the observational Million Women Study (155, 252) found no significant differences in risk of breast cancer diagnosis between estrogen alone (RR = 1.30; 95% CI = 1.22–1.38) and estrogen combined with various progestins including tibolone (RR = 1.45; 95% CI = 1.25–1.67), estrogen/norethindrone (RR = 1.53; 95% CI = 1.35–1.75), estrogen/MPA (RR = 1.60; 95% CI = 1.33–1.93), and estrogen/norgestrel/levonorgestrel (RR = 1.97; 95% CI = 1.74–2.33). Moreover, until recently, micronized progesterone has not been included in such studies.
That changed with the publication of results from the French E3N Cohort Study (253), which assessed and compared the association between different HT regimens and breast cancer risk in 80,377 postmenopausal women, whose mean age was 53.1 yr. The women were followed up for an average of 8.1 postmenopausal years, during which they completed self-administered biannual questionnaires addressing medical history, menopausal status, and lifestyle characteristics. A total of 2354 cases of invasive breast cancer cases were identified primarily from the self-reports, 95.3% of which were confirmed by pathology reports. Information on lifetime use of hormonal treatments was also obtained from the questionnaires. The women were given a booklet listing the hormonal treatments marketed in France, complete with color photographs and products, to help them remember what preparations they had taken. Seventy percent of the women had used HT for a mean duration of 7 yr.
When RRs of invasive breast cancer associated with the most frequently used HTs were compared with never-used HT, RRs varied significantly between the different progestogens for any given route of estrogen administration (oral or transdermal). Estrogen-progesterone and estrogen-dydrogesterone combinations were associated with no or slight and nonsignificant increases in risk, whereas all other estrogen/progestogen combinations showed substantially increased risks, most of which were statistically significant but did not differ significantly between preparations. Other than progesterone and dydrogesterone, the progestogens included medrogestone, chlormadinone acetate, cyproterone acetate, promegestone, nomegestrol acetate, norethindrone acetate, and MPA. The latter two progestins were combined with oral estrogen compared with all the other progestogens, which were combined with transdermal estrogen. Thus, the estrogen delivery system was also a differential factor in the study (253).
Because of those findings, subsequent statistical evaluations included separate estimates for HTs containing progesterone or dydrogesterone, but the other progestogens were grouped together. When RRs for invasive breast cancer associated with type of HT and duration of exposure were compared with never-used HT, women in the estrogen-alone and estrogen/other progestogen groups had a significantly increased breast cancer risk, with RRs of 1.29 (95% CI = 1.02–1.65) and 1.69 (95% CI = 1.50–1.91), respectively. In contrast, estrogen-progesterone was associated with a RR of 1.00 (95% CI = 0.83–1.22) and estrogen-dydrogesterone with a RR of 1.16 (95% CI = 0.94–1.43). Estrogen-alone, estrogen-progesterone, and estrogen-dydrogesterone were associated with breast cancer risks that did not differ significantly from one another but were all significantly lower than the RR of estrogen-other progestogens (253).
Estrogen itself remains controversial regarding breast cancer risk. However, the epidemiological studies clearly delineate a different risk of diagnosis of breast cancer in estrogen-alone vs. estrogen plus progestogen treatments. Although the return to baseline risk in former HT users supports a promotional effect rather than an initiating effect of estrogen with progestogen in the risk of breast cancer diagnosis (254), the data do not support such a promotional effect from estrogen alone. It is only when the progestogen is added to the estrogen-primed breast tissue that there is an increase in diagnosis of breast cancer. The emerging clinical epidemiological data support the hypothesis that progestogens are not a uniform class and that progesterone and progestins have different effects, with distinctive impacts on the risk of breast cancer diagnosis in menopausal women using HT.
C. Effects on the cardiovascular system
The widespread prescription of HT to postmenopausal women was historically, in addition to relieving menopausal symptoms, intended to protect women from cardiovascular disease. However, results of the larger trials to test the benefits of HT in reducing cardiovascular events were disappointing. The 6.8-yr follow-up report on the Heart and Estrogen/Progestin Replacement Study found no significant reduction in primary or secondary coronary heart disease (CHD) events in the CEE/MPA group vs. the placebo group (255). Manson and co-workers (256) reported an increased risk of cardiovascular disease with CEE/MPA in the WHI trial, noting that this was most apparent only during the first year of use. Additional analysis of the WHI study concluded that women who initiated HT closer to menopause tended to have reduced CHD event risk vs. women more distant from menopause (257). Also, women who initiated therapy at a younger age had a lower CHD event risk compared with women who initiated therapy at an older age. Short-term use of HT in the immediate postmenopausal years has been advocated to protect against cardiovascular events in the long term, whereas initiating HT is not recommended for older women who are already at higher risk of cardiovascular disease (258).
Further examinations of those WHI study trends demonstrate a differential CHD event risk between the CEE/MPA group and the CEE-alone group (257). For example, women not stratified by age, who initiated HT within 10 yr of menopause in the CEE-alone group had decreased CHD events (HR = 0.48; 95% CI = 0.2–1.17) compared with the CEE/MPA group (HR = 0.88; 95% CI = 0.54–1.43). Also, the decrease in CHD events in the 50- to 59-yr group was more pronounced in the CEE-alone group (HR = 0.63; 95% CI = 0.36–1.09) when compared with the CEE/MPA group (HR = 1.29; 95% CI = 0.79–2.12). The decreased CHD events in the CEE arm vs. the CEE/MPA arm in these two patient groups suggest that it is the CEE/MPA that has the adverse impact. Therefore, there is a potential for cardiovascular protection with estrogen alone and perhaps by estrogen combined with other progestogens. This potential should be further explored.
Much has been written since the Heart and Estrogen/Progestin Replacement Study and WHI results were published about the cardiovascular implications of HT, largely concentrating on the type and route of administration of the estrogenic component because of its importance in modulating cardiovascular risk. Estrogen treatment improves lipid profiles and insulin sensitivity and has beneficial effects on mitigating central weight gain in menopausal women. Unfortunately, there are few long-term clinical studies comparing different progestogens used in HT with respect to cardiovascular outcomes. However, some aspects of potential cardiovascular risk have been examined, namely effects on lipids, vascular function/blood pressure, inflammation, thrombosis, and carbohydrate metabolism.
The most common comparison has been between progesterone and MPA, and Hermsmeyer et al. (259) have cautioned against a negative view of HT for cardiovascular protection based only on the results of trials involving MPA. When oral micronized progesterone was used in one group in the PEPI study in place of MPA, this group had significantly higher HDL cholesterol levels than the MPA group, indicating a more favorable effect on blood lipids (260), although there is no evidence that this improved cardiovascular outcomes. A small study of 18 women showed that progesterone vaginal gel produced an increase in exercise tolerance in postmenopausal women with coronary artery disease or previous myocardial infarction who were being treated with estradiol, whereas MPA did not, compared with estradiol alone (261). Primate studies have demonstrated a marked adverse effect of MPA on coronary artery hyperreactivity that is the opposite of the protective effect seen with progesterone (262). Furthermore, MPA, but not progesterone, negated the coronary vasospasm protective effects of estradiol, shown by measuring intracellular calcium and protein kinase C signals (263, 264), and progesterone reduced coronary hyperreactivity even in the presence of atherosclerosis in oophorectomized rhesus monkeys (265). Adverse effects on carbohydrate metabolism, namely higher fasting glucose and insulin levels, and higher insulin responses to glucose challenge have also been demonstrated in oophorectomized cynomolgus monkeys receiving CEE plus MPA compared with CEE alone or no HT (266).
A review of the effects of progestins on cardiovascular risk markers by Sitruk-Ware (159) showed that progesterone and its 19-norprogesterone derivatives, which have no androgenic effects, did not adversely impact the beneficial effects of estrogens on the lipid profile, notably in their ability to increase HDL cholesterol levels, whereas those with androgenic properties, namely the 19-nortestosterone derivatives and some 17-hydroxyprogesterone derivatives, including MPA, have shown negative effects on lipids. This observation has been confirmed in more recent studies, e.g. a comparison of trimegestone (either 0.25 or 0.5 mg) or norethindrone acetate combined with estradiol (267), which showed a 10% reduction in lipid markers of myocardial infarction risk with the trimegestone doses but no effect with norethindrone acetate. A meta-analysis of 248 studies published from 1974–2000 (268) examining effects of HT regimens on lipids found that estrogen-only regimens raised HDL and triglycerides and lowered low-density lipoprotein and total cholesterol; these effects were opposed by progestins to various degrees according to the progestin. Progestins placed in order from the least to the greatest opposing effects were dydrogesterone and medrogestone, progesterone, cyproterone acetate, MPA, transdermal norethindrone acetate, norgestrel, and oral norethindrone acetate. An intermittent regimen with estradiol plus norgestimate has been compared with estradiol plus norethindrone acetate in women who already had an unfavorable lipid profile; although both regimens improved lipid values after 12 months, a higher percentage of the women treated with norgestimate than with norethindrone acetate were found to have HDL cholesterol within recommended ranges (269).
Another advantage of the less androgenic progestins is their more positive impact on the hemostatic system. In a multicenter study of 186 postmenopausal women comparing estradiol plus either dydrogesterone or trimegestone for 6 months (270), a decrease in protein C activity and an increase in plasmin-antiplasmin complex were seen in the trimegestone group. This suggested an enhanced fibrinolytic response that could translate to a reduced risk of thrombosis and consequent reduced risk of stroke or myocardial infarction. Recent clinical studies outline the contribution of the progestogen component of HT to thrombotic risk. The French Study of Norpregnanes on Coagulation compared hemostatic parameters with no HT, transdermal estradiol plus micronized progesterone, and transdermal estradiol plus norpregnane derivatives (nomegestrol acetate or promegestone) (271). Thrombin generation in the presence or absence of activated protein C showed activated protein C resistance and therefore increased thrombotic potential in the norpregnane group but not in the micronized progesterone group compared with no HT. Also, data on thromboembolism incidence after an average follow-up of 10.1 yr in the E3N Cohort Study of 80,308 postmenopausal women (272) showed significantly increased thrombotic risk with norpregnanes (HR = 1.8) compared with progesterone (HR = 0.9), pregnanes (HR = 1.3), and 19-nortestosterone derivatives (HR = 1.4).
CEE plus MPA or CEE alone was also found to increase levels of high-sensitivity C-reactive protein, a marker of inflammation that is implicated in the development of atherosclerosis, whereas CEE plus nomegestrol acetate reduced high-sensitivity C-reactive protein levels in a randomized study in postmenopausal women (273); all HT groups showed a decline in homocysteine levels. On the other hand, a small, randomized comparison of MPA or oral micronized progesterone combined with CEE in 20 postmenopausal women found similar improvements in both groups with respect to flow-mediated dilator response to hyperemia as well as similar effects on markers of inflammation, hemostasis, and fibrinolysis (274). Also, a comparison of cyproterone acetate or MPA in 26 postmenopausal women receiving estradiol valerate (275) found that both progestins attenuated the beneficial effects of the estrogen on nitric oxide release, the mechanism by which estrogen is thought to exert its vascular endothelial effects.
The newer progestin, drospirenone, has antimineralocorticoid properties and therefore antihypertensive effects, which could translate to benefits in cardiovascular risk. A study of three doses of drospirenone plus estradiol, estradiol alone, or placebo for 8 wk in 750 postmenopausal women with hypertension found a significant improvement in systolic blood pressure with 2- and 3-mg doses of drospirenone plus estradiol compared with estradiol alone or a 1-mg dose of drospirenone (276). Similar results were seen when measuring early morning systolic blood pressure (277). Simoncini and Genazzani (278) have reviewed the preclinical experience with drospirenone, noting its antiandrogenic as well as antialdosterone activity, giving it potential advantages over MPA in its effects on the cardiovascular system.
Examination of the United Kingdom General Practice Research Database for risk of developing myocardial infarction, thrombotic stroke, or venous thromboembolism (VTE) in users of estradiol plus dydrogesterone (279) showed no higher risk of myocardial infarction for this treatment compared with other HT (CEE plus norgestrel, estradiol valerate plus norethindrone acetate, or CEE plus MPA) or nonusers of HT. Risk of thrombotic stroke and VTE was slightly decreased for estradiol/dydrogesterone compared with nonusers or other HT, although differences were not significant, whereas VTE risk was increased in users of other HT compared with nonusers. The multicenter case-control Estrogen and Thromboembolism Risk (ESTHER) study in France (280) found, in addition to a significantly increased risk of VTE with oral compared with transdermal estrogen, that there was no significant association of VTE with micronized progesterone and pregnane derivatives but a 4-fold increased risk of VTE when norpregnane derivatives were used in combination HT. Pregnane derivatives included dydrogesterone, medrogestone, chlormadinone acetate, cyproterone acetate, and MPA, whereas the norpregnane derivatives were either nomegestrol acetate or promegestone. Studies of combined OCs have indicated that third-generation progestins (desogestrel or gestodene) may carry a greater VTE risk than OCs containing levonorgestrel (281). Although a recent review indicated that the newer progestin drospirenone did not appear to show an increased VTE risk compared with other progestins when used in combined OCs (282), two new case-control studies have found an increased VTE risk with drospirenone compared with levonorgestrel (283, 284). Also, a Danish cohort study showed that OCs containing levonorgestrel were associated with a 3-fold increase in risk for VTE compared with nonusers of OCs, whereas users of OCs containing desogestrel, gestodene, or drospirenone had a 6- to 7-fold increase in VTE risk compared with nonusers (285), i.e. at least twice the risk for levonorgestrel.
Although progestins have differing effects on aspects of cardiovascular risk, in general, those more similar to progesterone have been associated with a lower impact than the more androgenic progestins on the beneficial effects of concomitant estrogen therapy. However, the limited number of long-term clinical studies makes it difficult to extrapolate the short-term effects on various markers of cardiovascular risk to long-term cardiovascular morbidity.
D. Effects on the brain
Progesterone has important functions in the nervous system and has been classified as a neurosteroid. Endogenously, progesterone is synthesized de novo from cholesterol in the brain, spinal cord, and peripheral nerves, and its actions may be mediated by local metabolism to allopregnanolone (286, 287). Recent clinical studies have demonstrated significant neuroprotective activity of high-dose progesterone treatment in subjects with traumatic brain injury (288, 289), leading to additional phase III trials that are ongoing. Progesterone is a promising therapy for acute neuroprotection in brain injuries and neurogenerative diseases, probably via multiple physiological mechanisms (290, 291).
Whether or not the neuroprotective benefits of progesterone can also be achieved by progestins has not been determined clinically (292). However, rat studies have demonstrated that the neuroprotective effects of progesterone are not seen with MPA (293, 294). In a model of estrogen-induced neuroprotection involving assessment of the effects of glutamate toxicity in rat hippocampal neuron cultures, both progesterone and 19-norprogesterone showed neuroprotection, either alone or in combination with estradiol, whereas MPA was not neuroprotective and even blocked the neuroprotective activity of estradiol in this model (295). The same group assessed the impact of progesterone and MPA on the excitotoxic glutamate-induced rise in intracellular calcium levels, a neurotoxic effect, which was attenuated by both progesterone and estradiol. MPA did not have an effect when administered alone to the hippocampal neurons, but when administered together with estradiol, MPA completely antagonized the attenuation by estradiol of the rise in intracellular calcium (296). The MAPK cascade is a mechanism involved in this estrogen-mediated neuroprotection, and the authors showed that MPA, but not progesterone, blocked the estradiol-induced nuclear translocation of extracellular receptor kinase, which is required for calcium regulation. Furthermore, when this group looked at survival of hippocampal neurons exposed to crystalline MPA or a pharmaceutical formulation containing MPA (Depo-Provera) in that model, both showed a lack of neuroprotective efficacy; also, medroxyprogesterone (without the acetate group) was as ineffective as MPA (297).
A study of acute MPA administration to brain-injured rats showed a dose-related reduction in cerebral edema but no improvement in performance on a spatial learning task, indicating some beneficial antiinflammatory effects but no functional improvement compared with progesterone (298). Thus, the differences between effects of progesterone and progestins that have been observed in other tissues may also pertain to neurobiological effects (287).
In the WHI memory study, greater brain atrophy, assessed by a reduction in hippocampal volume on brain magnetic resonance imaging, was seen with CEE either with or without MPA compared with the placebo group, although this was most apparent in women with cognitive defects before initiating HT (299).
A review of the effects of progesterone on the nervous system by Schumacher and co-workers (300) notes that it is a mistake to consider progestogens as a single class, because progestins have some very different properties from those of natural progesterone. The authors also note that the adverse clinical effects of some progestins should not discourage the use of natural progesterone or the development of new, safer progestins for use in postmenopausal HT to reverse age-dependent dysfunction of the nervous system.
E. Effects on bone
Estrogens are well known to prevent bone loss because of their physiological inhibitory effect on bone resorption through the osteoclasts. According to meta-analyses, randomized controlled trials of menopausal HTs consistently indicate improved bone density with estrogen use (301, 302). Norethindrone acetate has been shown to prevent bone resorption in postmenopausal women without added estrogen (303). High doses of MPA cause partial reduction in bone resorption (304). Cyclic MPA has been found to increase bone mineral density in premenopausal women with anovulatory or short luteal-phase menstrual cycles (305); however, clinical investigations of the effect of progesterone have been inconclusive (306). Other progestins have not been well studied. Thus, there is limited evidence for an independent effect of progestogens on bone.
VIII. Conclusions
This review compared progestogens with respect to their chemical structures, structure-function relationships, metabolism, pharmacokinetic parameters, intracellular mechanisms, potency, efficacy, and biological and clinical effects. The chemical structures of progestogens vary widely. Some are structurally related to progesterone, others to testosterone, and one to spironolactone. Progestogens also differ in their metabolism and pharmacokinetic profiles. Some are prodrugs and require transformation to active forms, and they have wide differences in their bioavailabilities and half-lives. Although progestogens are designed to be potent and high-affinity PR agonists that mimic the biological actions of the natural ligand, progesterone, many of them bind to other members of the steroid receptor family, which include the AR, GR, and MR. Furthermore, they exhibit considerable variation in their binding affinities, potencies, and efficacies as well as the resulting extent of agonist, partial agonist, or antagonist responses via these receptors. All these differences are most likely major determinants of differential actions and the lack of a class effect of progestogens. Relative concentrations of free progestogen and competing endogenous ligands reaching the target cell would depend on relative affinities for serum binding globulins in the blood (87, 307) as well as half-life, metabolism, route, and method of administration and dosage (5, 7, 308–310). Once inside the cell, the fractional occupancy of a particular receptor, and hence the relative response via that receptor, would depend on concentrations of metabolizing enzymes in the target cell as well as relative intracellular concentrations of steroids and their relative affinities for different binding proteins.
Another major determinant of differential actions by progestogens is most likely the cell-specific concentration of proteins that affect the final biological response. These include variations in concentration and isoform type of classical intracellular and membrane-bound steroid receptors, nonsteroidal receptors that cross talk with the steroid receptor pathway, steroid-metabolizing enzymes, and all interacting partners in the various steps leading to the progestogen-induced biological response. For example, the effects of PR ligands on proteasomal-mediated turnover of coactivators like steroid receptor coactivator-1 are likely to affect progestogen genomic responses (311). Cell-specific effects can be dramatic; some steroid receptor ligands can even switch from agonist to antagonist, depending on the milieu of cell-specific cofactors (312), whereas a particular progestogen can act either as an agonist, a partial agonist, or an antagonist, dependent solely on steroid receptor concentration (187). The relative tissue-specific distribution and variable levels of steroid receptors are likely to play a major role in differential progestogen actions and side effects, given the variation in affinities of progestogens for different receptors.
The explicit clinical effects of progestogens are difficult to determine. In postmenopausal women, there is scant information on the clinical effects of progestogens alone, because progestogens are generally prescribed when estrogen is being used to prevent endometrial cancer associated with unopposed estrogen. Until the WHI trial, progestogens did not have a major role in the general HT discussion because they were prescribed only for endometrial safety should estrogen be prescribed. There was minimal concern for their contribution to the risk/benefit profile. The WHI findings have highlighted the need to understand the clinical effects of progestogens as well as those of estrogen. The differential clinical outcomes in the epidemiological studies of estrogen alone compared with combined estrogen and progestogen regimens have generated questions regarding progestogens and the recognition that they have potentially distinctive risk/benefit profiles when combined with estrogen. However, the clinical outcome data available are confusing due to the use of different estrogens and progestogens, different doses, delivery systems, and treatment regimens, and different risk attributes for the users. Post-WHI epidemiology is focusing on comparing the clinical outcomes of estrogen alone with estrogen combined with different specific progestogens.
In breast and cardiovascular studies, the outcome data show different RRs and lack of a class effect of progestogens. Although the Million Women Study shows that “benefits for endometrial cancer associated with continuous combined progestin therapy may be outweighed by risks for breast cancer, which is adversely affected by the therapy” (252), only progestins, and not progesterone, were evaluated in the study. In the recent French E3N Cohort Study, estrogen plus progesterone or dydrogesterone regimens were associated with decreased invasive breast cancer risk compared with regimens consisting of estrogen combined with other progestogens. Although the endometrial protection from progestins and progesterone as prescribed in the PEPI Trial is accepted as the standard in the United States, the European Prospective Investigation results call for additional confirmation of endometrial protection with progesterone. It is essential to improve our understanding of the role of progestogens when added to estrogen therapy and the risk/benefit profile compared with estrogen alone in terms of the risks of diagnosis of endometrial and breast cancer. We hypothesize that estrogen acts via a mechanism that primes the tissue, and when progestins are added, there is a promotional effect resulting in increased breast cancer diagnosis. The clinical characterization of progesterone and its differentiation from the progestins is an important component of the decision to add it to estrogen therapy.
Breast cancer is five times more common than endometrial cancer, and with regular monitoring, endometrial hyperplasia can be diagnosed well before endometrial cancer develops (313). In addition, the protective role of estrogen against risk of breast cancer, supported by the epidemiological literature, should not be overlooked in light of a potentially deleterious effect of progestogen.
Evaluation of thrombotic and thromboembolism risk, as well as the fact that different progestogens affect cardiovascular markers differently, suggests that progesterone may have a decreased cardiovascular risk compared with other progestogens. The neuroprotective effect of progesterone is also being explored, whereas MPA has been shown to blunt estradiol's beneficial effects in the brain.
A thorough review of the properties of the various progestogens shows that there are considerable data confirming the different effects of progestogens, the distinctiveness of progesterone, and the lack of a progestogen class effect. The properties of each progestogen should be carefully evaluated on an individual basis to determine its utility in postmenopausal HT.
Acknowledgments
We thank Margaret Groves, M.Phil., E.L.S., for her help in preparation of the manuscript, and Dr. Chanel Avenant for the preparation of Fig. 7.
Disclosure Summary: The authors have no relevant financial disclosures.
Footnotes
- AR
- Androgen receptor
- CEE
- conjugated equine estrogen
- CHD
- coronary heart disease
- CI
- confidence interval
- DBD
- DNA-binding domain
- DHT
- dihydrotestosterone
- ER
- estrogen receptor
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HDL
- high-density lipoprotein
- HR
- hazard ratio
- HT
- hormone therapy
- LBD
- ligand-binding domain
- MPA
- medroxyprogesterone acetate
- mPR
- membrane PR
- MR
- mineralocorticoid receptor
- NFκB
- nuclear factor-κB
- NO
- nitric oxide
- OC
- oral contraceptive
- PEPI
- Postmenopausal Estrogen/Progestin Interventions
- PR
- progesterone receptor
- PRE
- progesterone response element
- RBA
- relative binding affinity
- RR
- relative risk
- SRE
- steroid-responsive element
- TAF-1
- transcriptional activation function-1
- VEGF
- vascular endothelial growth factor
- VTE
- venous thromboembolism
- WHI
- Women's Health Initiative.
References
- 1. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J; Writing Group for the Women's Health Initiative Investigators 2002. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 288:321–333 [DOI] [PubMed] [Google Scholar]
- 2. Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, et al. 2004. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291:1701–1712 [DOI] [PubMed] [Google Scholar]
- 3. Stanczyk FZ, Henzl MR. 2001. Use of the name “Progestin.” Contraception 64:1–2 [DOI] [PubMed] [Google Scholar]
- 4. North American Menopause Society 2003. Role of progestogen in hormone therapy for postmenopausal women: position statement of the North American Menopause Society. Menopause 10:113–132 [DOI] [PubMed] [Google Scholar]
- 5. Stanczyk FZ. 2003. All progestins are not created equal. Steroids 68:879–890 [DOI] [PubMed] [Google Scholar]
- 6. Henzl ME. 1978. Natural and synthetic female sex hormones. In: Yen SCC, Jaffe RB, eds. Reproductive endocrinology: physiology, pathophysiology and clinical management. Philadelphia: WB Saunders; 421–468 [Google Scholar]
- 7. Kuhl H. 2005. Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric 8(Suppl 1):3–63 [DOI] [PubMed] [Google Scholar]
- 8. Stanczyk FZ, Roy S. 1990. Metabolism of levonorgestrel, norethindrone, and structurally related contraceptive steroids. Contraception 42:67–96 [DOI] [PubMed] [Google Scholar]
- 9. Edgren RA, Stanczyk FZ. 1999. Nomenclature of the gonane progestins. Contraception 60:313. [DOI] [PubMed] [Google Scholar]
- 10. Stanczyk FZ. 2000. Pharmacokinetics of progesterone administered orally and parenterally. In: Sitruk-Ware R, Mishell DR, Jr, eds. Progestins and antiprogestins in clinical practice. New York: Marcel-Dekker; 393–400 [Google Scholar]
- 11. Kuhnz W, Heuner A, Hümpel M, Seifert W, Michaelis K. 1997. In vivo conversion of norethesterone and norethisterone acetate to ethinyl estradiol in postmenopausal women. Contraception 56:379–385 [DOI] [PubMed] [Google Scholar]
- 12. Chu MC, Zhang X, Gentzschein E, Stanczyk FZ, Lobo RA. 2007. Formation of ethinyl estradiol in women during treatment with norethindrone acetate. J Clin Endocrinol Metab 92:2205–2207 [DOI] [PubMed] [Google Scholar]
- 13. Hammond GL, Lähteenmäki PL, Lähteenmäki P, Luukkainen T. 1982. Distribution and percentage of non-protein bound contraceptive steroids in human serum. J Steroid Biochem 17:375–380 [DOI] [PubMed] [Google Scholar]
- 14. Kuhnz W, Pfeffer M, al-Yacoub G. 1990. Protein binding of the contraceptive steroids gestodene, 3-keto-desogestrel and ethinylestradiol in human serum. J Steroid Biochem 35:313–318 [DOI] [PubMed] [Google Scholar]
- 15. Westphal U. 1974. Steroid-protein interactions. Berlin: Springler-Verlag [Google Scholar]
- 16. Simon JA, Robinson DE, Andrews MC, Hildebrand JR, 3rd, Rocci ML, Jr, Blake RE, Hodgen GD. 1993. The absorption of oral micronized progesterone: the effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertil Steril 60:26–33 [PubMed] [Google Scholar]
- 17. Victor A, Johansson ED. 1976. Pharmacokinetic observations on medroxyprogesterone acetate administered orally and intravaginally. Contraception 14:319–329 [DOI] [PubMed] [Google Scholar]
- 18. Farinha A, Bica A, Tavares P. 2000. Improved bioavailability of a micronized megestrol acetate tablet formulation in humans. Drug Dev Ind Pharm 26:567–570 [DOI] [PubMed] [Google Scholar]
- 19. Kuhnz W, Staks T, Jütting G. 1993. Pharmacokinetics of cyproterone acetate and ethinylestradiol in 15 women who received a combination oral contraceptive during three treatment cycles. Contraception 48:557–575 [DOI] [PubMed] [Google Scholar]
- 20. Kuhl H. 1990. Pharmacokinetics of oestrogens and progestogens. Maturitas 12:171–197 [DOI] [PubMed] [Google Scholar]
- 21. Lin WJ, Her SJ, Chen PF, Chen RR. 1998. Determination of medrogestone in plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 714:263–268 [DOI] [PubMed] [Google Scholar]
- 22. Mueck AO, Sitruk-Ware R. 2011. Nomegestrol acetate, a novel progestogen for oral contraception. Steroids 76:531–539 [DOI] [PubMed] [Google Scholar]
- 23. Back DJ, Breckenridge AM, Crawford FE, Mciver M, Orme ML, Rowe PH, Smith E. 1978. Kinetics of norethindrone in women. II. Single-dose kinetics. Clin Pharmacol Ther 24:448–453 [DOI] [PubMed] [Google Scholar]
- 24. Back DJ, Bates M, Breckenridge AM, Hall JM, MacIver M, Orme ML, Park BK, Rowe PH. 1981. The pharmacokinetics of levonorgestrel and ethynylestradiol in women: studies with Ovran and Ovranette. Contraception 23:229–239 [DOI] [PubMed] [Google Scholar]
- 25. Bergink W, Assendorp R, Kloosterboer L, van Lier W, Voortman G, Qvist I. 1990. Serum pharmacokinetics of orally administered desogestrel and binding of contraceptive progestogens to sex hormone-binding globulin. Am J Obstet Gynecol 163:2132–2137 [DOI] [PubMed] [Google Scholar]
- 26. Back DJ, Grimmer SF, Shenoy N, Orme ML. 1987. Plasma concentrations of 3-keto-desogestrel after oral administration of desogestrel and intravenous administration of 3-keto-desogestrel. Contraception 35:619–626 [DOI] [PubMed] [Google Scholar]
- 27. Orme M, Back DJ, Ward S, Green S. 1991. The pharmacokinetics of ethinylestradiol in the presence and absence of gestodene and desogestrel. Contraception 43:305–316 [DOI] [PubMed] [Google Scholar]
- 28. Täuber U, Tack JW, Matthes H. 1989. Single dose pharmacokinetics of gestodene in women after intravenous and oral administration. Contraception 40:461–479 [DOI] [PubMed] [Google Scholar]
- 29. Oettel M, Bervoas-Martin S, Elger W, Golbs S, Hobe G, Kaufmann G, Mathieu M, Moore C, Schneider B, Puri C, Ritter P, Reddersen G, Schon R, Strauch G, Zimmermann H. 1995. A 19-norprogestin without a 17α-ethinyl group. II. Dienogest from a pharmacokinetic point of view. Drugs Today 31:499–516 [Google Scholar]
- 30. Blode H, Wuttke W, Loock W, Röll G, Heithecker R. 2000. A l-year pharmacokinetic investigation of a novel oral contraceptive containing drospirenone in healthy female volunteers. Eur J Contracept Reprod Health Care 5:256–264 [DOI] [PubMed] [Google Scholar]
- 31. Krattenmacher R. 2000. Drospirenone: pharmacology and pharmacokinetics of a unique progestogen. Contraception 62:29–38 [DOI] [PubMed] [Google Scholar]
- 32. Nillius SJ, Johansson EDB. 1971. Plasma levels of progesterone after vaginal, rectal, or intramuscular administration of progesterone. Am J Obstet Gynecol 110:470–477 [PubMed] [Google Scholar]
- 33. Miles RA, Paulson RJ, Lobo RA, Press MF, Dahmoush L, Sauer MV. 1994. Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertil Steril 62:485–490 [DOI] [PubMed] [Google Scholar]
- 34. Stanczyk FZ, Paulson RJ, Roy S. 2005. Percutaneous administration of progesterone: blood levels and endometrial protection. Menopause 12:232–237 [DOI] [PubMed] [Google Scholar]
- 35. Leonetti HB, Wilson KJ, Anasti JN. 2003. Topical progesterone cream has an antiproliferative effect on estrogen-stimulated endometrium. Fertil Steril 79:221–222 [DOI] [PubMed] [Google Scholar]
- 36. Wren BG, McFarland K, Edwards L, O'Shea P, Sufi S, Gross B, Eden JA. 2000. Effect of sequential transdermal progesterone cream on endometrium, bleeding pattern, and plasma progesterone and salivary progesterone levels in postmenopausal women. Climacteric 3:155–160 [DOI] [PubMed] [Google Scholar]
- 37. Product insert 2007. Climara Pro. Wayne, NJ: Bayer HealthCare Pharmaceuticals [Google Scholar]
- 38. Harrison LI, Zurth C, Gunther C, Karara AH, Melikian A, Lipp R. 2007. Simultaneous estradiol and levonorgestrel transdermal delivery from a 7-day patch: in vitro and in vivo drug deliveries of three formulations. Drug Dev Ind Pharm 33:373–380 [DOI] [PubMed] [Google Scholar]
- 39. Product insert 2006. CombiPatch. East Hanover, NJ: Novartis Pharmaceuticals [Google Scholar]
- 40. Lu NZ, Wardell SE, Burnstein KL, Defranco D, Fuller PJ, Giguere V, Hochberg RB, McKay L, Renoir JM, Weigel NL, Wilson EM, McDonnell DP, Cidlowski JA. 2006. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol Rev 58:782–797 [DOI] [PubMed] [Google Scholar]
- 41. Africander D, Verhoog N, Hapgood JP. 2011. Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception. Steroids 76:636–652 [DOI] [PubMed] [Google Scholar]
- 42. Moore NL, Hickey TE, Butler LM, Tilley WD. 2012. Multiple nuclear receptor signaling pathways mediate the actions of synthetic progestins in target cells. Mol Cell Endocrinol 357:60–70 [DOI] [PubMed] [Google Scholar]
- 43. Lavery DN, McEwan IJ. 2005. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J 391:449–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Griekspoor A, Zwart W, Neefjes J, Michalides R. 2007. Visualizing the action of steroid hormone receptors in living cells. Nucl Recept Signal 5:e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang Y, Getzenberg RH, Vietmeier BN, Stallcup MR, Eggert M, Renkawitz R, DeFranco DB. 1998. The DNA-binding and τ2 transactivation domains of the rat glucocorticoid receptor constitute a nuclear matrix-targeting signal. Mol Endocrinol 12:1420–1431 [DOI] [PubMed] [Google Scholar]
- 46. Bourguet W, Germain P, Gronemeyer H. 2000. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci 21:381–388 [DOI] [PubMed] [Google Scholar]
- 47. Beato M, Klug J. 2000. Steroid hormone receptors: an update. Hum Reprod Update 6:225–236 [DOI] [PubMed] [Google Scholar]
- 48. Weatherman RV, Fletterick RJ, Scanlan TS. 1999. Nuclear-receptor ligands and ligand-binding domains. Ann Rev Biochem 68:559–581 [DOI] [PubMed] [Google Scholar]
- 49. Tanenbaum DM, Wang Y, Williams SP, Sigler PB. 1998. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci USA 95:5998–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williams SP, Sigler PB. 1998. Atomic structure of progesterone complexed with its receptor. Nature 393:392–396 [DOI] [PubMed] [Google Scholar]
- 51. Fuller PJ. 1991. The steroid receptor superfamily: mechanisms of diversity. FASEB J 5:3092–3099 [DOI] [PubMed] [Google Scholar]
- 52. Evans RM. 1988. The steroid and thyroid hormone receptor superfamily. Science 240:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Loosfelt H, Atger M, Misrahi M, Guiochon-Mantel A, Meriel C, Logeat F, Benarous R, Milgrom E. 1986. Cloning and sequence analysis of rabbit progesterone-receptor complementary DNA. Proc Natl Acad Sci USA 83:9045–9049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. 2002. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110:93–105 [DOI] [PubMed] [Google Scholar]
- 55. Kastner P, Bocquel MT, Turcotte B, Garnier JM, Horwitz KB, Chambon P, Gronemeyer H. 1990. Transient expression of human and chicken progesterone receptors does not support alternative translational initiation from a single mRNA as the mechanism generating two receptor isoforms. J Biol Chem 265:12163–12167 [PubMed] [Google Scholar]
- 56. Sartorius CA, Melville MY, Hovland AR, Tung L, Takimoto GS, Horwitz KB. 1994. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol 8:1347–1360 [DOI] [PubMed] [Google Scholar]
- 57. Wen DX, Xu YF, Mais DE, Goldman ME, McDonnell DP. 1994. The A and B isoforms of the human progesterone receptor operate through distinct signaling pathways within target cells. Mol Cell Biol 14:8356–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giangrande PH, Kimbrel EA, Edwards DP, McDonnell DP. 2000. The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol Cell Biol 20:3102–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oakley RH, Cidlowski JA. 2011. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286:3177–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou J, Cidlowski JA. 2005. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids 70:407–417 [DOI] [PubMed] [Google Scholar]
- 61. Mangal RK, Wiehle RD, Poindexter AN, 3rd, Weigel NL. 1997. Differential expression of uterine progesterone receptor forms A and B during the menstrual cycle. J Steroid Biochem Mol Biol 63:195–202 [DOI] [PubMed] [Google Scholar]
- 62. Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ, Lydon JP. 2005. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 41:58–66 [DOI] [PubMed] [Google Scholar]
- 63. Shyamala G, Schneider W, Schott D. 1990. Developmental regulation of murine mammary progesterone receptor gene expression. Endocrinology 126:2882–2889 [DOI] [PubMed] [Google Scholar]
- 64. Mulac-Jericevic B, Conneely OM. 2004. Reproductive tissue selective actions of progesterone receptors. Reproduction 128:139–146 [DOI] [PubMed] [Google Scholar]
- 65. Conneely OM, Mulac-Jericevic B, DeMayo F, Lydon JP, O'Malley BW. 2002. Reproductive functions of progesterone receptors. Recent Prog Horm Res 57:339–355 [DOI] [PubMed] [Google Scholar]
- 66. Khan JA, Amazit L, Bellance C, Guiochon-Mantel A, Lombès M, Loosfelt H. 2011. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol Endocrinol 25:1710–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mesiano S, Wang Y, Norwitz ER. 2011. Progesterone receptors in the human pregnancy uterus: do they hold the key to birth timing? Reprod Sci 18:6–19 [DOI] [PubMed] [Google Scholar]
- 68. Deroo BJ, Korach KS. 2006. Estrogen receptors and human disease. J Clin Invest 116:561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Courtin A, Communal L, Vilasco M, Cimino D, Mourra N, de Bortoli M, Taverna D, Faussat AM, Chaouat M, Forgez P, Gompel A. 2012. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat 131:49–63 [DOI] [PubMed] [Google Scholar]
- 70. Krozowski ZS, Funder JW. 1983. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA 80:6056–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Kloet ER, Van Acker SA, Sibug RM, Oitzl MS, Meijer OC, Rahmouni K, de Jong W. 2000. Brain mineralocorticoid receptors and centrally regulated functions. Kidney Int 57:1329–1336 [DOI] [PubMed] [Google Scholar]
- 72. Funder JW. 2004. Aldosterone, mineralocorticoid receptors and vascular inflammation. Mol Cell Endocrinol 217:263–269 [DOI] [PubMed] [Google Scholar]
- 73. Koubovec D, Ronacher K, Stubsrud E, Louw A, Hapgood JP. 2005. Synthetic progestogens used in HRT have different glucocorticoid agonist properties. Mol Cell Endocrinol 242:23–32 [DOI] [PubMed] [Google Scholar]
- 74. Philibert D, Bouchoux F, Degryse M, Lecaque D, Petit F, Gaillard M. 1999. The pharmacological profile of a novel norpregnane progestin (trimegestone). Gynecol Endocrinol 13:316–326 [DOI] [PubMed] [Google Scholar]
- 75. Phillips A, Demarest K, Hahn DW, Wong F, McGuire JL. 1990. Progestational and androgenic receptor binding affinities and in vivo activities of norgestimate and other progestins. Contraception 41:399–410 [DOI] [PubMed] [Google Scholar]
- 76. Juchem M, Pollow K, Elger W, Hoffmann G, Möbus V. 1993. Receptor binding of norgestimate: a new orally active synthetic progestational compound. Contraception 47:283–294 [DOI] [PubMed] [Google Scholar]
- 77. Bamberger CM, Else T, Bamberger AM, Beil FU, Schulte HM. 1999. Dissociative glucocorticoid activity of medroxyprogesterone acetate in normal human lymphocytes. J Clin Endocrinol Metab 84:4055–4061 [DOI] [PubMed] [Google Scholar]
- 78. Bergink EW, van Meel F, Turpijn EW, van der Vies J. 1983. Binding of progestagens to receptor proteins in MCF-7 cells. J Steroid Biochem 19:1563–1570 [DOI] [PubMed] [Google Scholar]
- 79. Deckers GH, Schoonen WG, Kloosterboer HJ. 2000. Influence of the substitution of 11-methylene, Δ15, and/or 18-methyl groups in norethisterone on receptor binding, transactivation assays and biological activities in animals. J Steroid Biochem Mol Biol 74:83–92 [DOI] [PubMed] [Google Scholar]
- 80. Chávez BA, Vilchis F, Pérez AE, García GA, Grillasca I, Pérez-Palacios G. 1985. Stereospecificity of the intracellular binding of norethisterone and its A-ring reduced metabolites. J Steroid Biochem 22:121–126 [DOI] [PubMed] [Google Scholar]
- 81. Teulings FA, van Gilse HA, Henkelman MS, Portengen H, Alexieva-Figusch J. 1980. Estrogen, androgen, glucocorticoid, and progesterone receptors in progestin-induced regression of human breast cancer. Cancer Res 40:2557–2561 [PubMed] [Google Scholar]
- 82. Schoonen WG, Deckers GH, de Gooijer ME, de Ries R, Kloosterboer HJ. 2000. Hormonal properties of norethisterone, 7α-methyl-norethisterone and their derivatives. J Steroid Biochem Mol Biol 74:213–222 [DOI] [PubMed] [Google Scholar]
- 83. Kemppainen JA, Langley E, Wong CI, Bobseine K, Kelce WR, Wilson EM. 1999. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Mol Endocrinol 13:440–454 [DOI] [PubMed] [Google Scholar]
- 84. Pérez-Palacios G, Chávez B, Escobar N, Vilchis F, Larrea F, Lince M, Pérez AE. 1981. Mechanism of action of contraceptive synthetic progestogens. J Steroid Biochem 15:125–130 [DOI] [PubMed] [Google Scholar]
- 85. Hackenberg R, Hawighorst T, Filmer A, Nia AH, Schulz KD. 1993. Medroxyprogesterone acetate inhibits the proliferation of estrogen- and progesterone-receptor negative MFM-223 human mammary cancer cells via the androgen receptor. Breast Cancer Res Treat 25:217–224 [DOI] [PubMed] [Google Scholar]
- 86. Bentel JM, Birrell SN, Pickering MA, Holds DJ, Horsfall DJ, Tilley WD. 1999. Androgen receptor agonist activity of the synthetic progestin, medroxyprogesterone acetate, in human breast cancer cells. Mol Cell Endocrinol 154:11–20 [DOI] [PubMed] [Google Scholar]
- 87. Schindler AE, Campagnoli C, Druckmann R, Huber J, Pasqualini JR, Schweppe KW, Thijssen JH. 2003. Classification and pharmacology of progestogens. Maturitas 46(Suppl 1):S7–S16 [DOI] [PubMed] [Google Scholar]
- 88. Sitruk-Ware R. 2004. Pharmacological profile of progestogens. Maturitas 47:277–283 [DOI] [PubMed] [Google Scholar]
- 89. Fotherby K. 1990. Interactions with oral contraceptives. Am J Obstet Gynecol 163:2153–2159 [DOI] [PubMed] [Google Scholar]
- 90. Fuhrmann U, Krattenmacher R, Slater EP, Fritzemeier KH. 1996. The novel progestin drospirenone and its natural counterpart progesterone: biochemical profile and antiandrogenic potential. Contraception 54:243–251 [DOI] [PubMed] [Google Scholar]
- 91. Wambach G, Higgins JR, Kem DC, Kaufmann W. 1979. Interaction of synthetic progestagens with renal mineralocorticoid receptors. Acta Endocrinol (Copenh) 92:560–567 [DOI] [PubMed] [Google Scholar]
- 92. Rafestin-Oblin ME, Lombes M, Couette B, Baulieu EE. 1992. Differences between aldosterone and its antagonists in binding kinetics and ligand-induced hsp90 release from mineralocorticosteroid receptor. J Steroid Biochem Mol Biol 41:815–821 [DOI] [PubMed] [Google Scholar]
- 93. Quinkler M, Diederich S. 2002. Difference of in vivo and in vitro antimineralocorticoid potency of progesterone. Endocr Res 28:465–470 [DOI] [PubMed] [Google Scholar]
- 94. Genazzani A, Gambacciani M, Simoncini T, Anniverno R, Becorpi AM, Biglia N, Brandi ML, Guaschino S, Lello S, Massobrio M, Melis GB, Mencacci C, Modena MG, Nappi C, Nappi RE, Pecorelli S, Petraglia F, Rosano GM, Serra GB, Sismondi P, Taddei S, Tonelli F. 2007. Italian position statement on hormone replacement therapy following the National Conference on Menopause and Hormone Replacement Therapy, Villa Tuscolana, Frascati (Rome), May 8–9, 2007. Gynecol Endocrinol 23:436–444 [DOI] [PubMed] [Google Scholar]
- 95. Winneker RC, Bitran D, Zhang Z. 2003. The preclinical biology of a new potent and selective progestin: trimegestone. Steroids 68:915–920 [DOI] [PubMed] [Google Scholar]
- 96. Elger W, Beier S, Pollow K, Garfield R, Shi SQ, Hillisch A. 2003. Conception and pharmacodynamic profile of drospirenone. Steroids 68:891–905 [DOI] [PubMed] [Google Scholar]
- 97. Sasagawa S, Shimizu Y, Kami H, Takeuchi T, Mita S, Imada K, Kato S, Mizuguchi K. 2008. Dienogest is a selective progesterone receptor agonist in transactivation analysis with potent oral endometrial activity due to its efficient pharmacokinetic profile. Steroids 73:222–231 [DOI] [PubMed] [Google Scholar]
- 98. Koubovec D, Vanden Berghe W, Vermeulen L, Haegeman G, Hapgood JP. 2004. Medroxyprogesterone acetate downregulates cytokine gene expression in mouse fibroblast cells. Mol Cell Endocrinol 221:75–85 [DOI] [PubMed] [Google Scholar]
- 99. Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Jr, Louw A, Hapgood JP. 2009. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol 299:219–231 [DOI] [PubMed] [Google Scholar]
- 100. Kontula K, Paavonen T, Luukkainen T, Andersson LC. 1983. Binding of progestogens to the glucocorticoid receptor. Correlation to their glucocorticoid-like effects on in vitro functions of human mononuclear leukocytes. Biochem Pharmacol 32:1511–1518 [DOI] [PubMed] [Google Scholar]
- 101. Fuhrmann U, Slater EP, Fritzemeier KH. 1995. Characterization of the novel progestin gestodene by receptor binding studies and transactivation assays. Contraception 51:45–52 [DOI] [PubMed] [Google Scholar]
- 102. McPhail MK. 1934. The assay of progestin. J Physiol 83:145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Edgren RA. 1994. Issues in animal pharmacology. In: Goldzieher JW, Fotherby K, eds. Pharmacology of the contraceptive steroids. New York: Raven Press; 81–97 [Google Scholar]
- 104. Bray JD, Jelinsky S, Ghatge R, Bray JA, Tunkey C, Saraf K, Jacobsen BM, Richer JK, Brown EL, Winneker RC, Horwitz KB, Lyttle CR. 2005. Quantitative analysis of gene regulation by seven clinically relevant progestogens suggests a highly similar mechanism of action through progesterone receptors in T47D breast cancer cells. J Steroid Biochem Mol Biol 97:328–341 [DOI] [PubMed] [Google Scholar]
- 105. Ghatge RP, Jacobsen BM, Schittone SA, Horwitz KB. 2005. The progestational and androgenic properties of medroxyprogesterone acetate: gene regulatory overlap with dihydrotestosterone in breast cancer cells. Breast Cancer Res 7:R1036–R1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hyder SM, Murthy L, Stancel GM. 1998. Progestin regulation of vascular endothelial growth factor in human breast cancer cells. Cancer Res 58:392–395 [PubMed] [Google Scholar]
- 107. Mueller MD, Vigne JL, Pritts EA, Chao V, Dreher E, Taylor RN. 2003. Progestogens activate vascular endothelial growth factor gene transcription in endometrial adenocarcinoma cells. Fertil Steril 79:386–392 [DOI] [PubMed] [Google Scholar]
- 108. Fu XD, Giretti MS, Goglia L, Flamini MI, Sanchez AM, Baldacci C, Garibaldi S, Sitruk-Ware R, Genazzani AR, Simoncini T. 2008. Comparative actions of progesterone, medroxyprogesterone acetate, drospirenone and nestorone on breast cancer cell migration and invasion. BMC Cancer 8:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lello S. 2010. Nomegestrol acetate: pharmacology, safety profile and therapeutic efficacy. Drugs 70:541–559 [DOI] [PubMed] [Google Scholar]
- 110. van Diepen HA, Lam TW, Kuil CW. 2011. Nomegestrol acetate: steroid receptor transactivation profile in Chinese hamster ovary cells and ovulation inhibition in rat and monkey. Contraception 84:199–204 [DOI] [PubMed] [Google Scholar]
- 111. Greenblatt RB, Jungck EC, Barfield WE. 1958. A new test of efficacy of progestational compounds. Ann NY Acad Sci 71:717–721 [DOI] [PubMed] [Google Scholar]
- 112. Swyer GI, Little V. 1968. Clinical assessment of relative potency of progestogens. J Reprod Fertil Suppl 5:63–68 [PubMed] [Google Scholar]
- 113. Dorflinger LJ. 1985. Relative potency of progestins used in oral contraceptives. Contraception 31:557–570 [DOI] [PubMed] [Google Scholar]
- 114. Whitehead MI, Townsend PT, Pryse-Davies J, Ryder TA, King RJB. 1981. Effects of estrogens and progestins on the biochemistry and morphology of the postmenopausal endometrium. N Engl J Med 305:1599–1605 [DOI] [PubMed] [Google Scholar]
- 115. Whitehead MI, Townsend PT, Pryse-Davies J, Ryder T, Lane G, Siddle NC, King RJB. 1982. Effects of various types and dosages of progestins on the postmenopausal endometrium. J Reprod Med 27:539–548 [PubMed] [Google Scholar]
- 116. Lane G, Siddle NC, Ryder TA, Pryse-Davies J, King RJ, Whitehead MI. 1983. Dose dependent effects of oral progesterone on the oestrogenised postmenopausal endometrium. Br Med J (Clin Res Ed) 287:1241–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Whitehead MI, Lane G, Siddle NC, Townsend PT, King RJ. 1983. Avoidance of endometrial hyperstimulation in estrogen-treated postmenopausal women. Semin Reprod Endocrinol 1:41 [Google Scholar]
- 118. Whitehead MI, Siddle NC, Townsend PT, Lane G, King RJ. 1983. The use of progestins and progesterone in the treatment of climacteric and postmenopausal symptoms. In: Bardin CW, Milgrom E, Mauvais Jarvis P, eds. Progesterone and progestins. New York: Raven Press; 277 [Google Scholar]
- 119. Lane G, Siddle NC, Ryder TA, Pryse-Davies J, King RJ, Whitehead MI. 1986. Effects of dydrogesterone on the oestrogenized postmenopausal endometrium. Br J Obstet Gynaecol 93:55–62 [DOI] [PubMed] [Google Scholar]
- 120. Lane G, Siddle NC, Ryder TA, Pryse-Davies J, King RJ, Whitehead MI. 1986. Is Provera the ideal progestin for addition to postmenopausal estrogen therapy? Fertil Steril 45:345–352 [PubMed] [Google Scholar]
- 121. King RJ, Whitehead MI. 1986. Assessment of the potency of orally administered progestins in women. Fertil Steril 46:1062–1066 [DOI] [PubMed] [Google Scholar]
- 122. Vicent GP, Nacht AS, Zaurín R, Ballaré C, Clausell J, Beato M. 2010. Role of kinases and chromatin remodeling in progesterone signaling to chromatin. Mol Endocrinol 24:2088–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Biddie SC, John S, Hager GL. 2010. Genome-wide mechanisms of nuclear receptor action. Trends Endocrinol Metab 21:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Leslie KK, Stein MP, Kumar NS, Dai D, Stephens J, Wandinger-Ness A, Glueck DH. 2005. Progesterone receptor isoform identification and subcellular localization in endometrial cancer. Gynecol Oncol 96:32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lim CS, Baumann CT, Htun H, Xian W, Irie M, Smith CL, Hager GL. 1999. Differential localization and activity of the A- and B-forms of the human progesterone receptor using green fluorescent protein chimeras. Mol Endocrinol 13:366–375 [DOI] [PubMed] [Google Scholar]
- 126. Pratt WB, Toft DO. 1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18:306–360 [DOI] [PubMed] [Google Scholar]
- 127. Hammes SR, Levin ER. 2007. Extranuclear steroid receptors: nature and actions. Endocr Rev 28:726–741 [DOI] [PubMed] [Google Scholar]
- 128. Perissi V, Rosenfeld MG. 2005. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554 [DOI] [PubMed] [Google Scholar]
- 129. Beato M. 1989. Gene regulation by steroid hormones. Cell 56:335–344 [DOI] [PubMed] [Google Scholar]
- 130. Ballaré C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M. 2003. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol 23:1994–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. 2011. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145:224–241 [DOI] [PubMed] [Google Scholar]
- 132. Kassel O, Herrlich P. 2007. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol Cell Endocrinol 275:13–29 [DOI] [PubMed] [Google Scholar]
- 133. De Bosscher K, Haegeman G. 2009. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 23:281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. 2001. Mechanisms of estrogen action. Physiol Rev 81:1535–1565 [DOI] [PubMed] [Google Scholar]
- 135. De Bosscher K, VandenBerghe W, Haegeman G. 2003. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488–522 [DOI] [PubMed] [Google Scholar]
- 136. Revollo JR, Cidlowski JA. 2009. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann NY Acad Sci 1179:167–178 [DOI] [PubMed] [Google Scholar]
- 137. Kobayashi S, Stice JP, Kazmin D, Wittmann BM, Kimbrel EA, Edwards DP, Chang CY, McDonnell DP. 2010. Mechanisms of progesterone receptor inhibition of inflammatory responses in cellular models of breast cancer. Mol Endocrinol 24:2292–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. McKay LI, Cidlowski JA. 1999. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev 20:435–459 [DOI] [PubMed] [Google Scholar]
- 139. Kalkhoven E, Wissink S, van der Saag PT, van der Burg B. 1996. Negative interaction between the RelA(p65) subunit of NF-κB and the progesterone receptor. J Biol Chem 271:6217–6224 [DOI] [PubMed] [Google Scholar]
- 140. Palvimo JJ, Reinikainen P, Ikonen T, Kallio PJ, Moilanen A, Jänne OA. 1996. Mutual transcriptional interference between RelA and androgen receptor. J Biol Chem 271:24151–24156 [DOI] [PubMed] [Google Scholar]
- 141. Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, Murray R, Manolagas SC. 1995. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. J Clin Invest 95:2886–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Liden J, Delaunay F, Rafter I, Gustafsson J, Okret S. 1997. A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J Biol Chem 272:21467–21472 [DOI] [PubMed] [Google Scholar]
- 143. Faivre EJ, Daniel AR, Hillard CJ, Lange CA. 2008. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol Endocrinol 22:823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Faivre EJ, Lange CA. 2007. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol 27:466–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Goldhar AS, Duan R, Ginsburg E, Vonderhaar BK. 2011. Progesterone induces expression of the prolactin receptor gene through cooperative action of Sp1 and C/EBP. Mol Cell Endocrinol 335:148–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Nettles KW, Greene GL. 2005. Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol 67:309–333 [DOI] [PubMed] [Google Scholar]
- 147. McKenna NJ, Lanz RB, O'Malley BW. 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344 [DOI] [PubMed] [Google Scholar]
- 148. Heinlein CA, Chang C. 2002. Androgen receptor (AR) coregulators: an overview. Endocr Rev 23:175–200 [DOI] [PubMed] [Google Scholar]
- 149. Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19:2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hakim O, Sung MH, Hager GL. 2010. 3D shortcuts to gene regulation. Curr Opin Cell Biol 22:305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Xi H, Shulha HP, Lin JM, Vales TR, Fu Y, Bodine DM, McKay RD, Chenoweth JG, Tesar PJ, Furey TS, Ren B, Weng Z, Crawford GE. 2007. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet 3:e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Santos GM, Fairall L, Schwabe JW. 2011. Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol Metab 22:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD. 2008. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA 105:19199–19204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Beral V; Million Women Study Collaborators 2003. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet [Erratum (2003) 362:1160] 362:419–427 [DOI] [PubMed] [Google Scholar]
- 156. Kaunitz AM. 2006. Hormone therapy and breast cancer risk: trumping fear with facts. Menopause 13:160–163 [DOI] [PubMed] [Google Scholar]
- 157. Colditz GA. 2005. Estrogen, estrogen plus progestin therapy, and risk of breast cancer. Clin Cancer Res 11:909s–917s [PubMed] [Google Scholar]
- 158. Rosano GM, Fini M. 2001. Comparative cardiovascular effects of different progestogens in menopause. Int J Fertil Womens Med 46:248–256 [PubMed] [Google Scholar]
- 159. Sitruk-Ware R. 2000. Progestins and cardiovascular risk markers. Steroids 65:651–658 [DOI] [PubMed] [Google Scholar]
- 160. Brunelli R, Frasca D, Perrone G, Pioli C, Fattorossi A, Zichella L, Doria G. 1996. Hormone replacement therapy affects various immune cell subsets and natural cytotoxicity. Gynecol Obstet Invest 41:128–131 [DOI] [PubMed] [Google Scholar]
- 161. Malarkey WB, Burleson M, Cacioppo JT, Poehlmann K, Glaser R, Kiecolt-Glaser JK. 1997. Differential effects of estrogen and medroxyprogesterone on basal and stress-induced growth hormone release, IGF-1 levels, and cellular immunity in postmenopausal women. Endocrine 7:227–233 [DOI] [PubMed] [Google Scholar]
- 162. Wakatsuki A, Okatani Y, Ikenoue N, Fukaya T. 2002. Effect of medroxyprogesterone acetate on vascular inflammatory markers in postmenopausal women receiving estrogen. Circulation 105:1436–1439 [DOI] [PubMed] [Google Scholar]
- 163. Stopińska-Głuszak U, Waligóra J, Grzela T, Głuszak M, Jóźwiak J, Radomski D, Roszkowski PI, Malejczyk J. 2006. Effect of estrogen/progesterone hormone replacement therapy on natural killer cell cytotoxicity and immunoregulatory cytokine release by peripheral blood mononuclear cells of postmenopausal women. J Reprod Immunol 69:65–75 [DOI] [PubMed] [Google Scholar]
- 164. Ohkura T, Isse K, Akazawa K, Hamamoto M, Yaoi Y, Hagino N. 1995. Long-term estrogen replacement therapy in female patients with dementia of the Alzheimer type: 7 case reports. Dementia 6:99–107 [DOI] [PubMed] [Google Scholar]
- 165. Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, 3rd, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. 2003. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA 289:2651–2662 [DOI] [PubMed] [Google Scholar]
- 166. Valdivia I, Campodónico I, Tapia A, Capetillo M, Espinoza A, Lavín P. 2004. Effects of tibolone and continuous combined hormone therapy on mammographic breast density and breast histochemical markers in postmenopausal women. Fertil Steril 81:617–623 [DOI] [PubMed] [Google Scholar]
- 167. Odmark IS, Bäckström T, Jonsson B, Bixo M. 2004. Long-term effects of two different continuous combined regimens of hormone replacement therapy on well-being. Gynecol Endocrinol 18:305–317 [DOI] [PubMed] [Google Scholar]
- 168. Oelkers W. 2004. Drospirenone, a progestogen with antimineralocorticoid properties: a short review. Mol Cell Endocrinol 217:255–261 [DOI] [PubMed] [Google Scholar]
- 169. Liu L, Zhao L, She H, Chen S, Wang JM, Wong C, McClure K, Sitruk-Ware R, Brinton RD. 2010. Clinically relevant progestins regulate neurogenic and neuroprotective responses in vitro and in vivo. Endocrinology 151:5782–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhao D, Lebovic DI, Taylor RN. 2002. Long-term progestin treatment inhibits RANTES (regulated on activation, normal T cell expressed and secreted) gene expression in human endometrial stromal cells. J Clin Endocrinol Metab 87:2514–2519 [DOI] [PubMed] [Google Scholar]
- 171. Ramhorst R, Patel R, Corigliano A, Etchepareborda JJ, Fainboim L, Schust D. 2006. Induction of maternal tolerance to fetalalloantigens by RANTES production. Am J Reprod Immunol 56:302–311 [DOI] [PubMed] [Google Scholar]
- 172. Tatsumi H, Kitawaki J, Tanaka K, Hosoda T, Honjo H. 2002. Lack of stimulatory effect of dienogest on the expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 by endothelial cell as compared with other synthetic progestogens. Maturitas 42:287–294 [DOI] [PubMed] [Google Scholar]
- 173. Yasa M, Turkseven S. 2005. Vasoprotective effects of nitric oxide in atherosclerosis. FABAD J Pharm Sci 30:41–53 [Google Scholar]
- 174. Simoncini T, Mannella P, Fornari L, Caruso A, Willis MY, Garibaldi S, Baldacci C, Genazzani AR. 2004. Differential signal transduction of progesterone and medroxyprogesterone acetate in human endothelial cells. Endocrinology 145:5745–5756 [DOI] [PubMed] [Google Scholar]
- 175. Simoncini T, Mannella P, Pluchino N, Genazzani A. 2007. Comparative effects of dydrogesterone and medroxyprogesterone acetate in critical areas: the brain and the vessels. Gynecol Endocrinol 23(Suppl 1):9–16 [DOI] [PubMed] [Google Scholar]
- 176. Irwin RW, Yao J, Ahmed SS, Hamilton RT, Cadenas E, Brinton RD. 2011. Medroxyprogesterone acetate antagonizes estrogen up-regulation of brain mitochondrial function. Endocrinology 152:556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schneider HP. 2003. Androgens and antiandrogens. Ann NY Acad Sci 997:292–306 [DOI] [PubMed] [Google Scholar]
- 178. Muhn P, Fuhrmann U, Fritzemeier KH, Krattenmacher R, Schillinger E. 1995. Drospirenone: a novel progestogen with antimineralocorticoid and antiandrogenic activity. Ann NY Acad Sci 761:311–335 [DOI] [PubMed] [Google Scholar]
- 179. Zhang Z, Lundeen SG, Zhu Y, Carver JM, Winneker RC. 2000. In vitro characterization of trimegestone: a new potent and selective progestin. Steroids 65:637–643 [DOI] [PubMed] [Google Scholar]
- 180. García-Becerra R, Cooney AJ, Borja-Cacho E, Lemus AE, Pérez-Palacios G, Larrea F. 2004. Comparative evaluation of androgen and progesterone receptor transcription selectivity indices of 19-nortestosterone-derived progestogens. J Steroid Biochem Mol Biol 91:21–27 [DOI] [PubMed] [Google Scholar]
- 181. Africander D, Louw R, Verhoog N, Noeth D, Hapgood JP. 2011. Differential regulation of endogenous pro-inflammatory cytokine genes by medroxyprogesterone acetate and norethisterone acetate in cell lines of the female genital tract. Contraception 84:423–435 [DOI] [PubMed] [Google Scholar]
- 182. Birrell SN, Butler LM, Harris JM, Buchanan G, Tilley WD. 2007. Disruption of androgen receptor signaling by synthetic progestogens may increase risk of developing breast cancer. FASEB J 21:2285–2293 [DOI] [PubMed] [Google Scholar]
- 183. Hadley KE, Louw A, Hapgood JP. 2011. Differential nuclear localisation and promoter occupancy play a role in glucocorticoid receptor ligand-specific transcriptional responses. Steroids 76:1176–1184 [DOI] [PubMed] [Google Scholar]
- 184. Ishida Y, Ishida Y, Heersche JN. 2002. Pharmacologic doses of medroxyprogesterone may cause bone loss through glucocorticoid activity: an hypothesis. Osteoporos Int 13:601–605 [DOI] [PubMed] [Google Scholar]
- 185. Ishida Y, Mine T, Taguchi T. 2008. Effect of progestogens with different glucocorticoid activity on bone metabolism. Clin Endocrinol (Oxf) 68:423–428 [DOI] [PubMed] [Google Scholar]
- 186. Kurebayashi J, Otsuki T, Tanaka K, Yamamoto Y, Moriya T, Sonoo H. 2003. Medroxyprogesterone acetate decreases secretion of interleukin-6 and parathyroid hormone-related protein in a new anaplastic thyroid cancer cell line, KTC-2. Thyroid 13:249–258 [DOI] [PubMed] [Google Scholar]
- 187. Zhao Q, Pang J, Favata MF, Trzaskos JM. 2003. Receptor density dictates the behavior of a subset of steroid ligands in glucocorticoid receptor-mediated transrepression. Int Immunopharmacol 3:1803–1817 [DOI] [PubMed] [Google Scholar]
- 188. Thomas CP, Liu KZ, Vats HS. 2006. Medroxyprogesterone acetate binds the glucocorticoid receptor to stimulate α-ENaC and sgk1 expression in renal collecting duct epithelia. Am J Physiol Renal Physiol 290:F306–F312 [DOI] [PubMed] [Google Scholar]
- 189. Ouatas T, Halverson D, Steeg PS. 2003. Dexamethasone and medroxyprogesterone acetate elevate Nm23-H1 metastasis suppressor gene expression in metastatic human breast carcinoma cells: new uses for old compounds. Clin Cancer Res 9:3763–3772 [PubMed] [Google Scholar]
- 190. Zerr-Fouineau M, Chataigneau M, Blot C, Schini-Kerth VB. 2007. Progestogens overcome inhibition of platelet aggregation by endothelial cells by down-regulating endothelial NO synthase via glucocorticoid receptors. FASEB J 21:265–273 [DOI] [PubMed] [Google Scholar]
- 191. Spangelo BL, Gorospe WC. 1995. Role of the cytokines in the neuroendocrine-immune system axis. Front Neuroendocrinol 16:1–22 [DOI] [PubMed] [Google Scholar]
- 192. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. 2002. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 16:61–71 [DOI] [PubMed] [Google Scholar]
- 193. Herkert O, Kuhl H, Sandow J, Busse R, Schini-Kerth VB. 2001. Sex steroids used in hormonal treatment increase vascular procoagulant activity by inducing thrombin receptor (PAR-1) expression: role of the glucocorticoid receptor. Circulation 104:2826–2831 [DOI] [PubMed] [Google Scholar]
- 194. Moutsatsou P, Papavassiliou AG. 2008. The glucocorticoid receptor signalling in breast cancer. J Cell Mol Med 12:145–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Courtin A, Communal L, Vilasco M, Cimino D, Mourra N, de Bortoli M, Taverna D, Faussat AM, Chaouat M, Forgez P, Gompel A. 2012. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat 131:49–63 [DOI] [PubMed] [Google Scholar]
- 196. Wan Y, Nordeen SK. 2002. Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol 16:1204–1214 [DOI] [PubMed] [Google Scholar]
- 197. Oelkers WK. 1996. Effects of estrogens and progestogens on the renin-aldosterone system and blood pressure. Steroids 61:166–171 [DOI] [PubMed] [Google Scholar]
- 198. Rylance PB, Brincat M, Lafferty K, De Trafford JC, Brincat S, Parsons V, Studd JW. 1985. Natural progesterone and antihypertensive action. Br Med J (Clin Res Ed) 290:13–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Quinkler M, Meyer B, Bumke-Vogt C, Grossmann C, Gruber U, Oelkers W, Diederich S, Bahr V. 2002. Agonistic and antagonistic properties of progesterone metabolites at the human mineralocorticoid receptor. Eur J Endocrinol 146:789–799 [DOI] [PubMed] [Google Scholar]
- 200. Oelkers W. 2005. Drospirenone in combination with estrogens: for contraception and hormone replacement therapy. Climacteric 8(Suppl 3):19–27 [DOI] [PubMed] [Google Scholar]
- 201. Arias-Loza PA, Muehlfelder M, Elmore SA, Maronpot R, Hu K, Blode H, Hegele-Hartung C, Fritzemeier KH, Ertl G, Pelzer T. 2009. Differential effects of 17β-estradiol and of synthetic progestogens on aldosterone-salt-induced kidney disease. Toxicol Pathol 37:969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Seeger H, Wallwiener D, Mueck AO. 2009. Effects of drospirenone on cardiovascular markers in human aortic endothelial cells. Climacteric 12:80–87 [DOI] [PubMed] [Google Scholar]
- 203. Di Carlo F, Gallo E, Conti G, Racca S. 1983. Changes in the binding of oestradiol to uterine oestrogen receptors induced by some progesterone and 19-nor-testosterone derivatives. J Endocrinol 98:385–389 [DOI] [PubMed] [Google Scholar]
- 204. Markiewicz L, Gurpide E. 1994. Estrogenic and progestagenic activities coexisting in steroidal drugs: quantitative evaluation by in vitro bioassays with human cells. J Steroid Biochem Mol Biol 48:89–94 [DOI] [PubMed] [Google Scholar]
- 205. Mendoza-Rodríguez CA, Camacho-Arroyo I, García GA, Cerbón MA. 1999. Variations of progesterone receptor and c-fos gene expression in the rat uterus after treatment with norethisterone and its A-ring reduced metabolites. Contraception 59:339–343 [DOI] [PubMed] [Google Scholar]
- 206. Larrea F, García-Becerra R, Lemus AE, García GA, Pérez-Palacios G, Jackson KJ, Coleman KM, Dace R, Smith CL, Cooney AJ. 2001. A-ring reduced metabolites of 19-nor synthetic progestogens as subtype selective agonists for ERα. Endocrinology 142:3791–3799 [DOI] [PubMed] [Google Scholar]
- 207. Stellato C. 2004. Post-transcriptional and nongenomic effects of glucocorticoids. Proc Am Thorac Soc 1:255–263 [DOI] [PubMed] [Google Scholar]
- 208. Wierman ME. 2007. Sex steroid effects at target tissues: mechanisms of action. Adv Physiol Educ 31:26–33 [DOI] [PubMed] [Google Scholar]
- 209. Grossmann C, Gekle M. 2009. New aspects of rapid aldosterone signaling. Mol Cell Endocrinol 308:53–62 [DOI] [PubMed] [Google Scholar]
- 210. Zhu Y, Hanna RN, Schaaf MJ, Spaink HP, Thomas P. 2008. Candidates for membrane progestin receptors: past approaches and future challenges. Comp Biochem Physiol C Toxicol Pharmacol 148:381–389 [DOI] [PubMed] [Google Scholar]
- 211. Nilsen J, Brinton RD. 2003. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proc Natl Acad Sci USA 100:10506–10511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Boonyaratanakornkit V, Bi Y, Rudd M, Edwards DP. 2008. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulating gene transcription and cell cycle progression. Steroids 73:922–928 [DOI] [PubMed] [Google Scholar]
- 213. Hagan CR, Daniel AR, Dressing GE, Lange CA. 2012. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol 357:43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, Molinolo AA, Frahm I, Charreau EH, Schillaci R, Elizalde PV. 2005. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol 25:4826–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Béguelin W, Díaz Flaqué MC, Proietti CJ, Cayrol F, Rivas MA, Tkach M, Rosemblit C, Tocci JM, Charreau EH, Schillaci R, Elizalde PV. 2010. Progesterone receptor induces ErbB-2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB-2 function as a coactivator of Stat3. Mol Cell Biol 30:5456–5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Thomas P. 2008. Characteristics of membrane progestin receptor α (mPRα) and progesterone membrane receptor component 1 (PGMRC1) and their roles in mediating rapid progestin actions. Front Neuroendocrinol 29:292–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Thomas P, Pang Y, Dong J, Groenen P, Kelder J, de Vlieg J, Zhu Y, Tubbs C. 2007. Steroid and G protein binding characteristics of the sea trout and human progestin membrane receptor α subtypes and their evolutionary origins. Endocrinology 148:705–718 [DOI] [PubMed] [Google Scholar]
- 218. Dosiou C, Hamilton AE, Pang Y, Overgaard MT, Tulac S, Dong J, Thomas P, Giudice LC. 2008. Expression of membrane progesterone receptors on human T lymphocytes and Jurkat cells and activation of G-proteins by progesterone. J Endocrinol 196:67–77 [DOI] [PubMed] [Google Scholar]
- 219. Saitoh M, Ohmichi M, Takahashi K, Kawagoe J, Ohta T, Doshida M, Takahashi T, Igarashi H, Mori-Abe A, Du B, Tsutsumi S, Kurachi H. 2005. Medroxyprogesterone acetate induces cell proliferation through up-regulation of cyclin D1 expression via phosphatidylinositol 3-kinase/Akt/nuclear factor-κB cascade in human breast cancer cells. Endocrinology 146:4917–4925 [DOI] [PubMed] [Google Scholar]
- 220. Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz MA, Liao JK. 2002. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest 110:1729–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. 2008. Rapid estrogen signaling in the brain. Neurosignals 16:140–153 [DOI] [PubMed] [Google Scholar]
- 222. Walker WH. 2003. Nongenomic actions of androgen in Sertoli cells. Curr Top Dev Biol 56:25–53 [DOI] [PubMed] [Google Scholar]
- 223. Rarick L. 2007. United States regulatory considerations for intrauterine progestins for hormone replacement therapy. Contraception 75:S140–S143 [DOI] [PubMed] [Google Scholar]
- 224. Genant HK, Lucas J, Weiss S, Akin M, Emkey R, McNaney-Flint H, Downs R, Mortola J, Watts N, Yang HM, Banav N, Brennan JJ, Nolan JC. 1997. Low-dose esterified estrogen therapy: effects on bone, plasma estradiol concentrations, endometrium, and lipid levels. Estratab/Osteoporosis Study Group. Arch Intern Med 157:2609–2615 [DOI] [PubMed] [Google Scholar]
- 225. Ettinger B, Ensrud KE, Wallace R, Johnson KC, Cummings SR, Yankov V, Vittinghoff E, Grady D. 2004. Effects of ultralow-dose transdermal estradiol on bone mineral density: a randomized clinical trial. Obstet Gynecol 104:443–451 [DOI] [PubMed] [Google Scholar]
- 226. Beral V, Bull D, Reeves G; Million Women Study Collaborators 2005. Endometrial cancer and hormone-replacement therapy in the Million Women Study. Lancet 365:1543–1551 [DOI] [PubMed] [Google Scholar]
- 227. The Writing Group for the PEPI Trial 1996. Effects of hormone replacement therapy on endometrial histology in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 275:370–375 [DOI] [PubMed] [Google Scholar]
- 228. Allen NE, Tsilidis KK, Key TJ, Dossus L, Kaaks R, Lund E, Bakken K, Gavrilyuk O, Overvad K, Tjønneland A, Olsen A, Fournier A, Fabre A, Clavel-Chapelon F, Chabbert-Buffet N, Sacerdote C, Krogh V, Bendinelli B, Tumino R, Panico S, Bergmann M, Schuetze M, van Duijnhoven FJ, Bueno-de-Mesquita HB, Onland-Moret NC, et al. 2010. Menopausal hormone therapy and risk of endometrial carcinoma among postmenopausal women in the European Prospective Investigation Into Cancer and Nutrition. Am J Epidemiol 172:1394–1403 [DOI] [PubMed] [Google Scholar]
- 229. Cicinelli E, de Ziegler D, Galantino P, Pinto V, Barba B, Morgese S, Schonauer S. 2002. Twice-weekly transdermal estradiol and vaginal progesterone as continuous combined hormone replacement therapy in postmenopausal women: a 1-year prospective study. Am J Obstet Gynecol 187:556–560 [DOI] [PubMed] [Google Scholar]
- 230. Cicinelli E, de Ziegler D, Alfonso R, Nicoletti R, Bellavia M, Colafiglio G. 2005. Endometrial effects, bleeding control, and compliance with a new postmenopausal hormone therapy regimen based on transdermal estradiol gel and every-other-day vaginal progesterone in capsules: a 3-year pilot study. Fertil Steril 83:1859–1863 [DOI] [PubMed] [Google Scholar]
- 231. Hampton NR, Rees MC, Lowe DG, Rauramo I, Barlow D, Guillebaud J. 2005. Levonorgestrel intrauterine system (LNG-IUS) with conjugated oral equine estrogen: a successful regimen for HRT in perimenopausal women. Hum Reprod 20:2653–2660 [DOI] [PubMed] [Google Scholar]
- 232. Suvanto-Luukkonen E, Kauppila A. 1999. The levonorgestrel intrauterine system in menopausal hormone replacement therapy: five-year experience. Fertil Steril 72:161–163 [DOI] [PubMed] [Google Scholar]
- 233. Varila E, Wahlström T, Rauramo I. 2001. A 5-year follow-up study on the use of a levonorgestrel intrauterine system in women receiving hormone replacement therapy. Fertil Steril 76:969–973 [DOI] [PubMed] [Google Scholar]
- 234. Seeger H, Mueck AO. 2008. Are the progestins responsible for breast cancer risk during hormone therapy in the postmenopause? J Steroid Biochem Mol Biol 109:11–15 [DOI] [PubMed] [Google Scholar]
- 235. Stefanick ML, Anderson GL, Margolis KL, Hendrix SL, Rodabough RJ, Paskett ED, Lane DS, Hubbell FA, Assaf AR, Sarto GE, Schenken RS, Yasmeen S, Lessin L, Chlebowski RT; WHI Investigators 2006. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA 295:1647–1657 [DOI] [PubMed] [Google Scholar]
- 236. Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, Manson JE, Stefanick ML, Ockene J, Sarto GE, Johnson KC, Wactawski-Wende J, Ravdin PM, Schenken R, Hendrix SL, Rajkovic A, Rohan TE, Yasmeen S, Prentice RL; WHI Investigators 2010. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 304:1684–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M, Lane D, Rodabough RJ, Gilligan MA, Cyr MG, Thomson CA, Khandekar J, Petrovitch H, McTiernan A; WHI Investigators 2003. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. JAMA 289:3243–3253 [DOI] [PubMed] [Google Scholar]
- 238. Greendale GA, Reboussin BA, Sie A, Singh HR, Olson LK, Gatewood O, Bassett LW, Wasilauskas C, Bush T, Barrett-Connor E. 1999. Effects of estrogen and estrogen-progestin on mammographic parenchymal density. Postmenopausal Estrogen/Progestin Interventions (PEPI) Investigators. Ann Intern Med 130:262–269 [DOI] [PubMed] [Google Scholar]
- 239. Lundström E, Bygdeson M, Svane G, Azavedo E, von Schoultz B. 2007. Neutral effect of ultra-low-dose continuous combined estradiol and norethisterone acetate on mammographic breast density. Climacteric 10:249–256 [DOI] [PubMed] [Google Scholar]
- 240. Weiss LK, Burkman RT, Cushing-Haugen KL, Voigt LF, Simon MS, Daling JR, Norman SA, Bernstein L, Ursin G, Marchbanks PA, Strom BL, Berlin JA, Weber AL, Doody DR, Wingo PA, McDonald JA, Malone KE, Folger SG, Spirtas R. 2002. Hormone replacement therapy regimens and breast cancer risk (1). Obstet Gynecol 100:1148–1158 [DOI] [PubMed] [Google Scholar]
- 241. Olsson HL, Ingvar C, Bladström A. 2003. Hormone replacement therapy containing progestins and given continuously increases breast carcinoma risk in Sweden. Cancer 97:1387–1392 [DOI] [PubMed] [Google Scholar]
- 242. Ross RK, Paganini-Hill A, Wan PC, Pike MC. 2000. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst 92:328–332 [DOI] [PubMed] [Google Scholar]
- 243. Schairer C, Lubin J, Troisi R, Sturgeon S, Brinton L, Hoover R. 2000. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA [Erratum (2000) 284:2597] 283:485–491 [DOI] [PubMed] [Google Scholar]
- 244. Li CI, Malone KE, Porter PL, Weiss NS, Tang MT, Cushing-Haugen KL, Daling JR. 2003. Relationship between long durations and different regimens of hormone therapy and risk of breast cancer. JAMA 289:3254–3263 [DOI] [PubMed] [Google Scholar]
- 245. Chen CL, Weiss NS, Newcomb P, Barlow W, White E. 2002. Hormone replacement therapy in relation to breast cancer. JAMA 287:734–741 [DOI] [PubMed] [Google Scholar]
- 246. Jick SS, Hagberg KW, Kaye JA, Jick H. 2009. Postmenopausal estrogen-containing hormone therapy and the risk of breast cancer. Obstet Gynecol 113:74–80 [DOI] [PubMed] [Google Scholar]
- 247. Kerlikowske K, Miglioretti DL, Ballard-Barbash R, Weaver DL, Buist DS, Barlow WE, Cutter G, Geller BM, Yankaskas B, Taplin SH, Carney PA. 2003. Prognostic characteristics of breast cancer among postmenopausal hormone users in a screened population. J Clin Oncol 21:4314–4321 [DOI] [PubMed] [Google Scholar]
- 248. Collins JA, Blake JM, Crosignani PG. 2005. Breast cancer risk with postmenopausal hormonal treatment. Hum Reprod Update [Erratum (2006) 12:331] 11:545–560 [DOI] [PubMed] [Google Scholar]
- 249. Calle EE, Feigelson HS, Hildebrand JS, Teras LR, Thun MJ, Rodriguez C. 2009. Postmenopausal hormone use and breast cancer associations differ by hormone regimen and histologic subtype. Cancer [Erratum (2009) 115:1587] 115:936–945 [DOI] [PubMed] [Google Scholar]
- 250. Anderson GL, Chlebowski RT, Aragaki AK, Kuller LH, Manson JE, Gass M, Bluhm E, Connelly S, Hubbell FA, Lane D, Martin L, Ockene J, Rohan T, Schenken R, Wactawski-Wende J. 2012. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women's Health Initiative randomised placebo-controlled trial. Lancet Oncol 13:476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Chlebowski RT, Anderson GL. 2012. Changing concepts: menopausal hormone therapy and breast cancer. J Natl Cancer Inst 104:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Brinton LA, Lacey JV, Jr, Trimble EL. 2005. Hormones and endometrial cancer–new data from the Million Women Study. Lancet 365:1517–1518 [DOI] [PubMed] [Google Scholar]
- 253. Fournier A, Berrino F, Riboli E, Avenel V, Clavel-Chapelon F. 2005. Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer 114:448–454 [DOI] [PubMed] [Google Scholar]
- 254. Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, Feuer EJ, Edwards BK, Berry DA. 2007. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 356:1670–1674 [DOI] [PubMed] [Google Scholar]
- 255. Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N; HERS Research Group 2002. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA [Erratum (2002) 288:1064] 288:49–57 [DOI] [PubMed] [Google Scholar]
- 256. Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, Trevisan M, Black HR, Heckbert SR, Detrano R, Strickland OL, Wong ND, Crouse JR, Stein E, Cushman M; Women's Health Initiative Investigators 2003. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med 349:523–534 [DOI] [PubMed] [Google Scholar]
- 257. Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, Ko M, LaCroix AZ, Margolis KL, Stefanick ML. 2007. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 297:1465–1477 [DOI] [PubMed] [Google Scholar]
- 258. Rosano GM, Vitale C, Fini M. 2009. Cardiovascular aspects of menopausal hormone replacement therapy. Climacteric 12(Suppl 1):41–46 [DOI] [PubMed] [Google Scholar]
- 259. Hermsmeyer RK, Thompson TL, Pohost GM, Kaski JC. 2008. Cardiovascular effects of medroxyprogesterone acetate and progesterone: a case of mistaken identity? Nat Clin Pract Cardiovasc Med 5:387–395 [DOI] [PubMed] [Google Scholar]
- 260. Writing Group for the PEPI Trial 1995. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The postmenopausal estrogen/progestin interventions (PEPI) trial. JAMA 273:199–208 [PubMed] [Google Scholar]
- 261. Rosano GM, Webb CM, Chierchia S, Morgani GL, Gabraele M, Sarrel PM, de Ziegler D, Collins P. 2000. Natural progesterone, but not medroxyprogesterone acetate, enhances the beneficial effect of estrogen on exercise-induced myocardial ischemia in postmenopausal women. J Am Coll Cardiol 36:2154–2159 [DOI] [PubMed] [Google Scholar]
- 262. Mishra RG, Hermsmeyer RK, Miyagawa K, Sarrel P, Uchida B, Stanczyk FZ, Burry KA, Illingworth DR, Nordt FJ. 2005. Medroxyprogesterone acetate and dihydrotestosterone induce coronary hyperreactivity in intact male rhesus monkeys. J Clin Endocrinol Metab 90:3706–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Miyagawa K, Vidgoff J, Hermsmeyer K. 1997. Ca2+ release mechanism of primate drug-induced coronary vasospasm. Am J Physiol 272:H2645–H2654 [DOI] [PubMed] [Google Scholar]
- 264. Minshall RD, Stanczyk FZ, Miyagawa K, Uchida B, Axthelm M, Novy M, Hermsmeyer K. 1998. Ovarian steroid protection against coronary artery hyperreactivity in rhesus monkeys. J Clin Endocrinol Metab 83:649–659 [DOI] [PubMed] [Google Scholar]
- 265. Hermsmeyer RK, Mishra RG, Pavcnik D, Uchida B, Axthelm MK, Stanczyk FZ, Burry KA, Illingworth DR, Kaski JC, Nordt FJ. 2004. Prevention of coronary hyperreactivity in pre-atherogenic menopausal rhesus monkeys by transdermal progesterone. Arteriosclerosis Thromb Vasc Biol 24:955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Wagner JD, Martino MA, Jayo MJ, Anthony MS, Clarkson TB, Cefalu WT. 1996. The effects of hormone replacement therapy on carbohydrate metabolism and cardiovascular risk factors in surgically postmenopausal cynomolgus monkeys. Metabolism 45:1254–1262 [DOI] [PubMed] [Google Scholar]
- 267. Al-Azzawi F, Thompson J, Stevenson J. 2006. Which progestogen is more likely to increase the risk of fatal myocardial infarction: a combination of epidemiological and trial evidence. Maturitas 54:154–163 [DOI] [PubMed] [Google Scholar]
- 268. Godsland IF. 2001. Effects of postmenopausal hormone replacement therapy on lipid, lipoprotein, and apolipoprotein (a) concentrations: analysis of studies published from 1974–2000. Fertil Steril 75:898–915 [DOI] [PubMed] [Google Scholar]
- 269. Shulman LP. 2002. Effects of progestins in different hormone replacement therapy formulations on estrogen-induced lipid changes in postmenopausal women. Am J Cardiol 89:47E–54E; discussion 54E–55E [DOI] [PubMed] [Google Scholar]
- 270. Norris LA, Brosnan J, Bonnar J, Conard J, Kluft C, Hellgren M. 2008. Inhibitors and activation markers of the haemostatic system during hormone therapy: a comparative study of oral estradiol (2 mg)/dydrogesterone and estradiol (2 mg)/trimegestone. Thromb Haemost 100:253–260 [PubMed] [Google Scholar]
- 271. Canonico M, Alhenc-Gelas M, Plu-Bureau G, Olié V, Scarabin PY. 2010. Activated protein C resistance among postmenopausal women using transdermal estrogens: importance of progestogen. Menopause 17:1122–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Canonico M, Fournier A, Carcaillon L, Olié V, Plu-Bureau G, Oger E, Mesrine S, Boutron-Ruault MC, Clavel-Chapelon F, Scarabin PY. 2010. Postmenopausal hormone therapy and risk of idiopathic venous thromboembolism: results from the E3N cohort study. Arterioscler Thromb Vasc Biol 30:340–345 [DOI] [PubMed] [Google Scholar]
- 273. Gol M, Akan P, Dogan E, Karas C, Saygili U, Posaci C. 2006. Effects of estrogen, raloxifene, and hormone replacement therapy on serum C-reactive protein and homocysteine levels. Maturitas 53:252–259 [DOI] [PubMed] [Google Scholar]
- 274. Koh KK, Jin DK, Yang SH, Lee SK, Hwang HY, Kang MH, Kim W, Kim DS, Choi IS, Shin EK. 2001. Vascular effects of synthetic or natural progestagen combined with conjugated equine estrogen in healthy postmenopausal women. Circulation 103:1961–1966 [DOI] [PubMed] [Google Scholar]
- 275. Imthurn B, Rosselli M, Jaeger AW, Keller PJ, Dubey RK. 1997. Differential effects of hormone-replacement therapy on endogenous nitric oxide (nitrite/nitrate) levels in postmenopausal women substituted with 17β-estradiol valerate and cyproterone acetate or medroxyprogesterone acetate. J Clin Endocrinol Metab 82:388–394 [DOI] [PubMed] [Google Scholar]
- 276. White WB, Hanes V, Chauhan V, Pitt B. 2006. Effects of a new hormone therapy, drospirenone and 17-β-estradiol, in postmenopausal women with hypertension. Hypertension 48:246–253 [DOI] [PubMed] [Google Scholar]
- 277. White WB, Hanes V, Mallareddy M, Chauhan V. 2008. Effects of the hormone therapy, drospirenone and 17-β estradiol, on early morning blood pressure in postmenopausal women with hypertension. J Am Soc Hypertens 2:20–27 [DOI] [PubMed] [Google Scholar]
- 278. Simoncini T, Genazzani AR. 2010. A review of the cardiovascular and breast actions of drospirenone in preclinical studies. Climacteric 13:22–33 [DOI] [PubMed] [Google Scholar]
- 279. Schneider C, Jick SS, Meier CR. 2009. Risk of cardiovascular outcomes in users of estradiol/dydrogesterone or other HRT preparations. Climacteric 12:445–453 [DOI] [PubMed] [Google Scholar]
- 280. Canonico M, Oger E, Plu-Bureau G, Conard J, Meyer G, Lévesque H, Trillot N, Barrellier MT, Wahl D, Emmerich J, Scarabin PY; Estrogen and Thromboembolism Risk (ESTHER) Study Group 2007. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 115:840–845 [DOI] [PubMed] [Google Scholar]
- 281. Gomes MP, Deitcher SR. 2004. Risk of venous thromboembolic disease associated with hormonal contraceptives and hormone replacement therapy: a clinical review. Arch Intern Med 164:1965–1976 [DOI] [PubMed] [Google Scholar]
- 282. Sehovic N, Smith KP. 2010. Risk of venous thromboembolism with drospirenone in combined oral contraceptive products. Ann Pharmacother 44:898–903 [DOI] [PubMed] [Google Scholar]
- 283. Parkin L, Sharples K, Hernandez RK, Jick SS. 2011. Risk of venous thromboembolism in users of oral contraceptives containing drospirenone or levonorgestrel: nested case-control study based on UK General Practice Research Database. BMJ 342:d2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Jick SS, Hernandez RK. 2011. Risk of non-fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: case-control study using United States claims data. BMJ 342:d2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Lidegaard Ø, Nielsen LH, Skovlund CW, Skjeldestad FE, Løkkegaard E. 2011. 2011 Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ 343:d6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Tsutsui K. 2008. Progesterone biosynthesis and action in the developing neuron. Endocrinology 149:2757–2761 [DOI] [PubMed] [Google Scholar]
- 287. Singh M. 2007. Progestins and neuroprotection: are all progestins created equal? Minerva Endocrinol 32:95–102 [PubMed] [Google Scholar]
- 288. Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC, Salomone JP, Dent LL, Harris OA, Ander DS, Lowery DW, Patel MM, Denson DD, Gordon AB, Wald MM, Gupta S, Hoffman SW, Stein DG. 2007. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med 49:391–402, 402.e1–e2 [DOI] [PubMed] [Google Scholar]
- 289. Xiao G, Wei J, Yan W, Wang W, Lu Z. 2008. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care 12:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Hu Z, Li Y, Fang M, Wai MS, Yew DT. 2009. Exogenous progesterone: a potential therapeutic candidate in CNS injury and neurodegeneration. Curr Med Chem 16:1418–1425 [DOI] [PubMed] [Google Scholar]
- 291. Stein DG, Wright DW. 2010. Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin Investig Drugs 19:847–857 [DOI] [PubMed] [Google Scholar]
- 292. Schumacher M, Sitruk-Ware R, De Nicola AF. 2008. Progesterone and progestins: neuroprotection and myelin repair. Curr Opin Pharmacol [Erratum (2009) 9:227] 8:740–746 [DOI] [PubMed] [Google Scholar]
- 293. Ciriza I, Carrero P, Frye CA, Garcia-Segura LM. 2006. Reduced metabolites mediate neuroprotective effects of progesterone in the adult rat hippocampus. The synthetic progestin medroxyprogesterone acetate (Provera) is not neuroprotective. J Neurobiol 66:916–928 [DOI] [PubMed] [Google Scholar]
- 294. Singh M. 2006. Progesterone-induced neuroprotection. Endocrine 29:271–274 [DOI] [PubMed] [Google Scholar]
- 295. Nilsen J, Brinton RD. 2002. Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143:205–212 [DOI] [PubMed] [Google Scholar]
- 296. Nilsen J, Brinton RD. 2003. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proc Natl Acad Sci USA 100:10506–10511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Nilsen J, Morales A, Brinton RD. 2006. Medroxyprogesterone acetate exacerbates glutamate excitotoxicity. Gynecol Endocrinol 22:355–361 [DOI] [PubMed] [Google Scholar]
- 298. Wright DW, Hoffman SW, Virmani S, Stein DG. 2008. Effects of medroxyprogesterone acetate on cerebral oedema and spatial learning performance after traumatic brain injury in rats. Brain Inj 22:107–113 [DOI] [PubMed] [Google Scholar]
- 299. Resnick SM, Espeland MA, Jaramillo SA, Hirsch C, Stefanick ML, Murray AM, Ockene J, Davatzikos C. 2009. Postmenopausal hormone therapy and regional brain volumes: the WHIMS-MRI Study. Neurology 72:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Schumacher M, Guennoun R, Ghoumari A, Massaad C, Robert F, El-Etr M, Akwa Y, Rajkowski K, Baulieu EE. 2007. Novel perspectives for progesterone in hormone replacement therapy, with special reference to the nervous system. Endocr Rev 28:387–439 [DOI] [PubMed] [Google Scholar]
- 301. Nelson HD. 2002. Hormone replacement therapy and osteoporosis. Systematic Evidence Reviews No. 12. Rockville, MD: Agency for Healthcare Research and Quality; http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=es12 [PubMed] [Google Scholar]
- 302. Dören M, Nilsson JA, Johnell O. 2003. Effects of specific post-menopausal hormone therapies on bone mineral density in post-menopausal women: a meta-analysis. Hum Reprod 18:1737–1746 [DOI] [PubMed] [Google Scholar]
- 303. Abdalla HI, Hart DM, Lindsay R, Leggate I, Hooke A. 1985. Prevention of bone mineral loss in postmenopausal women by norethisterone. Obstet Gynecol 66:789–792 [PubMed] [Google Scholar]
- 304. Gallagher JC, Kable WT, Goldgar D. 1991. Effect of progestin therapy on cortical and trabecular bone: comparison with estrogen. Am J Med 90:171–178 [PubMed] [Google Scholar]
- 305. Prior JC, Vigna YM, Barr SI, Rexworthy C, Lentle BC. 1994. Cyclic medroxyprogesterone treatment increases bone density: a controlled trial in active women with menstrual cycle disturbances. Am J Med 96:521–530 [DOI] [PubMed] [Google Scholar]
- 306. Prior JC, Tremollieres F, Forsmo S, Seifert-Klauss V. 2006. Unsuccessful attempt to demonstrate progesterone's bone formation actions. Am J Obstet Gynecol 194:1502–1503; author reply 1503–1504 [DOI] [PubMed] [Google Scholar]
- 307. Hammond GL. 2002. Access of reproductive steroids to target tissues. Obstet Gynecol Clin North Am 29:411–423 [DOI] [PubMed] [Google Scholar]
- 308. Sitruk-Ware R. 2007. Routes of delivery for progesterone and progestogens. Maturitas 57:77–80 [DOI] [PubMed] [Google Scholar]
- 309. Sitruk-Ware R. 2005. Pharmacology of different progestogens: the special case of drospirenone. Climacteric 8(Suppl 3):4–12 [DOI] [PubMed] [Google Scholar]
- 310. Sitruk-Ware R. 2006. Contraception: an international perspective. Contraception 73:215–222 [DOI] [PubMed] [Google Scholar]
- 311. Charles NJ, Thomas P, Lange CA. 2010. Expression of membrane progesterone receptors (mPR/PAQR) in ovarian cancer cells: implications for progesterone-induced signaling events. Horm Cancer 1:167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Shang Y, Brown M. 2002. Molecular determinants for the tissue specificity of SERMs. Science 295:2465–2468 [DOI] [PubMed] [Google Scholar]
- 313. Kurman RJ, Kaminski PF, Norris HJ. 1985. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer 56:403–412 [DOI] [PubMed] [Google Scholar]
- 314. Africander D. 2010. Comparative study of the molecular mechanism of action of the synthetic progestins, MPA and norethisterone acetate. PhD thesis, University of Stellenbosch, South Africa [Google Scholar]