

Abstract
Chromosomal instability (CIN) is a common feature in human cancer, and highly aneuploid tumors are frequently associated with poor prognosis; however, the molecular and cellular mechanisms underlying CIN-induced tumorigenesis are poorly understood. Here we review recent findings about the role of CIN in driving tumor-like growth and host invasiveness in Drosophila epithelia and discuss the commonalities of CIN-induced tumors with other Drosophila-based cancer models. We also discuss possible scenarios that can account for the participation of CIN in tumorigenesis and propose that, alternatively to the classical role of aneuploidy in promoting the accumulation of mutations in cancer cells, aneuploidy can be a source of stress that may contribute to cancer initiation and/or progression.
Keywords: CIN, p53, JNK, delamination, Wingless, MMP1, stress
CIN-Induced Tumorigenesis in Drosophila: A Valid Model for Cancer Research
Chromosomal instability (CIN), defined as an elevated rate of gains and losses of whole chromosomes, is a common trait in human cancer and aneuploidy, a karyotype that is not a multiple of the haploid complement and that is frequently associated with poor prognosis. However, the molecular and cellular mechanisms underlying CIN-induced tumorigenesis are not well understood.1 Recently, we developed a Drosophila model to address the contribution of CIN to tumorigenesis2 and presented evidence that the molecular and cellular events underlying CIN-induced tumorigenesis reproduce key aspects of cancer initiation and progression. Chromosome segregation defects caused by mutations in genes involved in the spindle assembly checkpoint [bub3 and rough deal (rod)], spindle assembly [abnormal spindle (asp)], chromatin condensation (orc2) and cytokinesis [diaphanous (dia)] have been shown to recapitulate the genomic defects most frequently associated with human cancer, including chromosome rearrangements and aneuploidy.3 We knocked down the expression levels of these genes and analyzed the impact in epithelial cells of Drosophila. In particular, we used the larval primordium of the adult wing, a highly proliferative mono-layered epithelium.4 We first showed that CIN leads to apoptotic cell death. This cell death depended on the activation of the stress-response Jun N-terminal kinase (JNK) signaling pathway, and, in contrast to mammalian cells,1 CIN-induced apoptosis was independent of the activity of Dp53, the Drosophila ortholog of the tumor suppressor gene p53 5-7. Maintenance of aneuploid cells in the tissue by blocking cell death at different levels of the apoptotic pathway, including the pro-apoptotic genes hid, reaper and grim (the Drosophila functional orthologs of the mammalian genes Smac/DIABLO), the initiator caspase Dronc (Drosophila caspase-9) and the effector caspases DrICE and Dcp1 (Drosophila caspase-3), led to tissue overgrowth, basement membrane degradation and host invasiveness (micro-metastasis in allograft experiments).
A detailed analysis of the CIN-induced tumors identified two well-defined cell populations in these tumors (Fig. 1). Highly aneuploid cells mislocalized DE-cadherin, lost their apicobasal polarity and delaminated from the epithelium, whereas cells with a low level of aneuploidy were maintained in the epithelium. The characterization of these two cell populations helped us to unveil a cell-autonomous and a non-autonomous contribution of CIN to tumorigenesis. While aneuploidy-induced delaminating cells became motile, expressed the secreted matrix-metalloproteinase-1 (MMP1) and degraded the basement membrane, thus supporting a cell-autonomous role of CIN in driving tissue invasiveness, delaminating cells expressed Wingless (the Drosophila ortholog of mammalian Wnts), which promotes hyperplasia of the non-delaminated tissue. This hyperplastic growth is expected to increase aneuploidy levels in the non-delaminating cell layer and, consequently, augment the number of aneuploidy-induced delaminated cells. The self-reinforcement mechanism between these two cell populations, which produces a net increase in the number of delaminating and non-delaminating cell populations, might have implications in tumor initiation and progression in human cancer.

Figure 1. Cellular and molecular mechanisms underlying CIN-induced tumorigenesis in Drosophila epithelia. (A) CIN-induced tumorigenesis reproduces key aspects of human cancer, including cell delamination, hyperplasia, basement membrane (BM) degradation and invasiveness. Aneuploid cells (blue) lose their apicobasal polarity and delaminate from the epithelium (red). Delaminating cells activate the stress-responsive JNK signaling pathwa,y which, in turn, leads to the expression of at least two downstream targets genes, wingless and the matrix-metalloproteinase-1 (MMP1). Wingless contributes to the hyperplastic overgrowth of the monolayer epithelium (B), while MMP1 has an active role in degrading the BM, a critical step in tissue invasiveness (C).
The expression of MMP1 and Wingless in delaminated cells depends on the activity of the JNK pathway. The restricted activation of JNK to delaminated cells was required for CIN-induced tumorigenesis, as blocking the activity of this kinase completely rescued CIN-induced tumor-like overgrowth. These results reinforce the subversive role of JNK in driving tumorigenesis in Drosophila epithelia.8-10 While JNK is used primarily to remove highly aneuploid cells through apoptosis, this kinase plays a major role in driving tumor growth and basement membrane degradation when apoptosis is inhibited.
The invasiveness of tumor cells is generally preceded in human cancers by the delamination of tumor cells, followed by loss of the basement membrane.11 Cell delamination usually relies on compromised E-cadherin-mediated cell interactions, which prevent dissociation of individual epithelial cells from their neighbors. Furthermore, in tumor cells MMPs contribute to basement membrane degradation, and MMP1 has been shown to play a major role in this process in Drosophila epithelial cells both during development and disease.12 Thus, the molecular and cellular events that occur during CIN-induced tumorigenesis in Drosophila epithelia reproduce key aspects of cancer initiation and progression (Fig. 2).
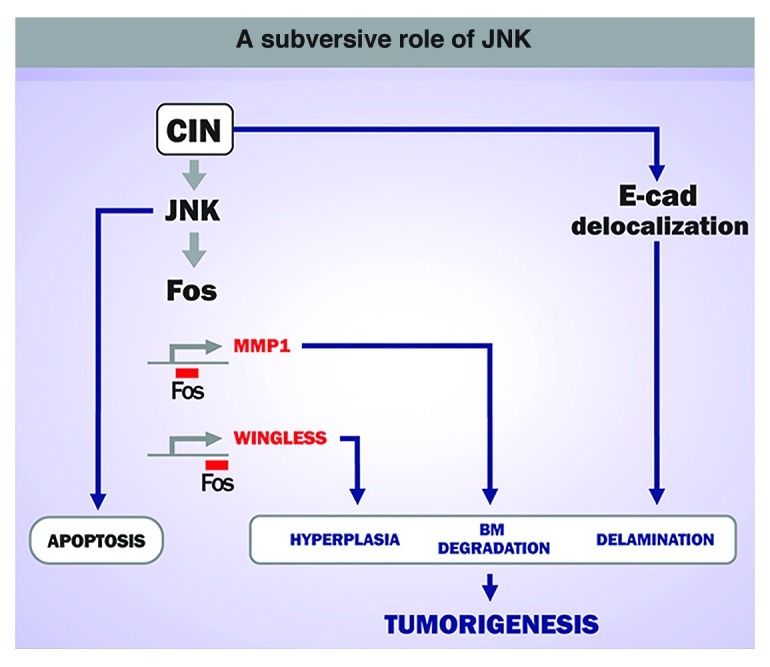
Figure 2. A subversive role of JNK in CIN-induced tumorigenesis. CIN leads to a JNK-dependent apoptotic response to eliminate potentially dangerous cells. When maintained in the tissue, aneuploid cells induce neoplastic growth in a JNK-dependent manner.
CIN-Induced Tumorigenesis in the Context of Other Drosophila Cancer Models
Drosophila epithelial cells have been previously used as model systems to elucidate the molecular mechanisms underlying the tumorigenic response and metastatic behavior of the tissue upon expression of the RasV12 oncogene and depletion of the conserved tumor suppressor genes that form the Scribble polarity complex (scribble, Discs large and Lethal giant larvae).8-10,13,14 Interestingly, these tumors share many features with those induced by CIN, including loss of DE-Cadherin, activation of JNK, expression of mitogenic molecules, basement membrane degradation and tissue invasiveness. Inactivation of the Scribble polarity complex alone or in combination with other tumor-initiating mutations (such as the proto-oncogene dMyc) does not produce metastatic tumors. This observation strongly suggests a highly specific cooperation between these genetic alterations. A subversive role of JNK in driving tumorigenesis has also been reported in these tumors.8-10 While JNK-mediated apoptosis eliminates cells mutant for the Scribble polarity complex, JNK promotes tumors in these cells when they express RasV12. JNK exerts its functions by inducing the expression of cytokines and MMP1, which are responsible for tissue overgrowth and basal membrane degradation, a pre-requisite for tissue invasiveness. JNK activation alone is not sufficient to drive tumorigenesis, and disruption of the apical-basal polarity appears to play a fundamental role in this process. The identification of the delaminating cell population in CIN-induced tumors as one with a critical contribution to promoting JNK-dependent deregulated growth and host invasiveness is consistent with these observations. Oncogene-induced DNA replication stress has previously been shown to induce genomic instability and to contribute to tumorigenesis.15 Whether RasV12 generates genomic instability in Drosophila epithelial cells and, if so, the extent of the contribution of oncogene-induced aneuploidies to the RasV12 /Scribble tumor model remains to be elucidated.
Aneuploidy-Induced Stress and Tumorigenesis
Several scenarios can account for the participation of CIN in tumorigenesis. CIN can be a source of mutability to specific tumor-prone aneuploid cells.16,18 Alternatively, aneuploidy can be a source of stress, and aneuploidy-induced stress may contribute to cancer initiation and/or progression.17,19 CIN may act as an initiator or late event by promoting the loss of heterozygosis of tumor suppressor genes. According to this hypothesis, resistance to apoptosis would be a direct consequence of CIN, and loss of heterozygosis of chromosomes that contain the remaining wild type copy of genes required for eliminating aneuploid cells (i.e., pro-apoptotic genes) might contribute to the survival of aneuploid cells and to cancer development. Consistently, CIN generated by haploinsufficient spindle assembly checkpoint (SAC) mutants increases the frequency and number of tumors in heterozygous mice for the p53 tumor suppressor gene.20 Remarkably, a mechanistic link between aneuploidy and genomic instability has also been recently identified both in yeast and mammals.18,19 In this scenario, aneuploidy-induced genomic instability could facilitate the acquisition of genetic alterations that drive malignant growth.
In addition, our results reveal a general and rapid response of the tissue upon induction of CIN, characterized by the loss of apical-basal polarity, cell delamination, JNK activation and basement membrane degradation. It is highly unlikely that this quick response is caused solely by CIN-dependent loss of tumor-suppressor genes or the accumulation of oncogenic mutations, and it supports the notion that aneuploidy-induced stress might also contribute to tumorigenesis. A recent analysis of gene expression data from aneuploid cells in several organisms, including yeast, plants, mice and humans, has unraveled a consistent upregulation of genes involved in the stress response in all these species.23 Protein stoichiometry imbalances as a source of stress have been verified to be responsible for some aneuploidy-related phenotypes.21,23 The formation of protein aggregates in aneuploid cells has been proposed to be caused by limiting protein quality-control systems, including the proteasome and the chaperone systems, and increasing proteosomal degradation by deleting the gene encoding the de-ubiquitinating enzyme Ubp6 improves the proliferative capacity of aneuploid cells.22 Whether aneuploidy-induced protein stoichiometry imbalances contribute to JNK activation and tumor-like overgrowth in Drosophila epithelial tissues remains to be elucidated (Fig. 3).
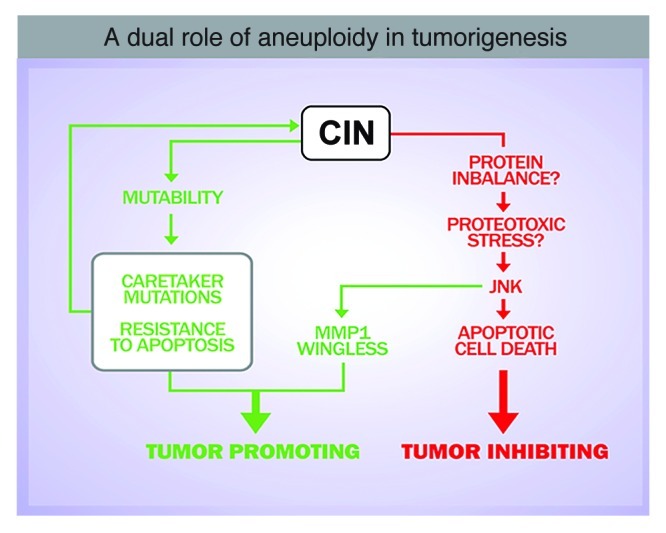
Figure 3. Aneuploidy-induced stress may contribute to tumorigenesis. Several scenarios can account for a role of CIN in driving tumorigenesis: CIN can be a source of mutability, which might facilitate the acquisition of genetic alterations that drive malignant growth. Alternatively, aneuploidy can be a source of stress and aneuploidy-induced stress might contribute to tumor initiation and/or progression.
Acknowledgments
We thank L. Barrio and M. Clemente for comments and T. Yates for help in editing the manuscript. A.D. is funded by a Juan de la Cierva post-doctoral contract. M.M. is an ICREA Research Professor and his lab is funded by grants from the Spanish Ministerio de Ciencia e Innovación (BFU2010-21123 and CSD2007-00008), the Generalitat de Catalunya (2005 SGR 00118) and the EMBO Young Investigator Programme. We thank J.M. Pochettino for graphic design. We apologize to colleagues whose work could not be cited because of space restrictions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23949
References
- 1.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA. 2010;107:14188–93. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekanty A, Barrio L, Muzzopappa M, Auer H, Milán M. Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc Natl Acad Sci USA. 2012;109:20549–54. doi: 10.1073/pnas.1206675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castellanos E, Dominguez P, Gonzalez C. Centrosome dysfunction in Drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol. 2008;18:1209–14. doi: 10.1016/j.cub.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 4.Cohen SM. Imaginal disc development. In: Martinez-Arias A, Bate M, eds. Drosophila Development. Cold Spring Harbor: Cold Spring Harbor Press, 1993:747-841 [Google Scholar]
- 5.Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–13. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 6.Jin S, Martinek S, Joo WS, Wortman JR, Mirkovic N, Sali A, et al. Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc Natl Acad Sci USA. 2000;97:7301–6. doi: 10.1073/pnas.97.13.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 8.Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–31. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- 9.Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006;25:5294–304. doi: 10.1038/sj.emboj.7601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–79. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012;139:3471–86. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci USA. 2007;104:2721–6. doi: 10.1073/pnas.0611666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, Igaki T. Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature. 2012;490:547–51. doi: 10.1038/nature11452. [DOI] [PubMed] [Google Scholar]
- 14.Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell. 2010;18:999–1011. doi: 10.1016/j.devcel.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 2012;13:515–27. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–43. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chi YH, Ward JM, Cheng LI, Yasunaga J, Jeang KT. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int J Cancer. 2009;124:1483–9. doi: 10.1002/ijc.24094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26:2696–708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA. 2012;109:12644–9. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, et al. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci USA. 2012;109:21010–5. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]