

Abstract
CTNNBL1 is an armadillo-repeat protein that associates with the CDC5L/Prp19 complex of the spliceosome. Unlike the majority of spliceosomal proteins (and despite having no obvious homologs), CTNNBL1 is inessential for cell viability as revealed by studies in both vertebrate B cell lines and in fission yeast. Here, however, we show that ablation of CTNNBL1 in the mouse germline results in mid-gestation embryonic lethality but that lineage-specific CTNNBL1 ablation in early B cell precursors does not affect the production and abundance of mature B lymphocytes. However, CTNNBL1-deficient resting B lymphocytes show sluggish exit from quiescence on cell activation, although once entry into cycle has initiated, proliferation and differentiation in response to mitogenic stimuli continue largely unaffected. A similar sluggish exit from quiescence is also observed on reprovision of nutrients to nitrogen-starved CTNNBL1-deficient yeast. The results indicate that, whereas other RNA splicing-associated factors have been connected to cell cycle progression, CTNNBL1 plays no essential role in cycling cells but does fulfill an evolutionarily conserved function in helping cells to undergo efficient exit from quiescence following activation.
Keywords: CTNNBL1, cell cycle, quiescence, RNA splicing, B lymphocyte, yeast, conditional knockout mice
Introduction
CTNNBL1 (catenin-β-like 1) is a widely expressed nuclear protein composed of multiple tandem armadillo domains that owes its name to the fact that it shows predicted structural homology (though little primary sequence homology) to β-catenin.1 CTNNBL1 likely functions in RNA splicing, since it is associated with the Prp19-CDC5L-PLRG1 complex (also known as the 19 or NTC complex) of the spliceosome, interacting directly with CDC5L.2-5 However, although present as a single-copy gene in vertebrates without any obvious sequence homologs, CTNNBL1 is inessential for cell viability, since the Ctnnbl1 gene has been disrupted in both chicken and mouse B cell lines without notably affecting cell proliferation or viability.3,6
The fact that CTNNBL1 is inessential for the viability of mammalian cell-lines and yet the gene is widely-expressed and well conserved among most eukaryotes led us to ask whether more subtle effects of CTNNBL1 deficiency might be observed in intact animals, as opposed to in B cell-lines. Here we show that germline disruption of the mouse Ctnnbl1 gene leads to mid-term embryonic lethality, whereas lineage-specific ablation of Ctnnbl1 in primary B cells results in substantially delayed cell enlargement and exit from quiescence following mitogenic stimulation without having a major detectable effect on cell proliferation once cycling has been initiated.
Results
Germline ablation of CTNNBL1 results in midterm embryonic lethality
Gene targeting was used to generate clones of embryonic stem cells that carry, on one Ctnnbl1 allele, an insertion of a β-gal/neo cassette into the second Ctnnbl1 intron together with LoxP sites flanking the linked Ctnnbl1 exon 3 (Fig. 1A and B). This targeted allele is designated Ctnnbl1-. The β-gal/neo cassette on this allele is itself flanked by flippase recognition target sequences and comprises (from 5′- to 3′-ends): an RNA splice acceptor site, an internal ribosomal entry site, a promoterless β-galactosidase gene and a neomycin-resistance gene that is driven by a phosphoglycerate kinase promoter. It is therefore anticipated that transcription initiated from the Ctnnbl1 promoter on the Ctnnbl1- allele will give rise to a truncated N-terminal CTNNBL1 polypeptide that terminates at codon 82 together with β-galactosidase, whose translation will be initiated from the IRES. These Ctnnbl1-/+ ES cells were injected into blastocysts isolated from C57BL/6 mice and the resultant chimaeras bred to obtain heterozygous mice carrying one targeted Ctnnbl1- allele in their germline.

Figure 1. Targeted inactivation of Ctnnbl1 results in mid-term embryonic lethality. (A) Targeting the mouse Ctnnbl1 locus. The top line depicts the mouse Ctnnbl1 locus (three exons: E1, E2 and E3 are depicted) aligned with the targeting construct which contains a promoterless β-galactosidase/pgk-neo cassette flanked by flippase recognition sites (oval shapes) integrated into intron 2 with LoxP sites (open triangles) flanking E3. The left and right homology arms are 4.6 and 4.8 kb, respectively. Restriction endonuclease sites Ase I (A) and Sap I (S) are indicated as are the locations of the left- and right-hand probes (LHP and RHP) used for the Southern blot analysis. (B) Southern blot of tail DNA from mice carrying the targeted β-gal/neo cassette insertion into Ctnnbl1 E3 on one allele as well as controls hybridized with probes LHP and RHP. (C) Genotypes of weaned animals born to Ctnnnbl1+/− intercrosses. A similar analysis is shown for the progeny born to intercrosses of AX0016 mice that carry a gene trap insertion into the first Ctnnbl1 intron. (D) Genotypes of embryos obtained at day 8.5 or 11.5 of gestation from Ctnnnbl1+/− intercrosses. (E) Broad expression of the Ctnnbl1 locus as revealed by staining of a day 14.5 Ctnnnbl1+/− embryo for β-galactosidase activity.
Interbreeding of the heterozygous mice failed to yield any weaned Ctnnbl1−/− homozygous offspring (Fig. 1C). A similar failure to obtain animals homozygous for an inactivated Ctnnbl1 allele was observed when interbreeding a different line of mice that carry a βgal/neo genetrap insertion into the first intron of Ctnnbl1 (Fig. 1C). Thus, germline deficiency in CTNNBL1 appears to be embryonically lethal. Analysis of embryos generated by intercrossing Ctnnbl1-/+ heterozygotes reveals that although Ctnnbl1−/− embryos can be obtained at day 8.5, their viability is already compromised by mid-gestation (Fig. 1D). Thus, CTNNBL1 deficiency is lethal around the embryonic midterm. We have not identified any specific lineage that is responsible for this effect: staining of Ctnnbl1-/+ heterozygous embryos (which carry the βgal/neo genetrap insertion on one Ctnnbl1 allele) for β-galactosidase activity indicates that Ctnnbl1 exhibits a broad expression pattern (Fig. 1E).
B cells develop in the absence of CTNNBL1
The embryonic lethality resulting from germline CTNNBL1 deficiency contrasts with the healthiness of CTNNBL1-deficient B lymphoid cell-lines.3,6 We therefore wondered if it would be possible to obtain primary B cells lacking CTNNBL1. Mice bearing the βgal/neo targeting on one Ctnnbl1 allele (Ctnnbl- in Fig. 2A) were crossed with mice that express Flip recombinase in the germline in order to yield offspring in which the βgal/neo cassette had been deleted through Flip-mediated recombination. The resulting Ctnnbl1flx allele is functional (in that Ctnnbl1flx/flx mice are viable and express CTNNBL1) but retains LoxP sites flanking Ctnnbl1 exon 3, meaning that the locus can then be inactivated by Cre-mediated recombination. Indeed, crossing Ctnnbl1flx mice with animals expressing Cre in the germline yielded a Ctnnbl1ΔE3 allele that, in homozygous form, resulted in embryonic lethality (five litters of Ctnnbl1flx/ΔE3 intercrosses yielded 20 Ctnnbl1flx/ΔE3 heterozygotes and seven Ctnnbl1flx/flx homozygotes but no Ctnnbl1ΔE3 /ΔE3 homozygotes).

Figure 2. Lineage-specific ablation of Ctnnbl1 has little effect on B cell development. (A) B cell-specific inactivation of Ctnnbl1. The top two lines depict the wild type locus and the derivative containing a targeted β-gal/neo insertion into intron 2 as in Figure 1A. In alignment with these are depicted the loci obtained following (i) Cre-mediated excision of E3 (Ctnnnbl1-ΔE3; which will be inactive); (ii) following Flippase-mediated excision of the β-gal/neo cassette (Ctnnnbl1flx; which will be functional but a ‘floxable’ substrate for Cre-mediated inactivation) and (iii) both Flippase – and cre-mediated excision (yielding Ctnnnbl1ΔE3; which will be inactive). (B) Southern blot analysis of DNA extracted from sorted splenic CD43- cells (which comprise > 95% resting B cells) as well as CD43+ cells (which comprise T cells, granulocytes/macrophages and activated B cells) from mice of the indicated Ctnnbl1 genotypes that also express Cre recombinase under control of the B-cell-specific mb1 promoter. Ase I-digested DNA was hybridized with a Ctnnbl1 E2 probe. The origins of the various hybridizing bands are indicated with the results revealing highly (> 95%) efficient deletion of Ctnnbl1 E3 in the resting B cells of mb1-Cre Ctnnbl1-/flx mice. Some deletion of Ctnnbl1 E3 is also observed within the CD43+ fraction, which probably reflects activated B cells within this population. (C) Loss of CTNNBL1 expression in the sorted splenic B cells of mb1-Cre Ctnnbl1-/flx mice as judged by (i) RT-PCR analysis of RNA using primers specific for Ctnnbl1 E3 relating to HPRT (as a control) and (ii) western blot analysis of protein using α-tubulin as a control. (D) Comparison of splenic B and T cell populations in mb1-Cre Ctnnbl1-/flx mice (squares) as compared with mb1-Cre Ctnnbl1+/flx controls (circles). T cells were identified as CD3+ and subdivided into CD4+ and CD8+ single-positive subpopulations. B cells were identified as B2220+ or CD19+ with B220+ CD19+ cells divided into B1 (CD23- CD21-); follicular (FO: CD23high CD21+) or marginal zone (MZ: CD23low CD21+) subpopulations. (E) Comparison of pre- and pro- B cell populations in the bone marrow of mb1-Cre Ctnnbl1-/flx mice (squares) as compared with mb1-Cre Ctnnbl1+/flx controls (circles). Pre-B cells were defined as CD43- B220+ IgM- whereas pro-B cells are as CD43- B220+ IgM+.
Mice were generated that carried a targeted inactivation of Ctnnbl1 on one allele, a flx-Ctnnbl1 locus on the other allele and which also expressed the Cre recombinase under control of the early B lymphocyte-specific mb1 promoter. Analysis of DNA, RNA and protein in the splenic B cells from these mb1-Cre Ctnnbl1-/flx mice revealed that the Cre-mediated deletion of Ctnnbl1 had been effective with the cells lacking CTNNBL1 RNA and protein (Fig. 2B and C). Splenic B cells were nevertheless present in normal (indeed, very slightly increased) numbers and were similarly distributed between follicular, marginal and B1 subsets as found in controls as judged by cell surface marker analysis (Fig. 2D). There was also no obvious perturbation of earlier stages of B cell differentiation as judged by analysis of B cell populations in the bone marrow (Fig. 2E). Thus, although CTNNBL1 is essential for embryonic development, it is dispensable for the generation and maturation of B cells.
CTNNBL1-deficient B cells showed reduced immunoglobulin class switching
Previous results have revealed that CTNNBL1 deficiency reduces the frequency of immunoglobulin gene conversion in the chicken DT40 B cell line whereas no effect of CTNNBL1 deficiency on immunoglobulin class switch recombination was observed in the mouse CH12 B cell line.3,6 We were therefore interested in ascertaining whether CTNNBL1 deficiency had any effect on immunoglobulin class switching in primary mouse B cells. Comparison of switching to IgG1 in splenic B cells from mb1-Cre, Ctnnbl1+/flx and mb1-Cre, Ctnnbl1-/flx mice that had been cultured for 3 d in the presence of LPS+IL4 revealed that the CTNNBL1-deficient B cells gave roughly one-third less switching (Fig. 3A). This reduced switching does not correlate with any change in the abundance of AID (Fig. 3B). Similar results regarding diminished class switching were obtained using B cells stimulated with anti-CD40 and IL4 (Fig. S1) as well with regard to switching to IgG3 (not shown).

Figure 3. CTNNBL1-deficient B cells give diminished immunoglobulin class-switch recombination. (A) Flow cytometric analysis of switching to IgG1 in spleen cells from mb1-Cre Ctnnbl1-/flx and control mice that had been cultured for 3 d in the presence of LPS+IL4. (i) Dot plots depict results from two pairs of littermates in independent experiments. (ii) Bar graphs on the right present the means ± sd from analysis of multiple mice as indicated. The values within each experiment are normalized to the average value of the control samples. (B) Abundance of AID in the day 3 LPS+IL4-activated B cells obtained from two littermate pairs of mb1-Cre Ctnnbl1+/flx and mb1-Cre Ctnnbl1-/flx mice analyzed by western blot using α-tubulin as a control. (C) Analysis of switching to IgG1 as a function of B cell clonal expansion in CFSE-labeled splenic cells from mb1-Cre Ctnnbl1-/flx and control mice that had been cultured for 3 d in the presence of LPS+IL4. (i) The CFSE-staining profiles for two pairs of litter-matched mice with the number of cell divisions undergone (inferred from dilution of the CFSE label) indicated. (ii) The percentage of cells that have switched to IgG1 as a function of number of cell divisions undergone in six independent experiments.
We were, however, concerned to find out whether the diminished switching correlated with any alteration in the rate of B cell proliferation and therefore performed the switching assay on B cells that had been labeled with CFSE, allowing cellular proliferation to be monitored by way of CFSE dilution. The results (Fig. 3Ci) reveal that the CTNNBL1-deficient B cells do indeed exhibit a perturbation in clonal expansion in that their CFSE fluorescence curves as analyzed at day 3 are shifted to the right as compared with the controls. The CTNNBL1-deficient B cells had, on average, undergone 1–2 fewer cell divisions than their wild type counterparts. Nevertheless, even when this reduced proliferation is taken into account, the CTNNBL1-deficient B cells show a significant (averaging 30%) reduction in the extent of switching to IgG1 that is observed in each cycle of clonal expansion (Fig. 3Cii).
Delayed entry into S phase on activation of resting CTNNBL1-deficient B cells
The CFSE profiles of the splenic B cells analyzed on day 3 of LPS/IL4 culture (Fig. 3C) suggested that CTNNBL1 deficiency largely affected the time required to complete the first round of cell division rather than affecting the subsequent rate of cell proliferation. This is supported by the CFSE profiles obtained at earlier time points (Fig. 4A). Measurement of cellular DNA content revealed that the delayed initiation of proliferation reflected a delay in initiating DNA synthesis, since the proportion of cells that have exited G0/G1 and entered S phase or G2/M after 24 h and 48 h of mitogen stimulation is substantially reduced in the CTNNBL1-deficient B cells (Fig. 4B). Indeed, culturing cells in the presence of BrdU and monitoring its incorporation into DNA revealed a similar delay (of approximately 6 h) in the initiation of DNA synthesis (Fig. 4C).

Figure 4. CTNNBL1-deficient B cells are slow to enter their first S-phase on LPS activation. (A) Clonal expansion of splenic B cells from three littermate pairs of mb1-Cre Ctnnbl1-/flx and control mice as monitored by CFSE dilution. (B) Cell cycle analysis of splenic B cells from multiple littermate pairs of mb1-Cre Ctnnbl1-/flx and control mice as analyzed at 24 and 48 h of incubation with LPS/IL4. (i) The propidium iodide staining profiles of the cells from two littermate pairs are shown on the left with (ii) the results from multiple animals summarized on the right. The proportion of cells per stage of the cell cycle is normalized to the average number of cells in the controls within each independent experiment (bars indicate mean and sd). (C) DNA synthesis in splenic B cells from littermate pairs of mb1-Cre Ctnnbl1-/flx and control mice cultured for the indicated lengths of time with bromodeoxyuridine (BrdU) and LPS. (i) Cells were analyzed for BrdU content by staining with anti-BrdU antibody and total DNA content by propidium iodide staining. The individual staining profiles are indicated (percent of cells in G0/G1 and S phase in black, with the ratio of cells in G1 vs. S that have entered a second cell division in gray) with (ii) a separate graph showing the percentage of cells that have incorporated BrdU at the various time points in four pairs of littermates.
Cell blasting is delayed in CTNNBL1-deficient B cells but immediate-early gene activation is unaffected
The delayed entry into S phase correlated with a delay in cell blasting as judged by light scatter analysis as well as with reduced expression of cyclin D3, CDK6 and phosphorylated Rb as monitored at 24 h of culture (Fig. 5A and B). However, the very early signaling events did not appear to be affected by CTNNBL1 deficiency. Thus, the upregulation of cell surface molecules CD69, CD86 and MHC class II proceeded similarly in mutant and control B cells as judged after 12 and 24 h of LPS/IL4 activation (Fig. 5C). It is notable, however, that the CTNNBL1-deficient B cells exhibit a significant reduction in the subsequent downregulation of these markers at the 48 and 72 h time points, an observation which might well correlate with the delayed blasting and increase in cell size. These results indicate that the delay exhibited by CTNNBL1-deficient B cells in the completion of the first round of cell division is a consequence of a delay that occurs after initial cell activation but prior to cell blasting and entry into S phase.

Figure 5. CTNNBL1 deficiency delays cell enlargement and S-phase entry but not the upregulation of early activation markers. (A) Comparison of blasting of splenic B cells from littermate pairs of mb1-Cre Ctnnbl1-/flx and control mice after 24 h of incubation with LPS as monitored by cell scatter analysis. (i) Individual contour plots of live cells from two of the littermates pairs with the gating for blasts vs. resting cells indicated. (ii) Percentage of large cells (blasts) gate in multiple B cell cultures 24 h post activation (mean and sd are indicated). (B) Induction of expression of cyclin D3, CDK6 and phosphorylated Rb after 24 h of incubation with LPS as monitored by western blot analysis. The abundance of lamin and α-tubulin served as loading controls. (C) Surface expression of CD69, CD86 and MHC class II on splenic B cells from littermate pairs of mb1-Cre Ctnnbl1-/flx and mb1-Cre Ctnnbl1+/flx control mice as analyzed after various times of incubation with LPS. (i) Histogram plots from a representative experiment and (ii) line graphs depicting the median fluorescence intensity at each time point derived from eight experiments. (Averages and sds are indicated). (D) Comparison of the blasting of splenic follicular and marginal zone B cells following LPS activation. The B cells were obtained from CD19-Cre Ctnnbl1-/flx and CD19-Cre Ctnnbl1+/flx control mice [with the CD19-Cre giving, like the mb1-Cre, efficient B cell-specific deletion of Ctnnbl1 (Fig. S1)]. (i) Histogram plots depicting the electronic volumes of sorted follicular and mantle zone B cells. The purity (assessed by flow cytometry of surface markers) of the sorted populations at the start of the cultures is indicated. (ii) The proportion of marginal zone B cells with diameter > 9 µ at different times post stimulation in multiple samples (means and sem shown).
CTNNBL1 deficiency affects follicular more than marginal zone B cells
Although most B cells in the spleen (the small follicular B cells) are quiescent in that they take more than 24 h to complete their first division following stimulation, a small proportion (the larger marginal zone B cells) are more readily activated.7,8 Separation of follicular and marginal zone B cells revealed that rapid blasting of marginal zone B cells is still observed regardless of whether CTNNBL1 is present or not (Fig. 5D).
No major perturbation of RNA splicing patterns in CTNNBL1-deficient B cells
Although CTNNBL1 cannot be essential for RNA splicing, since it is not required for cell viability, we wondered in light of its association with the CDC5L complex of the spliceosome whether the sluggish exit from quiescence might reflect that CTNNBL1 is required to achieve efficient enhancement of specific RNA splicing on cell activation. At 24 h of activation, control and knockout B cells already show considerable difference in cell size (Fig. 5A) and total RNA content as judged by acridine orange staining (not shown). We therefore focused on an earlier time point to see if we could discern major differences in the extent of splicing. RNaseq was performed on total RNA extracted from splenic follicular B cells prior to and following 12 h of LPS stimulation. The results reveal that the 12 h period of activation is accompanied by a marked increased in the ratio of total exonic to intronic reads (Fig. 6A). However, not only is there no evident difference in the extent of this overall increase when comparing control and CTNNBL1-deficient B cells, we have also not been able to discern any individual transcripts whose splicing is affected by CTNNBL1 deficiency (Fig. 6B). Thus, although there is some sample-to-sample variation in the read ratios in individual genes, the results do not reveal any individual gene whose read ratio is consistently altered as a consequence of CTNNBL1 deficiency by an amount beyond the level of variability attributable to the observed noise of the data set (i.e., by an amount greater than 2 standard deviations, where the standard deviation is deduced from sample data with similar read ratios and assuming a normal distribution). Furthermore, the B cells from control and knockout mice show a remarkably similar pattern of gene activation at the 12 h time point. The most notable difference (apart from those in CTNNBL1 itself) is an increase in the reads for haem oxygenase (Hmox1) in the activated CTNNBL1-deficient sample, which likely reflects small differences in the efficiency of red blood cell lysis during sample preparation since the inducible form of Hmox1 is very sensitive to byproducts of erythrocyte clearance9 (Fig S2). Thus, although we cannot exclude the possibility that CTNNBL1 deficiency has a small effect on the splicing of one or several specific RNAs, the RNaseq data do not reveal any genes that have suffered a major discernible splicing perturbation.
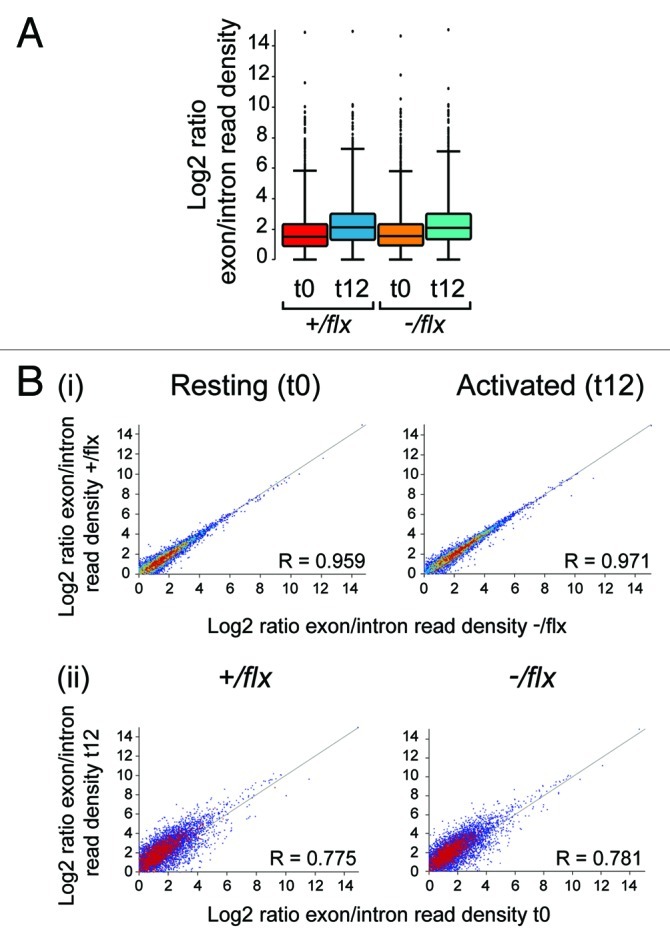
Figure 6. Comparison of RNA splicing in CTNNBL1-proficient and –deficient B cells. (A) Box Whisker plots depicting the distribution of splice indices defined as the ratio of the log2-transformed density of exonic vs. intronic reads per gene. The data include only expressed genes (defined as genes with exon/intron read densities ≥ 1). The boxes indicate the 25th to 75th percentile range with the line across the middle indicating the median and the lines above and below the box indicating the 1st and 99th percentiles. (B) Comparison of the splicing indices of individual expressed transcripts in (i) CTNNBL1-proficient vs. CTNNBL1-deficient B cells in both pre-activation (left) and post-activation (right) samples; (ii) Activated vs. resting B cells in both CTNNBL1-proficient (left) and CTNNBL1-deficient (right) samples. Whereas CTNNBL1 deficiency has no clear effect on individual splicing indices in either resting or activated samples (the correlation between the two genotypes in both sets is > 0.95), cell activation results in many genes giving altered splice indices (R < 0.79). Data on the total expression (as opposed to splicing index) of individual genes in the samples analyzed is provided in Table S1.
Ctnnbl1- S. pombe exhibit delayed exit from starvation-induced quiescence
CTNNBL1 is conserved as a single-copy gene from Schizosaccharomyces pombe through to man. If potentiating timely exit from quiescence is a core function of CTNNBL1, then this function might well be preserved through evolution. Following extended nitrogen starvation, S. pombe exhibits a quiescence that is in many respects similar to that observed in mammalian cells.10,11 We therefore asked whether inactivation of the ctnnbl1 locus in S. pombe affected the speed of exit from quiescence. Although deletion of the ctnnbl1 locus did not have any detectable effect on the doubling time of S. pombe during log-phase growth in rich medium as monitored in multiple experiments, deficiency in CTNNBL1 led to a considerable (3–4 h) and reproducible delay in the initiation of cell proliferation following nitrogen starvation which correlated with delayed exit from G0 (Fig. 7).
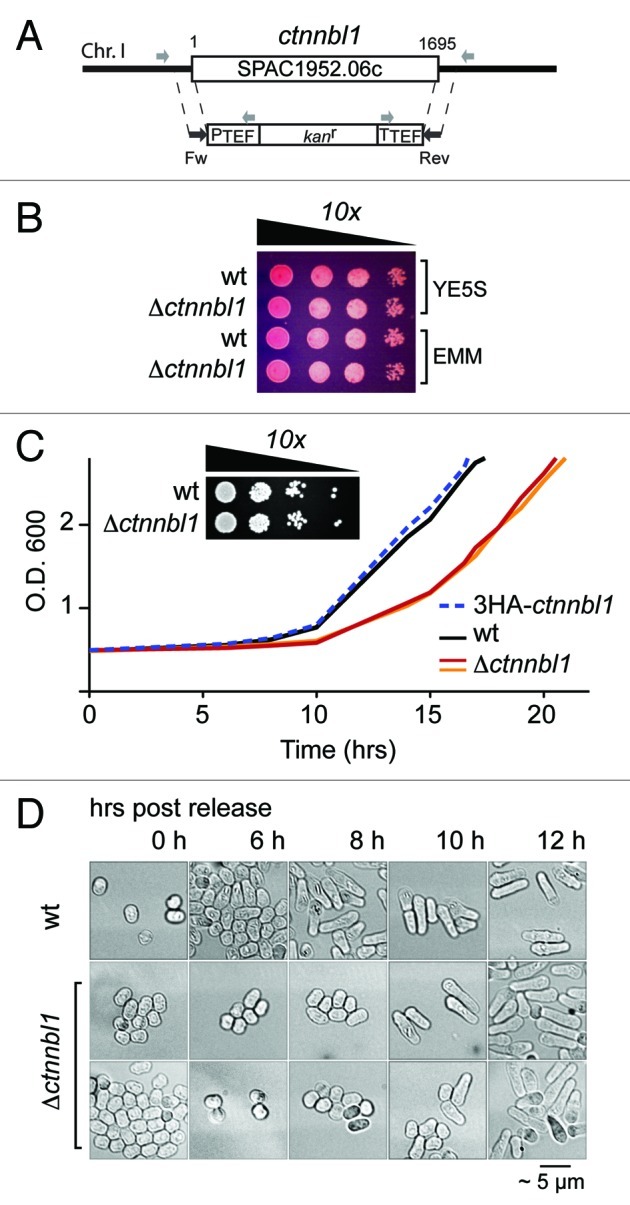
Figure 7. Loss of CTNNBL1 delays quiescence exit in S. pombe. (A) Targeting the yeast SPAC1952.06c (ctnnbl1) locus. The SPAC1952.06c gene was replaced by homologous recombination with a cassette comprising a kanamycin resistance marker under the control of translation elongation factor 1A regulatory signals as described by Bahler et al. (1998). Correct targeting was confirmed by PCR using primers whose locations are indicated. (B) Δctnnbl1 S. pombe grow similarly to controls after spotting on to YES plates. Viability of cells in log-phase growth in either yeast extract (YE) or minimal medium (EMM) from serial 10-fold dilutions spotted onto YE plates containing 5 mg/ml phloxine B. (C) Nitrogen-starved Δctnnbl1 S. pombe showed delayed initiation of growth compared with controls after transfer into rich medium. Growth curves after release from nitrogen starvation (time zero) are shown for wild type S. pombe (wt; black line), two-independent Δctnnbl1 mutants (red and orange lines) in which the ctnnbl1 locus had been replaced by a kanr cassette as well as of S. pombe carrying a control ctnnbl1 targeting in which the ctnnbl1 locus had been replaced by 3HA-tagged CTNNBL1 driven from the nmt1 promoter (blue dashed line). Similar results were obtained in four independent experiments. Growth of serial 10-fold dilutions of 3 week-starved S. pombe cultures on YE plates revealed that CTNNBL1 deficiency did not affect their viability. (D) Starved CTNNBL1-deficient S. pombe exhibit delayed exit from G0 following release from nitrogen starvation. Quiescent S. pombe, which adopt a small, round shape on nitrogen starvation, elongate prior to their first cell division on release into rich medium.23 Cells were fixed in 70% ethanol at the times indicated and visualized by phase-contrast microscopy.
Discussion
The results presented here reveal that although CTNNBL1 is not needed for the maturation or ongoing proliferation of primary mouse B cells, it is required for their timely exit from quiescence as well as for mouse embryonic development. A similar requirement for CTNNBL1 for timely exit from quiescence is also observed in fission yeast.
The embryonic lethality of CTNNBL1 deficiency manifests itself around the midterm of pregnancy, by which time most of the major cell lineages have been established. Although gross inspection of mid-term embryos has not revealed any organ that is specifically affected by CTNNBL1 deficiency, it remains possible that there is a particular cell type whose function or development is critically dependent on CTNNBL1 and that which accounts for the lethality of CTNNBL1 deficiency. Alternatively it may be that the phenotype observed in the B cells (sluggish exit from quiescence) has a severe effect on embryonic viability when recapitulated in multiple lineages during synchronized embryonic development and lineage expansion. It will therefore be interesting to cross our floxed CTNNBL1 allele into mouse lines carrying Cre recombinases active in multiple different lineages to ascertain whether there is any specific lineage or developmental stage at which loss of CTNNBL1 leads to a more severe phenotype than that observed in lymphocytes.
The molecular associations of CTNNBL1 suggest that it likely functions in RNA splicing. Although clearly not essential for splicing, a possibility suggested by this work is that CTNNBL1 may function in linking RNA splicing with timely exit from quiescence. This would be by no means the first occasion on which a link has been proposed between deficiency in an RNA splicing factor and the control of cell cycle progression.12-15 However, the suggestion here is that CTNNBL1 may function in connection with the timely “bulking up” of RNA splicing that must occur on exit from quiescence and allow progression through to S phase. Thus although CTNNBL1 deficiency does not affect the expression of early activation markers, CTNNBL1 deficiency does manifest itself in a delay to the subsequent cell enlargement/blasting. Such blasting must be accompanied by a substantial increase in the RNA processing and protein translation machinery of the cell. In contrast, a requirement for CTNNBL1 might not be manifest in cells already undergoing rapid division since such cells will inherit a substantial endowment of transcription and translation factors from their parents. Indeed, the need for bulking up may especially apply to lymphoid cells. The adaptive immune system is notable in comprising very long-lived quiescent cells with little cytoplasm, which, following antigen triggering, take time to blast and then undergo multiple sequential rounds of rapid division. It is notable that the effect of CTNNBL1 deficiency on delaying cell blasting is especially marked with respect to the small follicular B cells as compared with their larger marginal zone counterparts.
The phenotype revealed by CTNNBL1 deficiency is an intriguing one and suggests that there may be other genes whose primary function is to assist cell blasting and exit from quiescence. Several studies have been performed to identify pathways associated with cell quiescence—in budding and fission yeast, in nematode worms as well as in mammalian lymphocytes.11,16-18 Such studies have identified genes and pathways functioning in the maintenance of the quiescent state but have not, so far as we are aware, revealed any genes whose deficiency results in a phenotype similar to that seen with CTNNBL1. We suspect that this does not reflect that CTNNBL1 is unique with respect to functioning specifically in the blasting/“bulking up” phase of cell activation without being needed for immediate early gene activation or subsequent ongoing cell proliferation. Rather we suspect that such a phenotype might not have been easily scored in previous work, especially if deficiency in such genes leads to embryonic lethality, since it would only be readily apparent by following the blasting of resting cells obtained from animals carrying a lineage-specific gene ablation. It will be interesting therefore in future work not only to establish the molecular mechanism by which CTNNBL1 deficiency leads to sluggish exit from quiescence without affecting initial cell activation but also, by interrogation of mouse lines carrying other conditional gene ablations, to ascertain whether a similar phenotype is obtained with deficiencies in other genes.
Materials and Methods
Generation of Ctnnbl1-targeted mice
The targeting construct for conditional Ctnnbl1 inactivation (Fig. 1A) was obtained from EUCOMM (construct PG00008_A_1_E07). E14(129/Ola) ES cells were transfected with AsiS1-digested linear construct and 84 G418-resistant clones tested for targeted integration by Southern analysis using PCR-generated probes (5′-GGAAAGGTGATAACCCTAGACACTTTGC and 5′-TCCCACCCATCCTCGGCCTCAG-TGC for probe LHP; 5′-TTCCCAGAATCAGTTTCTCTGTCCC and 5′-TCTCACCTGAAAG-GAAGGCCATCT for probe RHP). From 26 correctly targeted clones, two were used to generate blastocyst chimeras that led to germ line transmission. The selection cassette on the targeted allele in these Ctnnbl1+/− mice was removed in vivo by crossing with a FLPase-expressing transgenic mouse line19 to produce Ctnnbl1+/flx animals. Breeding with mice in which Cre recombinase expression was driven from within the B cell-specific mb1 gene20 (kindly provided by Michael Reth) was used to generate Ctnnbl1flx/- mb1Cre/+ experimental animals that carry a B cell-specific deletion of Ctnnbl1 exon3 on one allele and a null Ctnnbl1-knockout on the other, as well as Ctnnbl1flx/+ mb1Cre/+ control littermates, which retain one intact Ctnnbl1 allele with the other allele carrying a B cell-specific exon3 deletion. Mice expressing Cre from within the cd19 gene21 were also used for B-cell specific gene deletion (Fig. S1).
Mice in which the Ctnnbl1 locus on one allele had been inactivated by integration of a gene-trapped promoterless β-Gal/neomycin cassette into the first Ctnnbl1 intron were generated using ES cell line AX0016 obtained from the Sanger Institute Gene Trap Resource (GenBank accession number CZ259087).
Embryos (d14.5 post-mating) were dissected, fixed in 4% paraformaldehyde and stained with 2 ml of X-gal (2.25 mg/ml), 1 mM MgCl2, 45 ml PBS, 5 mM potassium ferrocyanide and 5 mM potassium ferricyanide for 48 h at 37°C prior to washing in PBS and refixing in 4% paraformaldehyde .
Cellular analyses
For analysis of lymphoid subpopulations, cell suspensions from bone marrow and spleen were depleted of erythrocytes by incubation in RBC lysis buffer (Ebioscience) and stained using fluorescent rat mAbs to mouse cell surface markers (CD8-FITC, CD4-PE, CD19-Pacific Blue, PE-CD45R(B220), CD21-FITC, CD23-PE, CD43-FITC and Ig-M-FITC from BD PharMingen; B220-APC and IgM-APC from Invitrogen; CD3-PECY7 from Ebioscience). For analysis of cell activation, resting splenic B cells were magnetically separated from CD43+ cells using MACS/LD columns (Miltenyi) and splenic marginal zone and follicular B cells sorted from (B200+, CD19+)-gated splenocytes on the basis of their CD23lowCD21high or CD23+CD21+ phenotype. Cells in RPMI/10%FBS/0.05 mM 2-mercaptoethanol were cultured in the presence of either 50 μg/ml E. coli LPS or 1 μg/ml anti-CD40 (HM40–3; PharMingen) together with 50 ng/ml IL-4 (R&D Systems). Cell activation and immunoglobulin class switching were then monitored by staining with antibodies to CD69, CD86 or MHC class II or with biotinylated anti-mouse IgG3 or IgG1 and APC-streptavidin together with PE-conjugated anti-mouse CD45R(B220). CFSE labeling (Invitrogen) was performed according to manufacturer's instructions. To monitor cellular DNA synthesis, cells were cultured in the presence of 10 µM BrdU, fixed, permeabilized, treated with DNase and stained with anti FITC-conjugated anti-BrdU antibody according to manufacturer instructions (FITC BrdU flow kit, BD PharMingen). For cell cycle analysis, ethanol-fixed cells were permeabilized on 0.1% Triton in the presence of RNase (0.02 mg/ml) and stained with propidium iodide. Flow cytometry analysis was done on LSRII (BD) or Eclipse (i-Cyt) (for six color analysis or electronic volume measurements, respectively).
Biochemical analyses
For RNA analyses, total RNA was extracted with PureLink RNA purification kit (Ambion) using a DNase treatment step. For Q-PCR, 200 ng total RNA was reverse-transcribed using random primers and the cDNA amplified using SYBR Green qPCR SuperMix Universal (Invitrogen) according to manufacturer’s instructions. Expression of Ctnnbl1 exon 3 was monitored using primers 5′-TCTCCGGATTGTCTGGAAAC and 5′-GCTGGATGAAAGCTCAGTGAA. HPRT and β2-microglobulin served as controls.
For protein expression analysis, cells (2 × 106) were lysed in RIPA buffer containing Protease and PhosStop inhibitors (Roche) and the clarified Benzonase- (Novagen) treated lysate then subjected to SDS-PAGE. Western blot analysis was performed using a rabbit antiserum to CTNNBL13 or using antibodies to AID (mAb 94.16; gift from Hans-Martin Jäck), CDK6 (antibody DCS-83; Invitrogen) or Rb and cyclin D3 (both rabbit antisera from Santa Cruz Biotechnology), detecting with HRP-conjugated anti-mouse (Abcam) or anti-rabbit Ig with ECLplus (Amersham GE Healthcare). Loading controls were provided by staining with rabbit antisera to α-tubulin and lamin-B (Abcam).
RNA-Seq analysis
Follicular B cells were obtained by cell sorting from three control and three CTNNBL1-deficient mice and total RNA prepared from independent 8 × 106 cell samples obtained both pre-activation (0 h) and following 12 h of LPS+IL4 stimulation. RNA was quantified by RT-qPCR for expression of β2 min and validated for efficient deletion of Ctnnbl1 exon 3. The three biological replica RNA samples (for each time point and genotype) were pooled in equimolar amounts, treated with Ribo-Zero rRNA removal kit (Epicenter) and RNaseq libraries prepared according to the TrueSeq RNA sample preparation protocol without the mRNA purification step (Illumina). Sequencing of the barcoded multiplexed libraries and base calling was performed on a GA II by the CRUK CRI (Cambridge) service. RNA-Seq data was mapped to the mouse NCBIM37 genome assembly using TopHat (v1.4.1, options −g 1) in conjunction with gene models from Ensembl release 61. Mapped RNaseq data was compared and visualized using SeqMonk software.
For expression analysis, each data set was quantitated by counting the number of reads per transcript, corrected per million reads for all protein-coding transcripts. Differential expression was called on log2 transformed counts by selecting transcripts which changed with a significance of p < 0.05 after Benjamini and Hochberg correction using a null model constructed from the 1% of transcripts showing the closest average level of observation to estimate experimental noise.
For splicing efficiency analysis, merged sets of exonic regions were created from the combined transcripts of each gene. Read density (reads per kilobase) was then calculated for the exonic and intronic parts of each gene and the log2 ratio of the exonic to intronic densities calculated for each gene.
Yeast strains and methods
The yeast targeting construct pFA6a-KanMX622 was amplified with 100 nt primers homologous to the SPAC1952.06c locus (FW: 5′-TGGAGGCCAACGACTCTTACTACAGAATTGACGAAAAACCTATGGTTACTGTTTTACTTGGATCTATGCAAGCATCTGTGCGGATCCCCGGGTTAATTAA; REV: 5′-AAACACTCTTTATGTTCCATAATAACAAATGTACAACTCATTAAATTGAAAATATGTTATCAGAACAAGTCATGATTTTAGAATTCGAGCTCGTTTAAAC). This PCR product was transfected into S. pombe and selected on plates containing G418. Integration was confirmed by PCR on both left (forward: CGGAGGAAAAGAGTACCAAATG; reverse: CGGATGTGATGTGAGAACTG) and right (forward: CGCTATACTGCTGTCGATTCG; reverse: TCGAAACGTCTATTCCGAAGTT) sides of the insert. Primer design, transfection using lithium acetate and the construction of yeast carrying a SPAC1952.06c locus in which 3HA-tagged CTNNBL1 was expressed under the control of the strong nmt1 promoter were performed as described by Bähler et al.22 The effects of CTNNBL1 deficiency on cell growth was assessed by comparing isogenic strains in a h− 972 (prototrophic wild type) background. Cells in log phase growth in YE-rich medium were transferred to EMM-nitrogen at an OD600 of 0.8. After 3 weeks of nitrogen starvation, cells were released into YE and growth monitored over time.
Supplementary Material
Acknowledgments
We thank members of the LMB biomedical services for help with animal husbandry, Jernej Ule and Jan Attig for advice on preparing RNaseq libraries, Andrew Deonarine and Melis Kayikci for help with RNaseq analysis, Maria Daly for assistance with flow cytometry and Bill Skarnes for advice on the EUCOMM targeting constructs. We also thank Karuna Ganesh and Felix Dingler for helpful discussions. We are grateful to the Leukaemia and Lymphoma Research Fund for support of A.C. and the Netherlands Organisation for Scientific Research (NWO Rubicon) for support of F.v.M. This work was funded by the Medical Research Council (file reference number U105178806).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23594
References
- 1.Jabbour L, Welter JF, Kollar J, Hering TM. Sequence, gene structure, and expression pattern of CTNNBL1, a minor-class intron-containing gene--evidence for a role in apoptosis. Genomics. 2003;81:292–303. doi: 10.1016/S0888-7543(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 2.Makarova OV, Makarov EM, Urlaub H, Will CL, Gentzel M, Wilm M, et al. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004;23:2381–91. doi: 10.1038/sj.emboj.7600241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–84. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Grote M, Wolf E, Will CL, Lemm I, Agafonov DE, Schomburg A, et al. Molecular architecture of the human Prp19/CDC5L complex. Mol Cell Biol. 2010;30:2105–19. doi: 10.1128/MCB.01505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganesh K, Adam S, Taylor B, Simpson P, Rada C, Neuberger M. CTNNBL1 is a novel nuclear localization sequence-binding protein that recognizes RNA-splicing factors CDC5L and Prp31. J Biol Chem. 2011;286:17091–102. doi: 10.1074/jbc.M110.208769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han L, Masani S, Yu K. Cutting edge: CTNNBL1 is dispensable for Ig class switch recombination. J Immunol. 2010;185:1379–81. doi: 10.4049/jimmunol.1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopes-Carvalho T, Kearney JF. Development and selection of marginal zone B cells. Immunol Rev. 2004;197:192–205. doi: 10.1111/j.0105-2896.2004.0112.x. [DOI] [PubMed] [Google Scholar]
- 8.Lopes-Carvalho T, Foote J, Kearney JF. Marginal zone B cells in lymphocyte activation and regulation. Curr Opin Immunol. 2005;17:244–50. doi: 10.1016/j.coi.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–54. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 10.Sajiki K, Hatanaka M, Nakamura T, Takeda K, Shimanuki M, Yoshida T, et al. Genetic control of cellular quiescence in S. pombe. J Cell Sci. 2009;122:1418–29. doi: 10.1242/jcs.046466. [DOI] [PubMed] [Google Scholar]
- 11.Yanagida M. Cellular quiescence: are controlling genes conserved? Trends Cell Biol. 2009;19:705–15. doi: 10.1016/j.tcb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Ohi R, McCollum D, Hirani B, Den Haese GJ, Zhang X, Burke JD, et al. The Schizosaccharomyces pombe cdc5+ gene encodes an essential protein with homology to c-Myb. EMBO J. 1994;13:471–83. doi: 10.1002/j.1460-2075.1994.tb06282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potashkin J, Kim D, Fons M, Humphrey T, Frendewey D. Cell-division-cycle defects associated with fission yeast pre-mRNA splicing mutants. Curr Genet. 1998;34:153–63. doi: 10.1007/s002940050381. [DOI] [PubMed] [Google Scholar]
- 14.Kuhn AN, Käufer NF. Pre-mRNA splicing in Schizosaccharomyces pombe: regulatory role of a kinase conserved from fission yeast to mammals. Curr Genet. 2003;42:241–51. doi: 10.1007/s00294-002-0355-2. [DOI] [PubMed] [Google Scholar]
- 15.Shimada M, Namikawa-Yamada C, Nakanishi M, Murakami H. Regulation of Cdc2p and Cdc13p is required for cell cycle arrest induced by defective RNA splicing in fission yeast. J Biol Chem. 2005;280:32640–8. doi: 10.1074/jbc.M504746200. [DOI] [PubMed] [Google Scholar]
- 16.Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2004;68:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Padilla PA, Ladage ML. Suspended animation, diapause and quiescence: arresting the cell cycle in C. elegans. Cell Cycle. 2012;11:1672–9. doi: 10.4161/cc.19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24:380–6. doi: 10.1016/S1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–40. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 20.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A. 2006;103:13789–94. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–8. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–51. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 23.Su SS, Tanaka Y, Samejima I, Tanaka K, Yanagida M. A nitrogen starvation-induced dormant G0 state in fission yeast: the establishment from uncommitted G1 state and its delay for return to proliferation. J Cell Sci. 1996;109:1347–57. doi: 10.1242/jcs.109.6.1347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.