

Abstract
Fractionated radiotherapy (RT) is widely used in cancer treatment, because it preserves normal tissues. However, repopulation of radioresistant tumors during fractionated RT limits the efficacy of RT. We recently demonstrated that a moderate level of long-term fractionated radiation confers acquired radioresistance to tumor cells, which is caused by DNA-PK/AKT/GSK3β-mediated cyclin D1 overexpression. The resulting cyclin D1 overexpression leads to forced progression of the cell cycle to S-phase, concomitant with induction of DNA double-strand breaks (DSBs). In this study, we investigated the molecular mechanisms underlying cyclin D1 overexpression-induced DSBs during DNA replication in acquired radioresistant cells. DNA fiber data demonstrated that replication forks progressed slowly in acquired radioresistant cells compared with corresponding parental cells in HepG2 and HeLa cell lines. Slowly progressing replication forks were also observed in HepG2 and HeLa cells that overexpressed a nondegradable cyclin D1 mutant. We also found that knockdown of Mus81endonuclease, which is responsible for resolving aberrant replication forks, suppressed DSB formation in acquired radioresistant cells. Consequently, Mus81 created DSBs to remove aberrant replication forks in response to replication perturbation triggered by cyclin D1 overexpression. After treating cells with a specific inhibitor for DNA-PK or ATM, apoptosis rates increased in acquired radioresistant cells but not in parental cells by inhibiting the DNA damage response to cyclin D1-mediated DSBs. This suggested that these inhibitors might eradicate acquired radioresistant cells and improve fractionated RT outcomes.
Keywords: cyclin D1, DSBs, Mus81, Perturbation of DNA replication, radioresistance
Introduction
The most severe form of DNA damage induced by ionizing radiation is DNA double-strand breaks (DSBs), which can trigger chromosomal aberrations such as deletions, insertions and translocations. A series of DNA damage responses (DDRs) are induced in eukaryotic cells after irradiation to maintain genomic stability. Cell cycle checkpoints are activated after irradiation resulting in blockage of cell cycle progression to achieve proper repair of DNA damage.1 Cell death is induced in order to exclude abnormal cells in response to high doses of irradiation.2 The molecular mechanisms involved in DDR have been well studied using single radiation (SR) exposure regimes; however, DDRs after multiple fractionated radiation (FR) exposure regime remain to be elucidated.
It is well known that cyclin D1 is degraded following SR exposure, which arrests cells at the G1/S boundary as a G1/S checkpoint.3 Conversely, cyclin D1 is stabilized in human tumor cells after exposure to FR of X-ray at 0.5 Gy twice per day for 1 mo. This exposure regime confers acquired radioresistance to tumor cells.4 By binding to Cdk4, cyclin D1 becomes an important regulator of cell cycle progression at the G1/S transition. Cyclin D1-Cdk4 phosphorylates Rb, after which E2F is released to transactivate genes required for G1- to S-phase progression.5,6 Overexpression of cyclin D1/Cdk4 prevents FGF-mediated growth arrest by inhibiting downregulation of cyclin E/Cdk2 activity.7, 8 In addition to its role in activating Cdk4, cyclin D1 controls transcription of several genes in a Cdk-independent manner.9, 10
The cyclin D1 level is tightly controlled for normal cell cycle progression, and its deregulation is linked to the development of cancer.11-13 Cyclin D1 is implicated in induction of chromosomal instability in mammary gland tumors.14 Abundance of cyclin D1 is also associated with cellular senescence in response to replicative stress.15 Cyclin D1 accumulates during G1-phase progression and is degraded during the S-phase.16 During cell cycling, cyclin D1 expression is regulated both at the transcriptional and post-translational levels. Cyclin D1 expression is regulated by mitogenic signaling through small guanosine triphosphate-binding proteins such as Ras.17
Glycogen synthase kinase 3beta (GSK3β) is a protein kinase that phosphorylates cyclin D1 on threonine286 (Thr286) to facilitate its degradation. AKT-mediated phosphorylation of GSK3β on serine 9 decreases its kinase activity on cyclin D1 Thr286, which inhibits nuclear export and cytoplasmic proteasomal degradation of cyclin D1.18,19 Thus, AKT positively regulates G1/S cell cycle progression by inactivating GSK3β, which results in cyclin D1 accumulation in the nucleus. We previously reported that long-term FR-induced cyclin D1 overexpression was due to downregulation of cyclin D1 proteolysis via the activation of the DNA-PK/AKT/GSK3β pathway.4,20
Oncogene activation perturbs DNA replication and induces both DSBs and DDRs in nonmalignant cells during tumorigenesis.21-23 Overexpression of cell cycle regulators such as cyclin D1, cyclin A and cyclin E induces DSBs and DNA damage checkpoints in human and mouse fibroblasts.24-26 We recently reported that persistent cyclin D1 expression during S-phase induces DSBs in acquired radioresistant cells.4 However, the molecular mechanisms underlying cyclin D1-mediated DSBs during DNA replication have not been completely characterized.
In this study, we investigated the effect of cyclin D1 overexpression on DNA replication in acquired radioresistant cells. We found that Mus81 created DSBs in response to aberrant replication forks that were induced by cyclin D1 overexpression. These DSBs were efficiently repaired, because acquired radioresistant cells continued to grow without any remarkable delay. We also demonstrated that either a DNA-PK inhibitor or an ATM inhibitor could induce cell death in acquired radioresistant cells. Thus, we provide evidence for a new strategy to suppress tumor radioresistance by targeting DDRs in response to cyclin D1-mediated DSBs in acquired radioresistant cells.
Results
Cyclin D1-mediated DSBs in acquired radioresistant 31FR-31NR cells
We previously established acquired radioresistant 31FR-31NR cells by 31-d FR exposure followed by 31-d non-FR (Fig. 1). This radioresistant phenotype was irreversible without FR exposure for > 1 mo; therefore, we termed it “acquired radioresistance.”4 In this study, we used the neutral comet assays to quantify the DSB levels in 31FR-31NR cells derived from HepG2 and HeLa cell lines (Fig. 2A). A higher tail moment value in 31FR-31NR cells compared with the corresponding parental (0FR) cells indicated that acquired radioresistant cells harbored large amounts of DSBs (Fig. 2A).
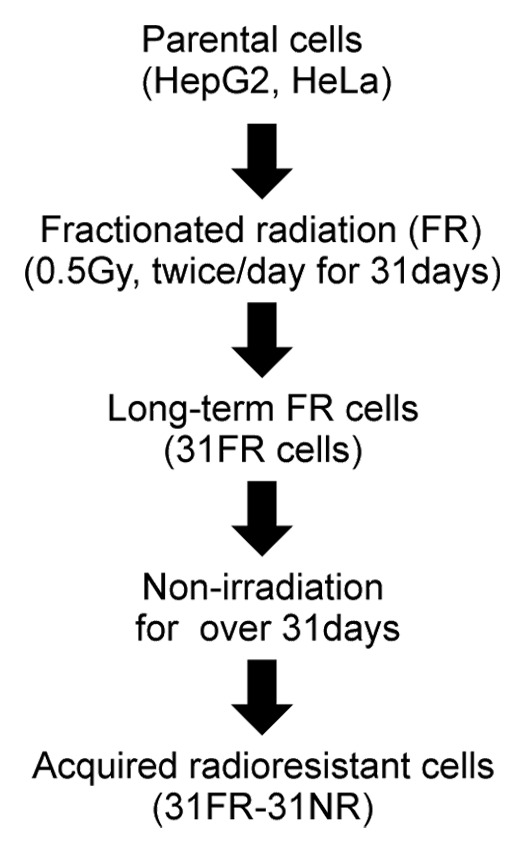
Figure 1. A flowchart describing the derivation of acquired radioresistant 31FR-31NR cells. Cells were exposed to FR consisting of a 0.5 Gy X-ray fraction dose at every 12 h, 6 d a week. The cells treated with this exposure scenario with 62 fractions for 31 d were referred to as 31FR cells. The 31FR cells were further cultured without irradiation for more than 31 d and the resulting cells were designated as 31FR-31NR cells.

Figure 2. Results for neutral comet assays. (A) Distributions of tail moment values for 0FR and 31FR-31NR cells. Results for HepG2 and HeLa cells are shown in the left and right panels, respectively. The median tail moment values are indicated at the bottom of the graph. (B) Distributions of tail moment values for 0FR and 31FR-31NR cells derived from the HeLa cell line. Samples were prepared at 48 h after transfection with control siRNA (siControl) or cyclin D1 siRNA (siCyclin D1). Median tail moment values are indicated at the bottom of the graph.
In a prior study, we found that cyclin D1 was overexpressed in 31FR-31NR cells derived from HepG2 and HeLa cell lines.4 We examined whether the amount of DSBs decreased after repressing cyclin D1 gene expression using siRNA (Fig. 2B). Knock down of cyclin D1 by using cyclin D1 siRNA was confirmed by western blot analyses in HeLa cells (Fig. 2C). Distributions of tail moment values were the same in HeLa 0FR cells with both control siRNA and cyclin D1 siRNA. Thus, cyclin D1 siRNA did not affect the amount of DSBs in parental 0FR cells. In contrast, transfection with cyclin D1 siRNA clearly decreased the tail moment value in HeLa 31FR-31NR cells compared with HeLa cells transfected with control siRNA. These results demonstrated that cyclin D1 overexpression induced DSBs in 31FR-31NR cells.
Cdk4-independent DSBs formation in 31FR-31NR cells
Cyclin D1-mediated DSBs may affect cell cycle progression in 31FR-31NR cells. However, the percentage of BrdU-positive S-phase 31FR-31NR cells with control siRNA was the same as 0FR cells with control siRNA. Thus, 31FR-31NR cells continued to grow without any remarkable delay compared with 0FR cells (Fig. 3A).

Figure 3. Cdk4-independent DSBs formation in 31FR-31NR cells (A) Cell cycle distributions for HeLa cells with either control siRNA (siControl) or Cdk4 siRNA (siCdk4). Percentages of G1-, G2/M- and S-phase cells are shown with the standard deviations in parentheses in the lower panel. (B) Western blotting results for Cdk4, γ-H2AX and actin in 0FR and 31FR-31NR cells derived from HeLa cell line. Cell extracts were prepared at 48 h after transfection with either control siRNA or Cdk4 siRNA. (C) Western blotting results for γ-H2AX and actin in the 0FR and 31FR-31NR cells derived from HeLa cell line. The cells were treated with 1.9 μM Cdk4-I for 24 h.
Cyclin D1 is a regulator of Cdk4 and Cdk6 during the G1/S transition of the cell cycle. We investigated whether Cdk4 was required for cyclin D1-mediated DSB formation in 31FR-31NR cells. In order to inactivate Cdk4, we used Cdk4 siRNA or a Cdk4 inhibitor (Cdk4-I). As we reported previously, 1.9 μM of Cdk4-I could suppress cyclin D1/Cdk4-dependent phosphorylation of Rb at Serine 795 in 0FR and 31FR-31NR cells of HeLa.4 In HeLa cells, human papillomavirus E7 disrupts the formation of RB-E2F complexes, which results in increased expression levels of E2F-responsive genes. Therefore, Cdk4 inactivation did not affect the G1/S transition in 0FR and 31FR-31NR cells derived from HeLa cells (Fig. 3A). The amounts of γ-H2AX did not decrease after treatment with either Cdk4 siRNA or a Cdk4-I in 31FR-31NR cells (Fig. 3B and C). These results demonstrated that cyclin D1/Cdk4 activity was unnecessary for DSB formation in 31FR-31NR cells.
Slowing down of replication fork progression due to cyclin D1 overexpression in 31FR-31NR cells
Persistent cyclin D1 expression during S-phase may perturb DNA replication in 31FR-31NR cells. We used the DNA fiber assay to determine if cyclin D1 affected the elongation stages of DNA replication. The cells were first pulse-labeled with 5-iodo-2′-deoxyuridine (IdU; detected by Cy3, red signal) and subsequently labeled with 5-chloro-2′-deoxyuridine (CldU; detected by Alexa 488, green signal). Replication fork elongation was detected as unidirectional red-green tracks (R–G, Fig. 4A). Short-length R–G tracks were evident in 31FR-31NR cells but not in 0FR cells (indicated by the arrowhead in Fig. 4A). Lengths of the green-labeled tracks in the R–G tracks were measured in 50 DNA tracks. The average replicating DNA tracks with the standard deviations was shown in Figure 4B and C. The lengths of replicating DNA in the 31FR-31NR cells were shorter than those in the 0FR cells derived from the HepG2 and HeLa cell lines (Fig. 4B). These results indicated that replication forks in 31FR-31NR cells progressed more slowly than those in 0FR cells.

Figure 4. Cyclin D1-mediated slowing down of replication forks. (A) Image of red- and green-labeled DNA tracks. Replication of DNA in 0FR cells is shown by the arrow. Short-labeled DNA tracks in 31FR-31NR cells are shown by arrowheads. (B) Histograms of labeled DNA lengths in 0FR and 31FR-31NR cells. Results for HepG2 and HeLa cells are shown in the left and right panels, respectively. The average lengths are indicated at the bottom of the graph. (C) Histograms of labeled DNA lengths in the HepG2 and HeLa cells that expressed wild-type cyclin D1(CD1-WT) or cyclin D1 mutants (CD1-T286A).
We have made cells overexpressing wild-type cyclin D1 (CD1-WT) and a nondegradable cyclin D1 mutant (CD1-T286A), mutated at the phosphorylation on Thr286, and confirmed their expression by western blotting in a prior study.4 Enforced expression of a CD1-T286A but not CD1-WT resulted in its overexpression. Short replicating DNA tracks increased by CD1-T286A overexpression in HepG2 and HeLa cells compared with parental cells (Fig. 4C). Thus, cyclin D1 overexpression disrupted DNA replication by suppressing replication fork progression.
Mus81-mediated DSBs in 31FR-31NR cells
Mus81 cleaves an aberrant fork structure to generate DSB, which removes a stalled fork from replication sites.27-29 Therefore, DSBs may be generated by Mus81 in response to replication perturbations caused by cyclin D1 overexpression. Mus81 expression was suppressed by its siRNA but not by random control siRNA in 0FR and 31FR-31NR cells (Fig. 5A). Mus81 knockdown decreased γ-H2AX signals in 31FR-31NR cells compared with the control siRNA cells. We further performed immunostaining of cyclin D1 and γ-H2AX in 0FR and 31FR-31NR cells derived from HeLa cells. γ-H2AX was observed in cyclin D1-positive 31FR-31NR cells as indicated by arrow on the lower panel in Figure 5B. Upon Mus81 depletion by using Mus81 siRNA, double-positive cells with γ-H2AX and cyclin D1 were disappeared in 31FR-31NR cells (Fig. 5B). These results demonstrated that cyclin D1-dependent DSBs were created by Mus81 endonuclease in response to aberrant replication forks triggered by cyclin D1 overexpression in 31FR-31NR cells.

Figure 5. Mus81-mediated DSBs in 31FR-31NR cells. (A) Western blotting results for Mus81, γ-H2AX and actin in 0FR and 31FR-31NR cells derived from HepG2 cell line. Cell extracts were prepared at 24 h after transfection with either control siRNA (Cont.) or Mus81 siRNA. The amounts of γ-H2AX were normalized by corresponding actin level. The values are expressed relative to the control value of 0FR cells with control siRNA. (B) Cyclin D1 (green) and γ-H2AX (red) in 0FR cells and 31FR-31NR cells of HeLa are shown. Immunofluoresence were performed 48 h after transfection with control siRNA or Mus81 siRNA. Double-staining cells with cyclin D1 and γ-H2AX are indicated by arrows.
Eradication of 31FR-31NR cells by inhibition of DNA-PK and ATM
We next investigated DDR in 31FR-31NR cells in response to cyclin D1-mediated DSBs. Activation of the DNA damage-activated serine/threonine protein kinases DNA-PK and ATM was examined in 31FR-31NR cells using an anti-phospho-DNA-PKcs-Thr2609 and anti-phospho-ATM-Ser1981 antibody, respectively. In order to identify cells in S phase, cells were stained with PCNA, which is a replication fork processivity protein at the ongoing replication fork during S phase. DNA-PK and ATM phosphorylation was not observed in the control 0FR cells but was observed in the PCNA-positive 31FR-31NR cells (Fig. 6A). These results indicated that cyclin D1-mediated DSBs activated DNA-PK and ATM during S-phase in 31FR-31NR cells.

Figure 6. DDR and cell death in acquired radioresistant cells. (A) Double immunostaining with p-DNA-PK and PCNA in HepG2 cells is shown in the upper panel. Double immunostaining with p-ATM and PCNA in HeLa cells is shown in the lower panel. (B) Colony survival of HeLa cells treated with 10 μM NU7026 or 1 μM KU55933. Asterisk indicates significant sensitivity to drugs by 31FR-31NR cells compared with 0FR cells (C) Percentage of annexin V-positive HeLa cells. The cells were treated with either NU7026 or KU55933. Asterisk indicates a significant difference in the frequency of apoptotic FR cells compared with 0FR cells.
We expected DNA-PK and ATM inactivation to prevent DNA repair of cyclin D1-mediated DSBs and induce 31FR-31NR cell death. Treatment with either a DNA-PK inhibitor NU7026 or an ATM inhibitor KU55236 did not affect 0FR cell survival; however, these inhibitors reduced the overall survival of the 31FR-31NR cells with cyclin D1-mediated DSBs (Fig. 5B). We also examined HeLa cell apoptosis using annexin V staining. After treatment with either the DNA-PK inhibitor or the ATM inhibitor, the proportions of the apoptotic 31FR-31NR cells increased, but that of the parental 0FR cells did not change. Thus, both these inhibitors could induce cell death only in 31FR-31NR cells with cyclin D1-mediated DSBs.
Discussion
Cyclin D1-mediated DSBs following long-term FR
We have been investigating the biological effects of long-term FR-induced cyclin D1 overexpression in human tumor cell lines. In response to long-term FR with a moderate dose, cyclin D1 behavior is completely different from that observed after SR with high doses. Cyclin D1 is degraded following SR and causes G1/S arrest through inactivating Cdk4, while cyclin D1 is stabilized by downregulating its proteolysis via the DNA-PK/AKT/GSK3β pathway in response to long-term FR.
Long-term FR-induced cyclin D1 overexpression is mediated by its protein expression level and not by its mRNA expression level.4 This epigenetic change in DNA-PK/AKT/GSK3β-mediated cyclin D1 overexpression is long-lasting, even after discontinuing FR for over 1 mo. In this study, we investigated the level of cyclin D1-mediated DSBs in 31FR-31NR cells using the neutral comet assay. Some of 31FR-31NR cells contained large amounts of DSBs, as shown by their high tail moment values compared with the values of the parental 0FR cells. Thus, DSBs are generated only at a specific stage of the cell cycle.
We previously reported that γ-H2AX-positive 31FR-31NR cells were also positive for IdU.4 In this study, we showed that activation of the DNA damage sensor kinases DNA-PK and ATM was observed in PCNA-positive 31FR-31NR cells. Thus, DSBs were generated during DNA replication and activated DNA damage signaling pathways in 31FR-31NR cells. Cyclin D1 knockdown by siRNA decreased the amounts of DSBs in 31FR-31NR cells, whereas inactivation of Cdk4 by either siRNA or Cdk4-I had no effect. Therefore, DSB formation was mediated by cyclin D1 itself but not by cyclin D1/Cdk4 in 31FR-31NR cells.
Cyclin D1 overexpression suppresses replication fork progression
It has been reported that the accumulation of cyclin D1 in the nucleus loads replicative MCM helicase onto the chromatin and triggers DNA re-replication, which is required for Cdk4 activity.24 Our results revealed that Cdk4 inactivation did not affect the amount of DSBs in the 31FR-31NR cells. Thus, cyclin D1-mediated DSBs are not induced by DNA re-replication in 31FR-31NR cells.
We also found that cyclin D1 overexpression perturbed DNA replication by downregulating replication fork progression. Cyclin D1 is associated with the replication factor PCNA, a clamp loader for DNA polymerase.30-32 Thus, PCNA may recruit cyclin D1 to replication forks and the interaction between cyclin D1 and PCNA may prevent replication fork movement in 31FR-31NR cells. However, further studies will be needed to determine the molecular mechanisms underlying slowing down on replication fork progression triggered by cyclin D1 overexpression.
Replication-associated DSBs are created by Mus81 endonuclease to remove stalled replication forks
Oncogene-induced replication stress is associated with induction of genomic instability and acceleration of tumor progression.21,22 The structure-specific Mus81/Eme1 complex is produced in response to this type of replication perturbation to control genomic stability during DNA replication.23 Mus81 creates DSBs by cleaving DNA to resolve stalled replication forks that are induced by treatments with DNA synthesis inhibitors such as aphidicolin and hydroxyurea.27-29 Our present results showed that Mus81 created DSBs in response to cyclin D1-mediated slowing down of replication forks in 31FR-31NR cells.
DSBs induced by radiation spread randomly over an entire genome and subsequently activate DNA damage signals involving DNA damage sensor kinases such as ATM and DNA-PK. In contrast, Mus81 cleaves aberrant fork structures to generate one-sided DSBs only at the sites of stalled replication forks.21-23 Mus81-induced DSBs are thought to be repaired more easily than radiation-induced DNA damage, because these DSBs are created during homologous recombination repair (HRR).27,29 Thus, cyclin D1-mediated DSBs are efficiently repaired in 31FR-31NR cells to promote the growth of these cells.
Eradication of acquired radioresistant cells by ATM and DNA-PK inhibition
Tumor radioresistance is one of the major obstacles in accomplishing complete cure of cancer with fractionated radiotherapy (RT).33,34 Thus, it is important to identify molecular targets to suppress tumor radioresistance during cancer treatment. Our findings indicated that treatment with either a DNA-PK inhibitor or an ATM inhibitor efficiently induces apoptosis of acquired radioresistant cells harboring cyclin D1-mediated DSBs but does not affect apoptosis of parental cells. Thus, both of these inhibitors may affect acquired radioresistant cells without harmful side effects to normal cells.
In conclusion, this is the first study to demonstrate that cyclin D1 overexpression perturbs DNA replication by suppressing replication fork progression. DSBs could be induced by Mus81 for the recovery of cyclin D1-mediated slowing down of replication forks. The combination of fractionated RT with a DNA-PK inhibitor or an ATM inhibitor can suppress tumor radioresistance by eradicating acquired radioresistant cells and may improve outcomes with fractionated RT.
Materials and Methods
Cell culture condition and drugs
The human liver cancer cell line HepG2 and the human cervical cancer cell line HeLa were obtained from the Cell Resource Center for Biomedical Research (IDAC, Tohoku University). Cells were grown in RPMI 1640 medium (NacalaiTesque) supplemented with 5% heat-inactivated fetal calf serum. A Cdk4 inhibitor,35 a DNA-PK inhibitor (NU7026) and an ATM inhibitor (KU55933) were purchased from Calbiochem. pFlex-cyclin D1 vectors36 were introduced into HepG2 and HeLa cells, as described previously.4
Neutral comet assay
The neutral comet assays were performed using CometAssay kits (Trevigen) following the manufacturer’s protocol, as described previously.37 Images were captured with a CCD camera attached to a fluorescence microscope. The tail moment was determined by multiplying the fraction of DNA in the tail by the length of the tail.
RNA interference
Cells were transfected with siRNA using Lipofectamine RNAiMAX reagent (Invitrogen). Cyclin D1 and control siRNAs were purchased from Santa Cruz Biotechnology. Mus81 siRNA was purchased from Invitrogen. Cells were incubated with 40 nM of these siRNAs for 24 h. The medium was then removed and replaced with fresh medium for another 24 h. Neutral comet assay, cell cycle analysis, western blotting and immunofluoresence staining were performed 48 h after siRNA transfection.
Cell cycle analysis
Cell cycle analysis was performed as described previously.37 The cells were pulse-labeled with 20 μM bromodeoxyuridine (BrdU) for 1 h, washed with PBS and then fixed in 70% ethanol overnight. BrdU-positive cells were quantified by a FACScan (Cytomics FC500, Becton Dickinson).
Western blot analyses
Western blotting was performed as described previously.38 Histone extracts were prepared as described by Tung and Winn.39 Proteins were separated by sodium-lauryl-sulfate-PAGE and transferred electrophoretically to PVDF membranes (Bio-Rad). The membranes were blocked with 5% (w/v) phospho-blocker (Cell Biolabs, Inc.) for 1 h and incubated with each primary antibody, including anti-β-actin (Sigma, A2066), anti-Cdk4 (Santa Cruz Biotechnology, SC-260), anti-cyclin D1 (Nichirei Bioscience) and anti-γ-H2AX (Upstate), either for 1 h at room temperature or overnight at 4°C. The membranes were then incubated for 1 h at room temperature with either HRP-conjugated goat anti-rabbit IgG (Nichirei Bioscience) or HRP-conjugated goat anti-mouse IgG (R&D Systems). Protein bands were visualized with Chemi-Lumi One L western blotting substrate (NacalaiTesquea). Band intensity was measured by densitometry using Image Lab Software (Bio-Rad).
Immunofluorescence staining
Immunofluorescence staining was performed as described previously.28 Cells were seeded onto coverslips placed in 10 mm tissue culture dishes. The coverslips were fixed with ice-cold acetone (5 min), ice-cold methanol (5 min) and then washed twice with PBS. The cells were permeabilized and blocked for 30 min at room temperature in 5% bovine serum albumin (BSA) and 0.1% Triton X-100 in PBS. Anti-PCNA antibody (Mouse IgG, PC10; Oncogen) (Rabbit IgG, SC-7007; Santa Cruz), anti-ATM-phosphoserine 1981 (Rockland), anti-DNA-PKcs-phosphothreonine 2609 (Thermo Scientific), anti-cyclin D1 (Nichirei) and anti-γ-H2AX (Trevigen) were diluted in PBS with 0.5% BSA and incubated with the coverslips for 1 h. The coverslips were then washed three times with 0.1% Triton X-100 in PBS, incubated for 1 h with secondary antibodies conjugated with Alex 488 (Molecular Probes for mouse IgG) or Cy-3 (Jackson Immuno Research Laboratories, Inc. for mouse IgG). The coverslips were washed three times with 0.1% Triton X-100 in PBS, counterstained for DNA with 4, 6-diamidino-2-phenylindole (DAPI) (4 μg/ml prepared in Vectashield mouting medium; Vector Laboratories). Images were captured with a CCD camera attached to a fluorescence microscope.
DNA fiber analysis
DNA fiber analysis was performed as described previously.38 Cells were labeled with 20 μM IdU for 10 min and then with 20 μM CldU for 20 min. The cells were trypsinized and resuspended in PBS at 1 × 106 cells/ml. The cells (2.5 μl) were then mixed with 7.5 μl lysis buffer (0.5% SDS in 200 mM TRIS-HCl, pH 7.4, 50 mM ethylene diaminetetraacetic acid) on a glass slide. After 8 min, DNA spreads were fixed in 3:1 methanol/acetic acid and then stored in 70% ethanol at 4°C. CldU and IdU staining used a previously described protocol.40 The length of fork extension was studied during second (green CldU, 20 min) labeling period. Signals were measured by using Photoshop software (Adobe Systems). The results of the analyses on 50 tracks are shown.
Clonogenic assay
Cells were treated with NU7026 or KU55933 for 24 h. They were then seeded in 60-mm dishes coated with 0.1% gelatin (Wako) at 1 × 103 cells per dish and incubated for 10 d until colonies were visible. Colonies were fixed with ethanol for 30 min and stained with Giemsa solution (Merck and Co., Inc.). Colonies with > 50 cells were counted under a light microscope (Olympus, SZX10).
Annexin V staining
Apoptotic cells were identified and quantified using the annexin V-FITC apoptosis detection kit (Bio Vision) following the manufacturer’s protocol. Cells were stained with annexin V-FITC and propidium iodide at 72 h after treatment with NU7026 or KU55933. Annexin V-positive apoptotic cells were analyzed by FACScan (Becton Dickinson).
Statistical analysis
All experiments were repeated at least three times with independent samples. Results are given as means + standard deviations. Group comparisons were made by Student’s t-test. A single asterisk and double asterisks indicate significance at p < 0.01 and p < 0.05, respectively.
Acknowledgments
This study was supported in part by research grants from the Japanese Ministry of Education and Science Kiban C (24510063), the Mishima Kaiun Memorial Foundation of Japan and grant for Cancer Research from National Cancer Institute (Japan).
Glossary
Abbreviations:
- BrdU
bromodeoxyuridine
- CDK4-I
Cdk4 inhibitor
- CldU
5-chloro-2′-deoxyuridine
- DDR
DNA damage response
- DSBs
double-strand breaks
- FR
fractionated irradiation
- GSK3β
glycogen synthase kinase 3beta
- IF
immunofluorescence
- IdU
5-Iodo-2′-deoxyuridine
- RT
radiotherapy
- SR
single radiation
- Thr286
threonine286
- CD1-WT
wild-type cyclin D1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23719
References
- 1.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–34. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 2.Szumiel I. Ionizing radiation-induced cell death. Int J Radiat Biol. 1994;66:329–41. doi: 10.1080/09553009414551271. [DOI] [PubMed] [Google Scholar]
- 3.Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102:55–66. doi: 10.1016/S0092-8674(00)00010-6. [DOI] [PubMed] [Google Scholar]
- 4.Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y, et al. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene. 2010;29:4826–37. doi: 10.1038/onc.2010.238. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 6.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 7.Kolupaeva V, Basilico C. Overexpression of cyclin E/CDK2 complexes overcomes FGF-induced cell cycle arrest in the presence of hypophosphorylated Rb proteins. Cell Cycle. 2012;11:2557–66. doi: 10.4161/cc.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolupaeva V, Laplantine E, Basilico C. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One. 2008;3:e3447. doi: 10.1371/journal.pone.0003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanse EA, Mashek DG, Becker JR, Solmonson AD, Mullany LK, Mashek MT, et al. Cyclin D1 inhibits hepatic lipogenesis via repression of carbohydrate response element binding protein and hepatocyte nuclear factor 4α. Cell Cycle. 2012;11:2681–90. doi: 10.4161/cc.21019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sladek FM. The yin and yang of proliferation and differentiation: cyclin D1 inhibits differentiation factors ChREBP and HNF4α. Cell Cycle. 2012;11:3156–7. doi: 10.4161/cc.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartkova J, Lukas J, Strauss M, Bartek J. The PRAD-1/cyclin D1 oncogene product accumulates aberrantly in a subset of colorectal carcinomas. Int J Cancer. 1994;58:568–73. doi: 10.1002/ijc.2910580420. [DOI] [PubMed] [Google Scholar]
- 12.Bartkova J, Lukas J, Müller H, Lützhøft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994;57:353–61. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- 13.Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, et al. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92:1316–21. doi: 10.1093/jnci/92.16.1316. [DOI] [PubMed] [Google Scholar]
- 14.Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget. 2012;3:224–5. doi: 10.18632/oncotarget.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11:4642–9. doi: 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol. 1999;9:1075–84. doi: 10.1016/S0960-9822(99)80476-X. [DOI] [PubMed] [Google Scholar]
- 17.Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–33. [PubMed] [Google Scholar]
- 18.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 19.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimura T. Acquired radioresistance of cancer and the AKT/GSK3β/cyclin D1 overexpression cycle. J Radiat Res. 2011;52:539–44. doi: 10.1269/jrr.11098. [DOI] [PubMed] [Google Scholar]
- 21.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 22.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 23.Murfuni I, Nicolai S, Baldari S, Crescenzi M, Bignami M, Franchitto A, et al. The WRN and MUS81 proteins limit cell death and genome instability following oncogene activation. Oncogene. 2012 doi: 10.1038/onc.2012.80. [DOI] [PubMed] [Google Scholar]
- 24.Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21:2908–22. doi: 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tane S, Chibazakura T. Cyclin A overexpression induces chromosomal double-strand breaks in mammalian cells. Cell Cycle. 2009;8:3900–3. doi: 10.4161/cc.8.23.10071. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, et al. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010;70:8802–11. doi: 10.1158/0008-5472.CAN-10-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 28.Shimura T, Torres MJ, Martin MM, Rao VA, Pommier Y, Katsura M, et al. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J Mol Biol. 2008;375:1152–64. doi: 10.1016/j.jmb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regairaz M, Zhang YW, Fu H, Agama KK, Tata N, Agrawal S, et al. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J Cell Biol. 2011;195:739–49. doi: 10.1083/jcb.201104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukami-Kobayashi J, Mitsui Y. Cyclin D1 inhibits cell proliferation through binding to PCNA and cdk2. Exp Cell Res. 1999;246:338–47. doi: 10.1006/excr.1998.4306. [DOI] [PubMed] [Google Scholar]
- 31.Prosperi E, Scovassi AI, Stivala LA, Bianchi L. Proliferating cell nuclear antigen bound to DNA synthesis sites: phosphorylation and association with cyclin D1 and cyclin A. Exp Cell Res. 1994;215:257–62. doi: 10.1006/excr.1994.1341. [DOI] [PubMed] [Google Scholar]
- 32.Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–14. doi: 10.1016/0092-8674(92)90518-H. [DOI] [PubMed] [Google Scholar]
- 33.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516–25. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen GH, Murph MM, Chang JY. Cancer Stem Cell Radioresistance and Enrichment: Where Frontline Radiation Therapy May Fail in Lung and Esophageal Cancers. Cancers (Basel) 2011;3:1232–52. doi: 10.3390/cancers3011232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL-/- cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci U S A. 2008;105:16484–9. doi: 10.1073/pnas.0806574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–72. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 37.Shimura T, Martin MM, Torres MJ, Gu C, Pluth JM, DeBernardi MA, et al. DNA-PK is involved in repairing a transient surge of DNA breaks induced by deceleration of DNA replication. J Mol Biol. 2007;367:665–80. doi: 10.1016/j.jmb.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimura T, Toyoshima M, Adiga SK, Kunoh T, Nagai H, Shimizu N, et al. Suppression of replication fork progression in low-dose-specific p53-dependent S-phase DNA damage checkpoint. Oncogene. 2006;25:5921–32. doi: 10.1038/sj.onc.1209624. [DOI] [PubMed] [Google Scholar]
- 39.Tung EW, Winn LM. Valproic acid-induced DNA damage increases embryonic p27(KIP1) and caspase-3 expression: a mechanism for valproic-acid induced neural tube defects. Reprod Toxicol. 2011;32:255–60. doi: 10.1016/j.reprotox.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 40.Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell. 1999;4:983–93. doi: 10.1016/S1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]