

Abstract
Immortality is a common characteristic of cancers, but its origin and purpose are still unclear. Here we advance a karyotypic theory of immortality based on the theory that carcinogenesis is a form of speciation. Accordingly, cancers are generated from normal cells by random karyotypic rearrangements and selection for cancer-specific reproductive autonomy. Since such rearrangements unbalance long-established mitosis genes, cancer karyotypes vary spontaneously but are stabilized perpetually by clonal selections for autonomy. To test this theory we have analyzed neoplastic clones, presumably immortalized by transfection with overexpressed telomerase or with SV40 tumor virus, for the predicted clonal yet flexible karyotypes. The following results were obtained: (1) All immortal tumorigenic lines from cells transfected with overexpressed telomerase had clonal and flexible karyotypes; (2) Searching for the origin of such karyotypes, we found spontaneously increasing, random aneuploidy in human fibroblasts early after transfection with overexpressed telomerase; (3) Late after transfection, new immortal tumorigenic clones with new clonal and flexible karyotypes were found; (4) Testing immortality of one clone during 848 unselected generations showed the chromosome number was stable, but the copy numbers of 36% of chromosomes drifted ± 1; (5) Independent immortal tumorigenic clones with individual, flexible karyotypes arose after individual latencies; (6) Immortal tumorigenic clones with new flexible karyotypes also arose late from cells of a telomerase-deficient mouse rendered aneuploid by SV40 virus. Because immortality and tumorigenicity: (1) correlated exactly with individual clonal but flexible karyotypes; (2) originated simultaneously with such karyotypes; and (3) arose in the absence of telomerase, we conclude that clonal and flexible karyotypes generate the immortality of cancers.
Keywords: Muller’s ratchet, aneuploidy proximate carcinogen, clonal and flexible cancer karyotypes, growth advantages of aneuploidy, karyotypes of immortal clones of telomerase-deficient mice, karyotypic linkage of immortality and tumorigenicity, long preneoplastic latency, low probability of speciation, selection for cancer-specific autonomy, sub-speciation via karyotypic drift
Introduction
Immortality is a common characteristic of cancers.1-5 But it is still unclear how immortal cancers originate from mortal somatic cells2-15 and why cancers are immortal, although normal somatic cells can grow into organs and organisms which contain many more cells than fatal cancers.5,6 Immortality is operationally defined by growth in excess of the Hayflick limit, which is about 50 generations in vitro.5,16,17
To answer these questions, one would need to know: (1) How cancers are generated from somatic cells, which is also still a matter of debate;5,8,9,11-13,18,19 (2) How cancer cells grow perpetually, despite the inevitable accumulation of spontaneous mutations of genes and chromosomes, termed “Muller’s ratchet.”13,20-26 According to the geneticist Herman Muller, asexual species, such as cancers,11-13 are doomed by extinction unless they have a mechanism to escape the ratchet; and (3) Why cancers are immortal, although immortality “cannot provide an immediate replicative advantage. Unless a cell can tell the future.”6
The currently prevailing immortality theory postulates that cells are immortalized by activation of telomerase.5,7,27-32 Since this enzyme is developmentally switched off in somatic cells, cancers are said to derive immortality from activation of telomerase. According to this theory, “Cells that have stabilized their telomeres through the actions of telomerase or the ALT mechanism proliferate indefinitely and are therefore said to be immortalized. Cell immortalization is a step that appears to govern the development of all human cancers.”5
But, even telomerase genes that are artificially overexpressed by a cytomegalovirus- and a retrovirus-derived promoter29,32-34 are not sufficient, and not even necessary to immortalize cells for the following reasons: mass cultures of “polyclonally”29,34 transfected cells are karyotypically unstable and thus not immortal for many, up to over 100 unstable, generations before they become immortal17,29,31,32,34-43 (see also Results below). Only < 1 in 105 cells of mass cultures transfected with artificially overexpressed telomerase genes (linked also to drug-resistance indicator genes) become clones of immortal cells.17,34,42,44,45 Studying carcinogenesis in telomerase-deficient mice with transgenic oncogenes, Argilla et al. found that, “Absence of telomerase had minimal impact on tumorigenesis…telomere numbers and relative lengths were maintained during progression, implicating a means for preserving telomere repeats and functionality in the absence of telomerase.” A search for these means, “revealed aneuploidy similar to that observed in human tumors.”46 It would thus appear that aneuploidy, rather than overexpressed telomerase, is necessary for immortalization.
Moreover, the telomerase theory does not explain how immortal cancers avoid the inevitably fatal consequences of accumulating spontaneous mutations over time in the non-telomeric sequences of their DNA, i.e., how they escape Muller’s ratchet.13,22-26
In view of these discrepancies with the hypothetical immortalizing function of telomerase, immortalization has been postulated to depend on “additional mutational events,” like the acquisition of an “oncogene,”29,30 undefined “rare events,”47,48“recombination” with unidentified telomere sources,49 “inactivation of tumor suppressors,”39 an “ALT-2” mechanism”46 and “genomic instability.”42 Since there are no consistent answers to these questions, we searched for an alternative theory of immortality.
Karyotypic theory of immortality
Here we advance a new karyotypic theory of immortality, which is based on the theory that carcinogenesis is a form of speciation.10-13,45 The speciation theory holds that cancers are generated by random rearrangements of the karyotypes of normal cells and selection for cancer-specific reproductive autonomy. Since such rearrangements unbalance long-established mitosis genes, cancer karyotypes vary spontaneously but are stabilized perpetually by clonal selections for autonomy. Immortality of cancers is thus a direct consequence of a dynamic equilibrium between spontaneous karyotypic variation and selection for reproductive autonomy—as it is in principle also for sexual species, although at a much lower rate.50-55 This dynamic equilibrium between variation and selection for autonomy also explains the constitutive heterogeneity of cancer karyotypes.56-58 The theory thus predicts that autonomous clonal but flexible karyotypes generate immortality.
To test this theory, we asked whether immortality of human cell lines, currently attributed to transfection with overexpressed telomerase, correlates with clonal flexible karyotypes, and whether immortality originates simultaneously with such karyotypes. Further we asked, whether in the absence of telomerase, SV40 tumor virus induces immortal neoplastic clones with flexible karyotypes in cells of a telomerase-deficient mouse.
Results
Immortal tumorigenic clones derived from human cells transfected with overexpressed telomerase have clonal flexible karyotypes
As a first test of the theory that karyotypic flexibility immortalizes we asked, whether flexible clonal karyotypes are present in three known immortal tumorigenic cell lines named HA1, HA1-2 and BJ.29 These lines arose from mass cultures of primary human kidney (HA1 and HA1-2) and skin cells (BJ), “approximately 60 population doublings” after transfection with artificially overexpressed telomerase and two artificially overexpressed oncogenes.29
Because of these long latent periods we have questioned in the past whether the transfected genes were sufficient for tumorigenicity and immortalization. In a first effort to find an alternative explanation, we looked for cancer-specific aneuploidy, and found that all three of these cell lines were highly aneuploid.59,60 An initial karyotype of HA1, prepared here as described in “Materials and Methods,” confirmed our earlier findings of aneuploidy (Fig. 1).

Figure 1. Karyotype of the immortal tumorigenic HA1 cell line, which originated late after transfection of human kidney cells with overexpressed telomerase. The chromosomes were prepared from metaphases and labeled by hybridization with chromosome-specific colors as described in “Materials and Methods.”
In view of this and our new karyotypic immortality theory, we ask now whether the three cell lines contain the clonal and flexible karyotypes predicted by this theory. For this purpose we used 3-dimensional arrays of 20 karyotypes—a technique, which we had recently developed to detect clonality within the karyotypic heterogeneity of cancers.11,45,61 Such karyotype arrays list chromosome numbers on the x-axis, chromosome copy numbers on the y-axis and the number of metaphases arrayed on the z-axis.
The karyotype arrays of HA1, HA1–2 and BJ, shown in Figure 2 and Table 1, clearly indicate that all three cell lines were clonal: the chromosome numbers of HA1 were 82 ± 1, those of HA1-2 were 47 ± 4, and those of BJ were 82 ± 5, and thus highly clonal. Moreover, the copy numbers of the individual chromosomes of HA1 were 85 to 100% clonal, those of HA1-2 were 85 to 90% clonal, and those BJ were 75 to 100% clonal. As a result of this high clonality, the copy numbers of most chromosomes of the three lines formed parallel lines in the arrays shown Figure 2. By contrast, a minority of chromosome numbers differed from the clonal values within narrow margins of ± 1 and thus stood out above or below the patterns formed by the parallel lines. The relative clonality and non-clonality can thus be seen in each array at a glance. In sum, all three cell lines contained the clonal and flexible karyotypes predicted by our theory.

Figure 2. Three-dimensional arrays of 20 karyotypes of the immortal tumorigenic human cell lines HA1 and HA1-2 and BJ based on the cytogenetic data listed in Table 1. HA1 and HA1-2 lines originated late from human kidney cells (Fig. 1), and BJ from human skin cells about 60 generations after transfection of mass cultures with artificially overexpressed telomerase and two overexpressed oncogenes (text). The karyotype arrays are three-dimensional tables, which show the chromosome numbers of 20 karyotypes on the x-axis, the copy numbers of each chromosome on the y-axis and the number of karyotypes arrayed on the z-axis. By forming parallel lines, the chromosome copy numbers of 20 karyotypes indicate that the three lines are highly clonal. A minority of non-clonal copy numbers differing from the clonal values in narrow margins of ± 1, stands out above or below these lines (Table 1). This indicates that all three lines have clonal but flexible karyotypes.
Table 1. Chromosome numbers and copy numbers of immortal, tumorigenic clones HA1, HA1–2 and BJ.
| Neoplastic clones |
HA1 |
HA1–2 |
BJ |
|---|---|---|---|
| Avg chromo no. ± SD |
82 ± 1 |
47 ± 4 |
82 ± 5 |
| Metaphases (no.) |
20 |
20 |
20 |
| Chromosomes | Copy number (% clonal) | ||
| 1 |
3(50) |
2(90) |
2(85) |
| 2 |
3(80) |
2(90) |
3(75) |
| 3 |
4(95) |
2(90) |
3(90) |
| 4 |
3(100) |
2(90) |
3(90) |
| 5 |
4(95) |
2(95) |
3(95) |
| 6 |
4(95) |
2(90) |
4(80) |
| 7 |
3(85) |
2(90) |
2(85) |
| 8 |
4(85) |
2(85) |
3(95) |
| 9 |
4(100) |
2(85) |
3(90) |
| 10 |
4(95) |
2(95) |
4(95) |
| 11 |
3(95) |
2(80) |
3(95) |
| 12 |
4(90) |
2(85) |
3(80) |
| 13 |
2(90) |
2(85) |
4(75) |
| 14 |
3(95) |
2(85) |
4(85) |
| 15 |
4(85) |
2(95) |
4(90) |
| 16 |
4(90) |
2(90) |
3(80) |
| 17 |
3(90) |
2(95) |
3(85) |
| 18 |
1(85) |
2(90) |
3(90) |
| 19 |
3(100) |
2(95) |
2(90) |
| 20 |
4(90) |
1(95) |
4(95) |
| 21 |
3(95) |
2(90) |
3(100) |
| 22 |
4(90) |
2(90) |
3(90) |
| X |
2(100) |
1(95) |
2(95) |
| Y |
1(100) |
1(15) |
2(95) |
| der(17;19) |
1(100) |
- |
- |
| der(1;19) |
1(90) |
- |
- |
| min(18) |
1(80) |
- |
- |
| der(18) |
1(95) |
- |
- |
| min(8) |
0(60) |
- |
- |
| der(1) |
1(10) |
- |
- |
| der(20;13) |
- |
1(95) |
- |
| der(3;17) |
- |
- |
1(50) |
| der(16;12) |
- |
- |
1(65) |
| der(17;22) |
- |
- |
1(100) |
| der(11;19) |
- |
- |
1(80) |
| der(1;3) |
- |
- |
1(90) |
| der(X;2) |
- |
- |
1(95) |
| der(4;8) |
- |
- |
1(100) |
| der(5;2?;8) |
- |
- |
1(90) |
| der(9;19) |
- |
- |
1(100) |
| der(11;7) |
- |
- |
1(95) |
| der(1;18) | - | - | 1(80) |
Further studies from other researchers including us have confirmed that other immortal tumorigenic lines derived from mass cultures of human cells transfected with artificially overexpressed telomerase, and various oncogenes also contain clonal and flexible karyotypes and also had evolved only late after transfection with overexpressed telomerase and additional oncogenes.42,45,61,62
Conclusions
(1) Based on exact correlations between tumorigenicity and immortality and clonal but flexible karyotypes in all neoplastic clones derived from cells transfected with overexpressed telomerase, we conclude that such karyotypes are necessary, perhaps even sufficient, for immortality.
(2) Based on the clonality of the three tumorigenic and immortal clones and the long pre-neoplastic and pre-immortal latencies of 60 generations from transfection with overexpressed genes, we conclude that tumorigenesis and immortalization are extremely rare and possibly simultaneous clonal events. It follows that overexpressed telomerase and overexpressed oncogenes are not sufficient for immortalization and transformation.
In the following we ask how overexpressed telomerase induces immortal tumorigenic clones, and whether it plays a lasting role in their immortality.
How does overexpressed telomerase induce immortal tumorigenic clones?
The speciation theory predicts that carcinogenesis is initiated by random aneuploidy10-13,45 (Introduction). According to this theory, biological carcinogens like oncogenic viruses and artificially overexpressed cellular genes or non-biological chemical and physical carcinogens, or spontaneous events induce this random aneuploidy.11,45,47,48,61-68 Since aneuploidy unbalances long-established mitosis genes, it self-catalyzes progressions of aneuploidy toward two ends: (1) lethal karyotypes that perish; and (2) new autonomous cancer karyotypes that are stabilized by clonal selection for cancer-specific reproductive autonomy11,45 (Introduction). Aneuploidy would thus be the proximal carcinogen. Because the probability of generating a new autonomous cancer or other species by random karyotypic rearrangements is very low, the latent periods from initiating aneuploidy to the origin of a new immortal species are very long and the resulting species are clonal, just as it was with the three cell lines described above.
To test the predicted role of random aneuploidy in the generation of an immortal neoplastic clone, we karyotyped human skin fibroblasts that had been immortalized by transfection with overexpressed telomerase by Mondello et al. in Pavia, Italy.32,40 Mondello et al. have studied the time course of immortalization of skin fibroblasts from a centenarian woman from transfection with overexpressed telomerase to over 1,000 generations or population doublings (PD) in vitro.32,40 We were particularly interested in this system, because Mondello et al. had already observed aneuploidy before and after immortalization.32,40
Transfection with overexpressed telomerase induces random aneuploidy prior to immortalization
In agreement with Mondello et al.,32,40 we found that after transfection with overexpressed telomerase, the skin cells from a centenarian woman, hence cen3tel, evolved as follows: 37 generations after transfection, at PD37, the cells had a typical fibroblastic, fusiform phenotype (Fig. 3A). Sixty-three generations later, at PD100, the cen3tel cells still had a basic fibroblastic phenotype. However, during that time, the cells had become less fusiform, had acquired a 2–3-fold higher growth rate and grew to a higher density per culture dish than their PD37 precursors (compare Fig. 3A and B).

Figure 3. Cellular phenotypes of human cen3tel skin cells from a centenarian female 37 (A), 100 (B), 166 (C) and 1014 (D) generations after transfection with overexpressed telomerase. The culture had a fibroblastic cellular phenotype at PD37 (A) and a less defined fibroblastic phenotype at generation PD100 (B). At generation PD166, the culture had acquired a polymorphic cellular phenotype typical of cancer cells and simultaneously became tumorigenic and immortal (C). This new polymorphic phenotype was unchanged at PD1014 (D).
The karyotypic analyses of the cen3tel culture at PD37 and PD100 show, in Tables 2 and 3 and Figure 4, that the culture was 60% aneuploid at 37 generations, and was 100% aneuploid at 100 generations after transfection with overexpressed telomerase. Further, while the chromosome copy numbers of aneuploid cells at PD37 were near-diploid, the chromosome copy numbers at PD100 ranged from 1 to 4 and beyond. Thus cen3tel at PD100 differed from PD37 not only in the percentage, but also in the degree of aneuploidy.
Table 2. Karyotypes of 20 cen3tel cells 37 generations after transfection with overexpressed telomerase.
| Metaphases |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome nos. |
47 |
47 |
47 |
47 |
47 |
47 |
47 |
47 |
44 |
45 |
44 |
42 |
44 |
44 |
44 |
46 |
46 |
45 |
27 |
65 |
|
| Chromosomes | near-diploid, native and aneuploid karyotypes | ||||||||||||||||||||
| 1 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
|
| 2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
|
| 3 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
|
| 4 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
3 |
2 |
2 |
2 |
2 |
|
| 5 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
2 |
2 |
2 |
2 |
1 |
2 |
|
| 6 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
1 |
2 |
2 |
2 |
2 |
2 |
4 |
|
| 7 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
2 |
2 |
2 |
2 |
5 |
|
| 8 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
0 |
1 |
|
| 9 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
|
| 10 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
0 |
4 |
|
| 11 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
|
| 12 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
2 |
2 |
2 |
2 |
2 |
3 |
2 |
2 |
0 |
3 |
|
| 13 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
0 |
2 |
|
| 14 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
3 |
|
| 15 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
|
| 16 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
0 |
1 |
|
| 17 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
0 |
4 |
|
| 18 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
0 |
1 |
|
| 19 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
0 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
|
| 20 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
1 |
2 |
2 |
2 |
1 |
2 |
|
| 21 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
3 |
|
| 22 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
2 |
2 |
2 |
4 |
|
| X |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
| Y |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
| t(6;10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
Table 3. Karyotypes of 20 cen3tel cells 100 generations after transfection with overexpressed telomerase.
| Metaphase |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome no. |
47 |
47 |
48 |
49 |
49 |
49 |
90 |
81 |
87 |
88 |
85 |
45 |
46 |
67 |
63 |
52 |
70 |
64 |
66 |
83 |
|
| Chromosomes | Chromosome copy numbers | ||||||||||||||||||||
| 1 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
3 |
4 |
4 |
2 |
2 |
3 |
3 |
4 |
4 |
4 |
4 |
4 |
|
| 2 |
1 |
1 |
1 |
1 |
1 |
2 |
2 |
3 |
3 |
2 |
3 |
2 |
2 |
2 |
4 |
0 |
2 |
2 |
3 |
3 |
|
| 3 |
2 |
2 |
2 |
2 |
2 |
2 |
7 |
4 |
4 |
3 |
3 |
2 |
2 |
3 |
2 |
2 |
3 |
7 |
4 |
4 |
|
| 4 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
3 |
4 |
4 |
1 |
2 |
2 |
1 |
1 |
2 |
2 |
1 |
3 |
|
| 5 |
1 |
2 |
2 |
2 |
2 |
2 |
3 |
4 |
4 |
3 |
4 |
2 |
2 |
3 |
4 |
2 |
3 |
3 |
4 |
4 |
|
| 6 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
3 |
3 |
3 |
3 |
2 |
2 |
2 |
2 |
2 |
3 |
4 |
2 |
3 |
|
| 7 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
6 |
6 |
6 |
2 |
1 |
2 |
2 |
2 |
2 |
2 |
2 |
5 |
|
| 8 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
4 |
4 |
4 |
2 |
2 |
4 |
3 |
2 |
3 |
4 |
3 |
4 |
|
| 9 |
2 |
1 |
2 |
2 |
2 |
1 |
4 |
3 |
4 |
4 |
3 |
2 |
2 |
2 |
2 |
2 |
3 |
4 |
2 |
3 |
|
| 10 |
1 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
4 |
3 |
2 |
1 |
1 |
2 |
2 |
2 |
1 |
2 |
3 |
|
| 11 |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
2 |
4 |
3 |
4 |
1 |
2 |
2 |
2 |
0 |
2 |
2 |
1 |
4 |
|
| 12 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
4 |
4 |
4 |
2 |
2 |
3 |
3 |
2 |
4 |
3 |
4 |
4 |
|
| 13 |
2 |
1 |
1 |
2 |
2 |
2 |
3 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
1 |
2 |
|
| 14 |
2 |
2 |
2 |
2 |
2 |
3 |
4 |
3 |
2 |
3 |
2 |
2 |
3 |
3 |
2 |
1 |
2 |
3 |
2 |
2 |
|
| 15 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
2 |
3 |
1 |
3 |
2 |
3 |
3 |
1 |
2 |
3 |
3 |
2 |
|
| 16 |
2 |
1 |
2 |
2 |
2 |
2 |
4 |
2 |
4 |
4 |
4 |
2 |
2 |
2 |
3 |
2 |
3 |
2 |
4 |
4 |
|
| 17 |
2 |
2 |
2 |
1 |
2 |
2 |
0 |
3 |
4 |
2 |
2 |
1 |
2 |
2 |
2 |
1 |
2 |
1 |
3 |
2 |
|
| 18 |
1 |
2 |
2 |
2 |
2 |
2 |
5 |
3 |
4 |
3 |
3 |
2 |
2 |
4 |
3 |
2 |
4 |
2 |
3 |
4 |
|
| 19 |
2 |
2 |
2 |
2 |
2 |
2 |
5 |
4 |
4 |
4 |
4 |
2 |
2 |
4 |
3 |
4 |
2 |
2 |
2 |
4 |
|
| 20 |
2 |
2 |
2 |
2 |
2 |
2 |
8 |
4 |
4 |
4 |
4 |
1 |
2 |
4 |
4 |
3 |
4 |
1 |
1 |
4 |
|
| 21 |
4 |
3 |
3 |
2 |
3 |
3 |
3 |
4 |
3 |
4 |
4 |
1 |
3 |
3 |
3 |
2 |
2 |
1 |
2 |
4 |
|
| 22 |
2 |
2 |
2 |
2 |
2 |
2 |
5 |
4 |
4 |
3 |
3 |
1 |
2 |
4 |
3 |
4 |
3 |
1 |
3 |
3 |
|
| X |
2 |
2 |
2 |
2 |
2 |
2 |
3 |
4 |
3 |
4 |
4 |
2 |
2 |
3 |
3 |
1 |
3 |
4 |
4 |
2 |
|
| Y |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
| der(14q;2) |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
|
| der(17) |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
2 |
2 |
1 |
0 |
2 |
0 |
1 |
2 |
0 |
0 |
2 |
|
| der(14p;2) |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
| der(11:21) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
2 |
1 |
0 |
1 |
0 |
1 |
0 |
|
| der(21) |
0 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
2 |
2 |
2 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
2 |
|
| der(10) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
2 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
2 |
1 |
2 |
0 |
|
| der(11) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
3 |
0 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
2 |
0 |
1 |
0 |
|
| der(2;X) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
|
| der(2:6) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
| der(21;2) |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
0 |
0 |
0 |
|
| der(8;14;2) |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
| der(5;10) | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| Non-clonal Markers | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| der(2;21) |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
| der(9:11) |
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(21;1) |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
| der(7) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
| der(22:6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
| der(18:19) |
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
| der(5;22) |
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
| der(11;12) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
| der(6:9) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
| der(10;21) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
| der(17:1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
| der(1;17) |
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(6;9) |
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(12;16) |
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(10;21) |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(1;17) short |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| der(4;7;22) |
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
| der(3;18) |
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
| der(9;18) |
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
| der(1) |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
| der(2;14;2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
| der(16) | 1 | ||||||||||||||||||||

Figure 4. Aneuploidy among karyotypes of 20 human cen3tel skin cells 37 (A) and 100 (B) generations after transfection with overexpressed telomerase. The 20 karyotypes were arrayed as described for Figure 2. The data show that 60% of the cen3tel cells were randomly aneuploid 37 generations after transfection (Table 2), and that 100% were randomly aneuploid 100 generations after transfection (Table 3). Moreover, the data show that the degree of aneuploidy increased sharply from generation 37 to generation 100 after transfection. The partial clonality of trisomy 7 was native of the cen3-skin biopsy studied here (see text).
Moreover, some aneusomies had reached low degrees of clonality during the progression of aneuploidy from PD37 to PD100. For example, 12 new marker chromosomes of PD100 were shared subclonally by 2–9 of 20 karyotypes, and one new monosomy 2 was shared by five karyotypes (Table 3). At the same time, 15 of 20 cen3tel cells at PD37 had retained a trisomy 7 that was subclonal in the original cen3 biopsy32 (Table 2 and Fig. 4A). This trisomy 7 was still present in 8 of 20 cen3tel cells at PD100 and in 3 of 20 PD100-cells in the form of a hexasomy 7 (Fig. 4B). Such subclonalities indicate that some randomly generated aneuploidies provide growth advantages.
The spontaneous increases in the percentages and the degrees of aneuploidies, and the evolution of subclonal aneuploidies of the cen3tel culture from PD37 to PD100 thus support the prediction of the speciation theory that aneuploidy progresses automatically based on newly generated growth advantages.
These results also confirm and extend earlier studies of Heng et al.69 showing that aneuploidies confer growth advantages to somatic cells from humans with genetic instability, and studies of Berman and Pavelka et al.15,70,71 showing that certain aneuploidies confer growth advantages to various strains of yeast. It seems probable, therefore, that such aneuploidy-specific growth advantages might also explain the pre-neoplastic human hyperplasias detected in vivo ever since Papanicolaou.72,73
Conclusions
(1) Overexpressed telomerase is not sufficient for immortalization, because the transfected cells have highly unstable, aneuploid karyotypes.
(2) Transfection of human cells with overexpressed telomerase induces random aneuploidies, which offer growth advantages. As a result of these advantages, the aneuploidies progress spontaneously and even form subclonal subsets. These aneuploidy-generated growth advantages thus explain the 2–3-fold increases in the growth rates of cen3tel cells following transfection with overexpressed telomerase.
New tumorigenic clone evolves late from randomly aneuploid cells
During the 66 cell generations from PD100 to PD166, the cen3tel culture acquired the neoplastic abilities to grow in agar gels and to form tumors in immunodeficient athymic mice.32,40 At the same time the cellular phenotype of the cen3tel cells changed completely from fibroblastic spindle-shaped to cubic-polymorphic, which is characteristic of cancer cells (Fig. 3A–C). Moreover the cen3tel cells at PD166 grew 3-dimensionally, which is also characteristic of cancer cells.74
To determine whether this cancer-specific phenotypic transformation coincided with the evolution of a new clonal and flexible cancer karyotype, we prepared an array of 20 cen3tel PD166 karyotypes, which is shown in Figure 5A and Table 4. It can be seen in Table 4 that the chromosome numbers of the tumorigenic cen3tel PD166 clone were 76 ± 3, and thus were highly clonal. The table also shows that the 76 clonal chromosomes included 14 new PD166-specific marker chromosomes. Further the array shows that the copy numbers of most chromosomes of the 20 karyotypes were clonal, forming parallel lines, which are characteristic of clonality (see above “Results,” “Immortal tumorigenic clones…”). At the same time a minority of chromosomes were non-clonal, forming non-parallel points within narrow ± 1 margins of variation above and below these parallel lines. The non-clonal copy numbers were thus near-clonal. The percentages of clonal vs. near-clonal chromosome copy numbers ranged from 60 to 100% (Table 4).
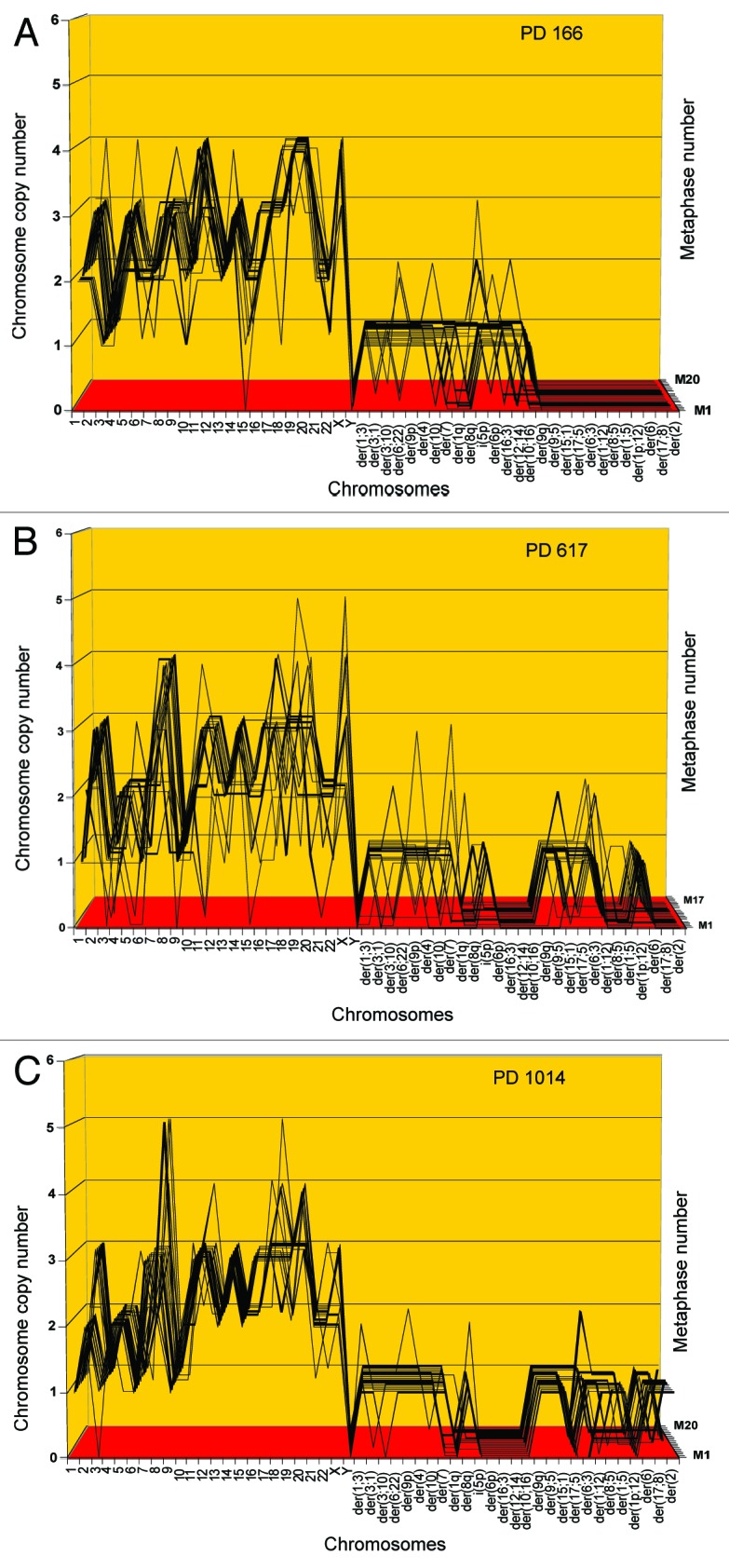
Figure 5. Karyotypic stability and drift of the tumorigenic clone cen3tel PD166 at the time of isolation (A) and after 451 (B) and 848 (C) unselected generations in vitro. As a test for immortalization, karyotype arrays prepared from the original clone cen3tel PD166 and descendents isolated 451 (PD617) and 848 (PD1014) unselected generations later were compared. These karyotype arrays show that the original cen3tel PD166 karyotype was mostly stable, but was also variable within narrow margins over 848 cell generations (Table 4). The copy numbers of 21 of 33 (64%) PD166-specific chromosomes including marker chromosomes were clone-specific during 848 generations, while 12 of 33 (36%) were generation-specific or cell-specific drifting within narrow margins of ± 1 over 848 generations (Table 4 and Fig. 6). Based on the high clonal stability over 848 unselected generations, we conclude that the cen3tel PD166 clone is immortal but flexible, exceeding the operational Hayflick threshold of 50 generations by 798 clonal generations.
Table 4. Chromosome numbers and copy numbers of the tumorigenic clone cen3tel PD166, and at subsequent generations PD617 and PD1014.
| Cen3tel clones |
PD166 |
PD617 |
PD1014 |
|---|---|---|---|
| Chromosome no. ± SD |
76 ± 3 |
72 ± 4 |
73 ± 1 |
| No. of metaphases |
20 |
17 |
20 |
| Chromosomes | Copy number (% clonal) | ||
| 1 |
2 (95) |
2 (65) |
1 (80) |
| 2 |
3 (70) |
3 (76) |
2 (60) |
| 3 |
1 (95) |
1 (82) |
1 (95) |
| 4 |
2 (95) |
2 (65) |
2 (100) |
| 5 |
3 (80) |
2 (65) |
2 (90) |
| 6 |
2 (95) |
2 (59) |
1 (75) |
| 7 |
2 (90) |
3 (65) |
3 (60) |
| 8 |
3 (80) |
4 (71) |
3 (60) |
| 9 |
3 (85) |
1 (94) |
1 (100) |
| 10 |
2 (70) |
2 (88) |
2 (85) |
| 11 |
4 (65) |
3 (65) |
3 (90) |
| 12 |
3 (80) |
3 (71) |
3 (80) |
| 13 |
2 (100) |
2 (88) |
2 (100) |
| 14 |
3 (95) |
3 (82) |
3 (100) |
| 15 |
2 (75) |
2 (76) |
2 (100) |
| 16 |
2 (65) |
3 (53) |
3 (60) |
| 17 |
3 (100) |
3 (59) |
3 (90) |
| 18 |
3 (65) |
3 (71) |
3 (75) |
| 19 |
4 (90) |
3 (59) |
3 (80) |
| 20 |
4 (95) |
3 (53) |
4 (65) |
| 21 |
2 (65) |
2 (82) |
2 (95) |
| 22 |
2 (80) |
2 (82) |
2 (85) |
| X |
4 (85) |
3 (35) |
3 (70) |
| Y |
0 |
0 |
0 |
| der(1;3) |
1 (100) |
1 (88) |
1 (95) |
| der(3;1) |
1 (90) |
1 (76) |
1 (95) |
| der(3;10) |
1 (95) |
1 (71) |
1 (95) |
| der(6;22) |
1 (80) |
1 (94) |
1 (100) |
| der(9p) |
1 (100) |
1 (76) |
1 (95) |
| der(4) |
1(100) |
1(82) |
1 (100) |
| der(10) |
1 (85) |
1 (65) |
1 (95) |
| der(7) |
1 (80) |
1 (65) |
1 (75) |
| der(1q) |
1 (75) |
1 (6) |
1 (5) |
| der(8q) |
1 (45) |
1 (12) |
1 (50) |
| i(5p) |
1 (85) |
1 (59) |
- |
| der(6p) |
1 (85) |
- |
- |
| der(16;3) |
1 (85) |
- |
- |
| der(12;14) |
1 (75) |
- |
- |
| der(10;16) |
1 (55) |
- |
- |
| der(9q) |
- |
1 (88) |
1 (100) |
| der(5;9) |
- |
1 (82) |
1 (100) |
| der(15;1) |
- |
1 (71) |
1 (100) |
| der(17;5) |
- |
1 (71) |
1 (35) |
| der(6;3) |
- |
1 (65) |
1 (90) |
| der(1;12) |
- |
1 (6) |
1 (55) |
| der(8;5) |
- |
1 (6) |
1 (55) |
| der(1;5) |
- |
1 (6) |
1 (55) |
| der(1p;12) |
- |
1 (76) |
- |
| der(6) |
- |
- |
1 (75) |
| der(17;8) |
- |
- |
1 (70) |
| der(2) | - | - | 1 (55) |
Given the rough chronological time points available, we estimate that the stem cell of cen3tel PD166 originated at or before PD146 as follows. Since we maintained cultures of at least 1 million cells, and since it takes about 20 cell generations to multiply one cell to 1 million (106 = 220), we deduce that the stem cell of PD166 must have arisen at PD146, at least 20 generations before PD166. In the following 20 generations it would have supplanted all non-clonal aneuploid precursors by the new tumorigenic PD166 cells (see also “Discussion” and Fig. 11).
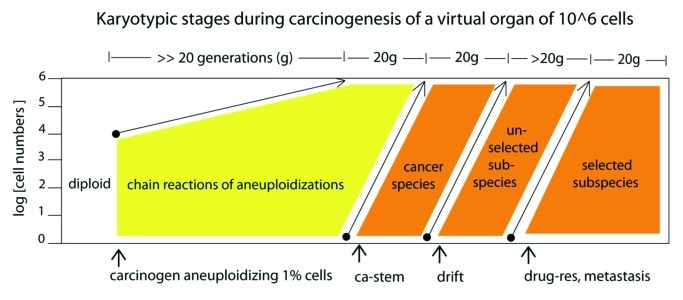
Figure 11. Karyotypic stages during carcinogenesis of a virtual organ of 106 cells: (1) The induction of random aneuploidy in an arbitrary 1% of normal cells by a carcinogen, such as overexpressed telomerase (Fig. 4) or SV40 virus (Figs. 9 and 10); (2) Self-catalyzed progression of aneuploidy over many cell generations up to 100% of the initiated cell population, as for example in the human skin cells 100 generations after aneuploidization by overexpressed telomerase shown in Figure 4; (3) The stochastic evolution of an autonomous cancer stem cell with a new, clonal flexible karyotype and phenotype. The new autonomous cancer cell supplants the 106 non-cancerous precursors in our Petri dish within 20 generations after its origin (Figs. 5 and 8); (4) Self-catalyzed karyotypic variations of cancer cells generate new unselected subspecies, spontaneously (Figs. 5and 8) and drug-resistant or metastatic subspecies, selectively (see text).
Conclusions
(1) Since neither tumorigenicity, nor clonal and flexible karyotypes, nor non-fibroblastic, polymorphic cell phenotypes were observed in the preneoplastic cen3tel mass culture before PD166, it is hard to escape the conclusion that the new clonal and flexible PD166 karyotype generated the new tumorigenic cen3tel PD166 clone. The recent identification of a new, cen3tel PD166-specific transcriptome by Ostano et al. lends independent support to this conclusion. They found that none of about 15 (except perhaps 1) cen3tel PD166-specific transcripts were shared with the preneoplastic cen3tel precursors at PD37 and PD97.75 It follows that the new PD166-transcriptome generates the new phenotypes of the cen3tel PD166 clone.
(2) The speciation theory also predicts the clonal heterogeneity of the neoplastic cen3tel PD166 clone, based on the underlying karyotypic flexibility of cancer karyotypes (Introduction).
(3) The random, preneoplastic aneuploidies of PD37 and PD100 followed by the new clonal, neoplastic karyotype of PD166 confirm the prediction of the speciation theory that aneuploidy is the proximate carcinogen.
In the following we ask whether the new tumorigenic cen3tel PD166 clone is indeed immortal, and, if so, whether it had acquired immortality together with tumorigenicity.
During 848 unselected generations the clonal chromosome number of cen3tel PD166 is stable, but the copy numbers of 36% of chromosomes drift ± 1
To determine whether the new, tumorigenic cen3tel PD166 clone is immortal in terms of the Hayflick definition, we compared the karyotypes of cen3tel PD166 during 848 unselected generations from PD166, when it was first identified, to its late descendants at PD617 and at PD1014.
These comparisons indicate that the chromosome numbers of cen3tel PD166 were 76 ± 3 at PD166, 72 ± 4 at PD617 and 73 ± 1 at PD1014 and thus were highly clonal and stable over 848 generations (Table 4). The copy numbers of 21 of 33 (64%) PD166 chromosomes were clonally conserved over 848 generations, while 12 of 33 (36%) chromosome numbers drifted within narrow margins of ± 1 (Table 4 and Fig. 5A–C). In addition to this quantitative drift of chromosome copy numbers, cen3tel at PD617 also differed from its precursor PD166 in four new generation-specific clonal markers, and cen3tel PD1014 differed in 10 generation-specific clonal markers (Table 4).
In Figure 6 we show the karyotypic consequences of 848 generations of unselected drift on two randomly selected karyotypes of PD166 and of PD1014 side-by-side. It can be seen that the two karyotypes differed from each other in generation-specific and cell-specific (non-clonal) copy numbers (Table 4) and marker chromosomes (Table 4, also marked in Fig. 6), which could only be distinguished as such with the statistics of the karyotype arrays.

Figure 6. Two individual karyotypes of the cen3tel clone PD166 at its origin (A) and after 848 generations of unselected growth in culture at PD1014 (B). About 64% of the chromosomes of the two karyotypes share PD166-specific copy numbers or are common markers (Table 4 and Fig. 5). The two karyotypes differ in generation-specific and cell-specific chromosomal copy numbers and markers [labeled in (A and B)] that could only be distinguished as such by the statistical analyses of the karyotype arrays shown in Figure 5.
But, despite such variations, the basic karyotype of PD166 was stably clonal and flexible over 848 cell generations. In agreement with the stability of this karyotype, the new transformation-specific phenotype of PD166 was also stable during 848 generations (Fig. 3C and D).
Conclusion
Based on its karyotypic and phenotypic stability over 848 generations, we conclude that cen3tel PD166 is immortal within clonal margins of variation. Indeed, the evidence for 848 generations of clonal stability and flexibility obtained here far exceeds the operational Hayflick threshold of immortality of 50 generations. This result confirms and extends the theory that the immortality of cancers is karyotypic.
Next, we asked whether tumorigenicity and immortality are also karyotypically united in independent tumorigenic clones, derived from the same primary cen3tel culture from which cen3tel PD166 was obtained.
An independent immortal and tumorigenic clone arises from preneoplastic cen3tel cells after an individual latency
In the following we test the prediction of the speciation theory that tumorigenicity and immortality are karyotypically linked in all neoplastic clones. By contrast, the telomerase theory predicts immortalization to be an independent genetic event and thus not inevitably linked to tumorigenesis.5,27
To distinguish between these alternatives, we studied an independent tumorigenic clone that arose from an aliquot of the same preneoplastic cen3tel cells from which cen3tel PD166 originated. The aliquot was taken only “a few” cell generations after transfection (Mondello, unpublished). This aliquot was termed cen3telS2 and was maintained and tested for tumorigenicity and immortality exactly like the parental cen3tel culture.
About 84 cell generations after transfection, at PD84, the independently growing cen3telS2 mass culture had become tumorigenic. The individual karyotype of cen3telS2 PD84 indicated that the new clone was indeed independent of cen3tel PD166 (Fig. 7A).

Figure 7. (A) Karyotype of the immortal tumorigenic clone cen3telS2 PD84, which arose from an independent preneoplastic aliquot of the cen3tel mass culture 84 generations after it was transfected with overexpressed telomerase. (See above Results, “How does overexpressed telomerase induce…”) (B) Karyotype of cen3telS2 PD143, which supplanted the PD84 clone 59 generations after it was identified at PD84.
To determine whether the new tumorigenic cen3telS2 culture had a new clonal and flexible karyotype, an array of 20 individual karyotypes was prepared (Table 5 and Fig. 8). This array shows that PD84 has a highly clonal chromosome number of 46 ± 1, which included three clonal S2-specific marker chromosomes. The new karyotype was thus pseudo-diploid. The array also shows that the chromosome copy numbers of the cen3telS2 PD84 culture were 75–90% clonal, except for three non-clonal marker chromosomes (Fig. 8A and Table 5). The presence of some non-clonal chromosomes is, of course, expected from the inherent flexibility of neoplastic karyotypes. Thus, cen3telS2 PD84 is a new clone with an individual, clonal and flexible karyotype that independently originated from the preneoplastic cen3tel mass culture described above.
Table 5. Chromosome numbers and copy numbers of immortal, tumorigenic clones cen3telS2 PD84 and subclone cen3telS2 PD143.
| Cen3telS2 clones |
PD84 |
PD143 |
|---|---|---|
| Chromosome no ± SD |
46 ± 1 |
85 ± 1 |
| Metaphases (no.) |
20 |
20 |
| Chromosomes | Copy number (% clonal) | |
| 1 |
2(85) |
4(100) |
| 2 |
2(75) |
4(90) |
| 3 |
2(85) |
4(95) |
| 4 |
1(80) |
2(100) |
| 5 |
2(85) |
4(100) |
| 6 |
2(85) |
4(95) |
| 7 |
2(85) |
6(90) |
| 8 |
2(80) |
2(80) |
| 9 |
2(85) |
4(85) |
| 10 |
1(90) |
3(95) |
| 11 |
2(75) |
4(95) |
| 12 |
2(85) |
4(100) |
| 13 |
2(75) |
2(100) |
| 14 |
2(75) |
3(95) |
| 15 |
1(85) |
3(100) |
| 16 |
2(90) |
4(95) |
| 17 |
1(90) |
2(90) |
| 18 |
2(75) |
4(95) |
| 19 |
2(85) |
4(80) |
| 20 |
2(80) |
4(95) |
| 21 |
2(85) |
2(85) |
| 22 |
2(85) |
2(95) |
| X |
2(80) |
3(95) |
| Y |
0 |
0 |
| der(4) |
1(90) |
1(85) |
| der(10;17) |
1(85) |
- |
| der(15;7) |
1(90) |
- |
| der(11;10) |
1(5) |
- |
| der(2;5) |
1(5) |
- |
| der(17;4) |
1(5) |
- |
| der(4;22) |
- |
1(95) |
| der(10) |
- |
1(100) |
| der(8;17) |
- |
2(85) |
| der(17) |
- |
2(85) |
| der(19;17) |
- |
1(5) |
| der(7) | - | 1(5) |
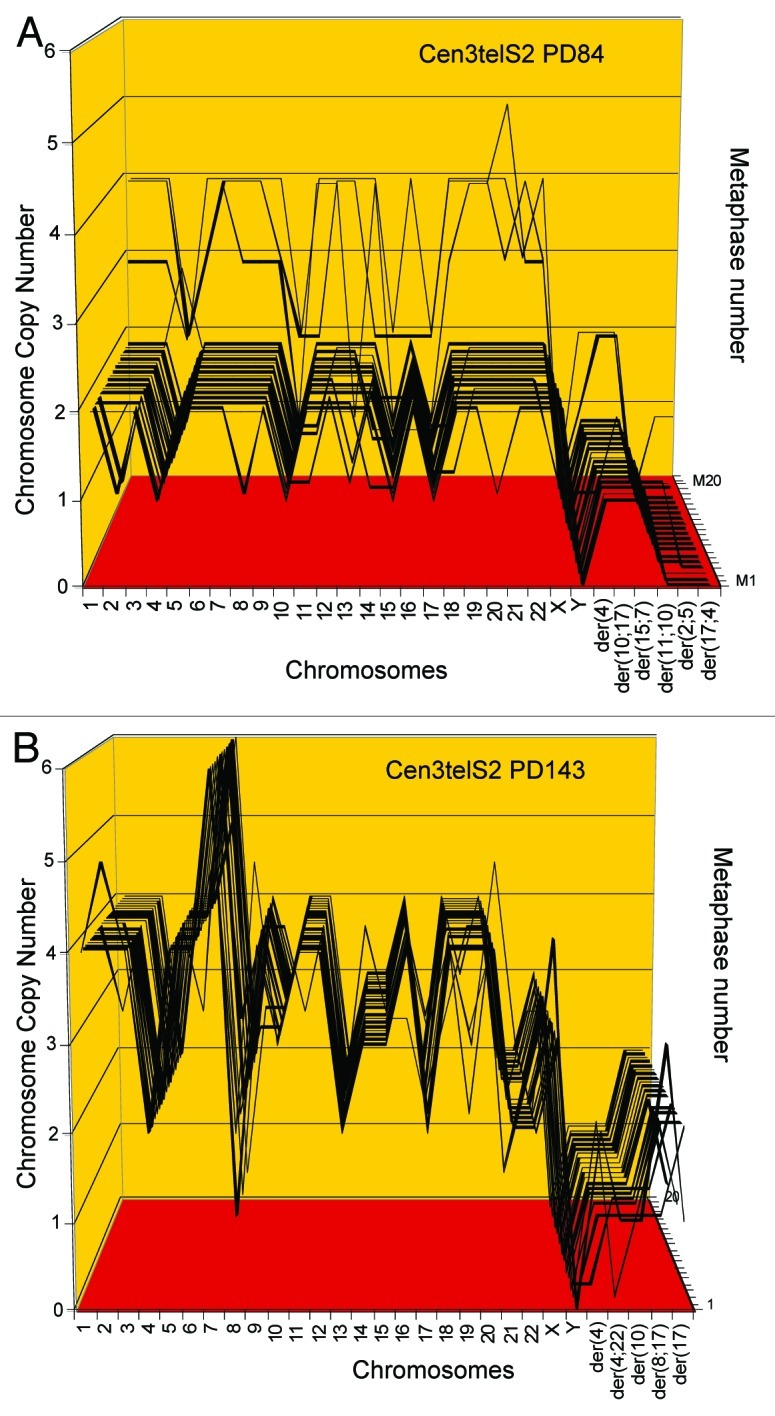
Figure 8. Karyotype arrays of the immortal, tumorigenic clones cen3telS2 PD84 (A) and a sub-clone cen3telS2 PD143 (B) based on data of Table 5. The cen3telS2 PD84 clone arose spontaneously in an independently growing preneoplastic aliquot of the cen3tel mass culture (from which also cen3tel PD166 originated) 84 generations after transfection with overexpressed telomerase. Upon further cultivation for 59 generations, the cen3telS2 PD84 clone was supplanted by a more transformed and faster growing variant, cen3telS2 PD143. Comparative karyotype arrays revealed that PD143 is a near-tetraploid, clonal variant of cene3teS2 PD84 (Table 5).
In agreement with the karyotypic individuality of cen3telS2 PD84, the clone differed from cen3tel PD166 in an individual growth rate, which was about two times lower than that of the cen3tel PD166. Moreover, the latent period from the time of transfection of the primary cen3-skin biopsy was much shorter, namely 84 cell generations, compared with the 166 cell generations of the cen3tel PD166 clone.
We infer from available data that the S2 PD84 clone is immortal in terms of the Hayflick definition as follows. In our studies we had passaged the clone for 25 cell generations. In addition, Mondello et al. have tested its stability by demonstrating that the clone is tumorigenic in athymic mice (Mondello, personal communication). Considering that the PD84 stem cell has survived about 30 cell generations to form an experimental tumor of about 1 ml, or 109 cells,5,74 and that the same PD84 clone has also survived 25 generations in our cultures, the PD84 clone has passed the 50 generation-threshold for immortality by the Hayflick limit. The clone is thus probably immortal, much like the PD166 clone derived from the same preneoplastic cen3tel precursor cells.
Conclusions
(1) The origins of two tumorigenic clones, with different individual clonal karyotypes and phenotypes, and different individual latent periods from the same aneuploid non-neoplastic cen3tel mass culture confirm the prediction of the speciation theory that immortality and tumorigenicity are intrinsic elements of clonal and flexible cancer-specific karyotypes
By contrast, the probability that a genetically independent,5,76 but non-essential immortalization event (Introduction and Discussion) would coincide with tumorigenicity by chance is only one in a million, because the rates of mutations in cancer cells are only about 10−6.77-79 Compare this to the ubiquity of immortality in cancer. One might argue, however, that cen3tel cells were already immortalized by transfection with overexpressed telomerase. But we have shown in Figure 4 that such cells are karyotypically highly unstable and thus not immortal in themselves.
(2) The origins of two karyotypically unrelated neoplastic clones from the same population of aneuploid cells supports the prediction of the speciation theory that cancers arise stochastically by independent karyotypic rearrangements. Heng et al. confirm this conclusion: “when performing multiple runs of evolutionary experiments using cell populations derived from the same initial cells, the end results are not the same (individual cancer cells) and thus are not predictable.”12
In the following, we show a new example of karyotypic-phenotypic linkage, namely the evolution of a new, more dominant-neoplastic and immortal subclone from cen3telS2 PD84.
Near-tetraploid tumorigenic subclone overgrows near-diploid precursor cen3telS2 PD84
The speciation theory predicts that owing to the inherent flexibility of cancer karyotypes, cancers can evolve subspecies with enhanced tumorigenicity at high rates compared with mutation.45,80 As a possible example of subspeciation, we have analyzed here a tumorigenic variant of cen3telS2 PD84 that had supplanted this clone 59 generations after its first appearance, at PD143 (Mondello, unpublished). Compared with cen3telS2 PD84, the new neoplastic PD143 variant grew faster, and its cells looked more transformed, i.e., more amorphic and polymorphic than those of its precursor PD84 (not shown).
As a test for a karyotypic basis for this evolution, we compared the karyotype of PD143 to that of its suspected predecessor PD84. It is shown in Figure 7B that the karyotype of the new PD143 was different from, but possibly a polyploid version of, PD84. Following this clue we compared arrays of 20 karyotypes of cen3telS2 PD143 and PD84 side-by-side in Table 5 and Figure 8. These comparisons indicated that PD143 is a karyotypic duplication of PD84 on the following grounds: (1) PD143 has a clonal, near-tetraploid chromosome number of 85 ± 1, compared with the chromosome number of 46 ± 1 of PD84; (2) The copy numbers of 15 of the 23 intact chromosomes of PD143 are exact duplicates of the copy numbers of the same PD84-chromosomes. The remaining eight intact PD84-chromosomes were either not amplified or amplified 1.5- or 3-fold in PD143; (3) Both tumorigenic clones share one specific marker chromosome, der4. Further, the early presence of three near-tetraploids in the karyotype array of cen3tel PD84 (Fig. 8A) already indicated the proclivity of PD84 for tetraploidization, which eventually generated PD143.
We deduce (1) from the high, 80–100% clonality of the chromosome copy numbers of cen3telS2 PD143, which is a proxy for stability (Table 5 and Fig. 8), and (2) from the immortality of its precursor that cen3tel PD143 has inherited karyotypic immortality from its precursor PD84.
Conclusions
(1) The duplication of the gene dosages of the cen3telS2 PD84 in the subspecies PD143 by polyploidization explains the enhanced tumorigenicity of cen3telS2 PD143 compared with PD84. It thus also explains its ability to supplant the whole PD84 culture within 20 to 40 generations (Figs. 8 and 11). Further the co-segregation of tumorigenicity and immortality over 59 unselected generations and during a major karyotypic remodeling lends new support to our theory that these characters are karyotypically linked.
(2) Since all six immortal and tumorigenic clones and subclones studied so far originated from cells transfected with overexpressed telomerase, it could be argued that clonal and flexible karyotypes were not sufficient to immortalize these clones, and hence also depend on telomerase for immortalization. In the next and last experiment we have addressed this question.
SV40 tumor virus induces immortal neoplastic clones with flexible karyotypes in cells of a telomerase-deficient mouse
To determine whether either endogenous or exogenous telomerase is necessary for immortalization, we asked whether SV40 tumor virus generates immortal neoplastic clones in cells of a telomerase-deficient mouse. The study of Argilla et al. with telomerase-deficient mice, introduced above, already suggested that neoplastic transformation is telomerase-independent, but did not identify a mechanism of immortalization46 (Introduction).
In view of this, we analyzed the karyotypes of foci of SV40-transformed lung and tail cells from a telomerase-deficient mouse for clonal and flexible, cancer-specific karyotypes. Such foci were obtained 2 to 3 mo after infection of primary cell cultures (“Materials and Methods”). Individual karyotypes of a lung cell focus, termed FL1 and of a tail cell focus, termed FT1, are shown in Figure 9, and arrays of 20 karyotypes are shown in Figure 10 and Table 6.

Figure 9. Karyotypes of immortal clones of SV40 virus-transformed lung cells, FL1 (A) and tail cells, FT1 (B) from a telomerase-deficient mouse (see also Table 6). The transformed clones arose together with their new individual karyotypes 2–3 mo after infection of the cells with SV40 virus.

Figure 10. Karyotype arrays of immortal, neoplastic clones of SV40 virus-transformed lung cells, FL1 (A and B) and tail cells, FT1 (C) from a telomerase-deficient mouse. The clones appeared 3 mo after infection by SV40 tumor virus together with their new clonal and flexible karyotypes. Karyotype arrays of the lung-derived focal culture, FL1 were determined 35 and 65 generations after isolation of the focal culture. The arrays show that the FL1 karyotype was near-triploid with near clonal FL1-specific chromosome numbers and chromosome copy numbers (see also Table 6). The karyotype array of the tail-derived focal culture, FT1 was determined 25 generations after isolation of the focal FT1 clone. The array shows that the FT1 karyotype was hypo-diploid with 94–100% clonal chromosome numbers and chromosome copy numbers.
Table 6. Chromosome numbers and copy numbers of two immortal SV40-transformed clones of telomerase-deficient mouse cells, FL1 and FT1.
| Neoplastic clones |
FT1 PD25 |
FL1 PD35 |
FL1 PD65 |
|---|---|---|---|
| Chromosome no. ± SD |
38 ± 1 |
61 ± 3 |
59 ± 3 |
| Metaphases (no.) |
16 |
20 |
20 |
| Chromosomes | Copy number (% clonal) | ||
| 1 |
2(100) |
3(100) |
3(80) |
| 2 |
2(100) |
3(85) |
3(95) |
| 3 |
2(100) |
2(75) |
3(85) |
| 4 |
2(100) |
1(95) |
1(95) |
| 5 |
2(100) |
4(85) |
2(35), 3(40), 4(20) |
| 6 |
2(100) |
2(60), 3(30) |
1(55), 2(45) |
| 7 |
2(100) |
3(70) |
3(95) |
| 8 |
2(100) |
3(35), 4(50) |
3(50), 4(40) |
| 9 |
2(100) |
3(70) |
3(85) |
| 10 |
2(94) |
3(35), 4(60) |
4(80) |
| 11 |
2(100) |
4(85) |
4(60) |
| 12 |
1(100) |
2(95) |
2(85) |
| 13 |
2(94) |
2(100) |
2(90) |
| 14 |
1(100) |
4(85) |
2(80) |
| 15 |
2(94) |
3(65) |
3(30), 4(45) |
| 16 |
2(100) |
3(85) |
2(80) |
| 17 |
2(100) |
3(80) |
1(65) |
| 18 |
2(100) |
3(75) |
2(70) |
| 19 |
2(100) |
3(35), 4(55) |
3(45), 4(15) |
| X |
1(100) |
2(100) |
2(70) |
| Y |
1(69), 2(31) |
0 |
0 |
| der(4) long |
- |
1(95) |
1(85) |
| der(4;12) |
- |
1(90) |
1(95) |
| der(6;3;19) |
- |
1(65) |
1(70) |
| der(13;15) |
- |
1(100) |
1(75) |
| i(17) |
- |
- |
1(55) |
| der(16;19) |
- |
- |
1(55) |
| i(6) |
- |
- |
1(35) |
| der(18;19) |
- |
- |
1(35) |
| der(5;14) | - | - | 1(25) |
These data indicate that FL1 has a near triploid karyotype with clonal chromosome numbers of 59 ± 3 (compared with the 40 chromosomes a normal mouse). The copy numbers of 16 intact and four FL1-specific marker chromosomes were 65 to 100% clonal and thus stable during 35 and 65 cell generations (Fig. 10 and Table 6). The copy numbers of the remaining four intact chromosomes oscillated between two or three complementary percentages with high clonality (Table 6).
In agreement with the theory that cancer karyotypes are clonal but variable within clonal margins, the FL1 clone included minor fractions (0–35%) of chromosomes with near-clonal copy numbers that oscillated ± 1 around clonal values. In addition the clone had evolved five new, marker chromosomes at intermediate degrees of clonalities (25–55%) between generations PD35 to PD65 (Table 6).
Since the percentages of chromosomal clonalities and near-clonalities of the FL1 clone were stable for over 65 generations (except for some new subclonal marker chromosomes), we concluded that the FL1 clone is immortal based of the Hayflick definition. In the meantime, we have found that the clone was stable for another 25 generations, for a total of 90 generations in culture, but a new karyotype analysis has not been performed.
The tail-derived FT1 clone was hypo-diploid with a clonal chromosome number of 38 ± 1 (Figs. 9 and 10 and Table 6). The clonality of the chromosome copy numbers of FT1 at generation PD25 was very high, ranging between 94 and 100%. Thus FT1 was highly stable and is therefore likely to be immortal, although it has not yet been passaged and karyotyped for 50 generations.
We suggest that two factors explain the relatively high flexibility of FL1 compared with that of FT1: (1) the karyotype-destabilizing function of the high, near-triploid aneuploidy of FL1; and probably (2) the ongoing aneuploidizing function of the SV40 T-antigen, knowing that the dosage and activity of the T-antigen of SV40-virus can be very high in some cells.38,68,81-83
Conclusions
(1) Since both SV40-induced neoplastic clones of cells from a telomerase-deficient mouse were immortal and carried clonal and flexible karyotypes, and since the new clones and their new clonal karyotypes both appeared late after infection with SV40 virus, we conclude that these new clonal and flexible karyotypes were sufficient to transform and immortalize cells of telomerase-free mice.
(2) Further, we conclude that the role of the SV40-T-antigen in this immortalization experiment was to initiate the evolution of these new karyotypes by inducing random aneuploidy.38,68,81-83 Thus SV40 T-antigen and overexpressed telomerase play the same indirect role in immortalization and neoplastic transformation, namely the induction of random aneuploidy (Results, “How does overexpressed telomerase induce…” and Fig. 4).
Discussion
Karyotypic theory of immortality
With the longitudinal analyses of cells transfected with overexpressed telomerase or infected with SV40-virus, we have now before us a complete sequence of the karyotypic stages that generate cancer and immortality. Figure 11 diagrams these karyotypic stages during carcinogenesis of a virtual organ of 106 cells: (1) the induction of random aneuploidy in an arbitrary 1% of normal cells by a carcinogen, such as overexpressed telomerase (Fig. 4) or SV40 virus T-antigen (Figs. 9 and 10); (2) self-catalyzed progression of random aneuploidy over many cell generations reaching up to 100% of the initiated cell population; the aneuploid human skin cells shown in Figure 4, 37 and 100 generations after transfection with overexpressed telomerase are examples; (3) the stochastic evolution of an autonomous cancer stem cell with a new, clonal flexible karyotype and phenotype; this new autonomous cancer cell supplants the 106 non-cancerous precursors in our virtual organ of 106 cells within 20 generations after its origin (Figs. 5 and 8); (4) self-catalyzed karyotypic variations of cancer cells generate new unselected subspecies, spontaneously (Fig. 5), and drug-resistant or metastatic subspecies, selectively.84,85
This sequence of events explains why in our experiments immortality and tumorigenicity: (1) correlated exactly with cancer-specific clonal and flexible karyotypes; (2) originated simultaneously with such karyotypes; and (3) progressed with subspecies of these karyotypes. Therefore, we conclude that clonal and flexible cancer karyotypes generate immortality and tumorigenicity simultaneously and independent of telomerase.
-
The literature supports this view on several grounds:
According to the “Mitelman Database of Chromosome Aberrations in Cancers,” an exact and huge correlation exists between immortality and individual, clonal yet flexible cancer karyotypes—namely over 61,000 cases!86
Levan and Biesele adduced independent evidence for the simultaneous origins of immortality and tumorigenicity in 1958: “One especially interesting change, demonstrated often to coincide with the establishment of the capacity for indeterminate growth in vitro, is the acquirement of malignant properties, made evident on inoculation of the cell strain in vivo.”63. Others have since described exact correlations between “indeterminate growth” in vitro and tumorigenicity.87
Several previous studies observed that immortalization and neoplastic transformation are independent of telomerase-function: (a) Mondello et al. observed that the immortalization the cen3tel PD166 clone described above coincided with the loss, instead of an expected gain of telomere length. The telomeres of cen3tel PD166 were “decreased and stabilized around values lower than those observed in senescent cen3 cells (at) 6 kb” during immortalization. By contrast, the telomeres of mortal cen3 precursor cells measured 8.5 kb;32 (b) Another study found that the lengths of telomeres are reduced in 40% of all tumors compared with the normal tissue of origin;88 (c) Others wrote, in agreement with Holliday,89 that “Approximately a quarter of in vitro immortalized cell lines so far examined have no detectable telomerase activity, yet have very long and heterogeneous telomeres”90.
In sum, the literature supports the theory that immortality is an integral part of the cancer karyotype, and is independent of telomerase.
Why are cancers immortal?
Current cancer researchers and biology textbooks faithfully report that immortality is one of the “hallmarks of cancer”5,8,9,29,76. There is, however, no compelling information on why cancers are immortal. According to cancer researcher Blagosklonny, immortality is in fact a “paradox,” because “it brings no immediate replicative advantage.”6.
The customary answer to the immortality question suggests that immortality is necessary for cancer cells to grow from single stem cells to the numbers of cells that generate fatal cancers.5 Since fatal cancers consist of 1 to 1,000 ml tissue or 109 to 1012 cells,5,91 about 30 to 40 cell generations are necessary to generate such cancers from a single cancer stem cell (230 = 109 and 240 = 1012).
But, according to the Hayfilck limit the reproductive capacity of somatic cells is 50 generations, enough to generate 250 or 1015 cells, the equivalent of 10 human bodies.5,92 As shown in this study and others cited above the reproductive capacity of human cells even from centenarians can actually be over 100 generations. There is also no consistent evidence that somatic cells must be "immortalized" by activated telomerase to produce sufficient numbers of cells for fatal cancers. For example, we found here that somatic cells of telomerase-deficient mice become immortal cancer cells without any telomerase. Moreover several studies have found normal telomere lengths in somatic human cells after a normal lifetime, even of 100 y.93-96 Such cells would also not depend on "immortalization" by activated telomerase to produce sufficient numbers of cells for carcinogenesis. So why are all cancers immortal, if immortality is not necessary to produce sufficient numbers of cells for fatal cancers?
Only the speciation theory of cancer offers a consistent explanation for the characteristic immortality of cancer cells: since cancers are indeed autonomous species of their own, they need a stable and, thus, immortal karyotype like all other species50-55—the price “for crossing the threshold of autonomy,” as Huxley pointed out in 1956.1 The dynamic equilibrium between inherent karyotypic variation and selection for cancer-specific autonomy advanced here is a stable yet flexible mechanism that satisfies this requirement.11-13 The same mechanism would also explain how cancers escape Muller’s ratchet.
This view confirms and explains Blagosklonny’s proposal that immortality must be “a byproduct of something that brings about an immediate replicative advantage”6—namely cancer-specific reproductive autonomy.
Flexibility of cancer karyotypes generating drug-resistance and metastasis at high rates
Immortality is just one of several consequences of the inherent karyotypic flexibility of cancer karyotypes. The karyotypic flexibility of cancers also explains the spontaneous karyotypic variations that generate subspecies that are drug-resistant80,85 or metastatic84—at rates of 10−1 to 10−5.80,84,85,97,98 These rates far exceed those of conventional gene mutations, which occur in cancer cells only at rates of 10−6 per dominant and 10−12 per recessive gene per mitosis.77-79 Thus the high, cancer-specific rates of phenotypic variations, to metastasis or drug-resistance, are consequences of inherently flexible cancer karyotypes, rather than of gene mutations.
The karyotypic variations that keep cancers immortal over thousands of cell generations11 also depend on these high rates of variation in order to exceed the rates of conventional lethal mutations of Muller’s ratchet.
In sum, the inherent karyotypic flexibility of cancer is the basis for its existence, considering that it originates with an unstable karyotype, and it is the common cause of its most notorious, if gratuitous acquisitions: drug-resistance and metastasis. In the words of cancer researcher and physician Mark Vincent, “Thus is constructed a machine in which the associated DNA instability is not just a pointless collection of errors, but in fact the principal means to the goal of cellular survival.”13.
Materials and Methods
Cells and cell culture
The immortal, tumorigenic human cell lines, HA1, HA1–2 and BJ, prepared as described previously,29 were kindly provided by Robert Weinberg (Massachusetts Institute of Technology). Chiara Mondello (Istituto di Genetica Molecolare, University of Pavia) kindly provided the cen3tel cultures of primary skin cells at various generations after transfection with artificially overexpressed telomerase analyzed here. The primary cen3-cells had been derived from a human centenarian, as described previously.32,40 The cultures studied are labeled following the generations or population doublings (PD) after transfection of the primary culture with overexpressed telomerase, namely PD37, PD100, PD166, PD617, PD1014 and cen3telS2 PD84 and PD143.
Primary lung and tail cultures were prepared from a young telomerase-minus mouse, strain B6.Cg-Terc, from the Jackson Laboratory, Bar Harbor, ME. For this purpose the lung and tail of the mouse were excised shortly after it was killed with CO2. The tissues were then minced with razor blades, washed free of blood cells and debris and then incubated in serum-free medium containing about 0.5% trypsin at 37°C on a shaker for 15 to 30 min. We monitored the dissociation of tissue nuggets into single cells by checking aliquots of the trypsin-digest under the microscope. Cells were then incubated in culture medium RPM1640 supplemented with 5% fetal calf serum to which 0.1 to 0.5 mg/ml collagenase (Sigma) was added to dissociate trypsin resistant cell nuggets for several up to 24 h, depending on the degree of dissociation. SV40 was kindly provided by Michele Carbone (Cancer Research Center of Hawaii, University of Hawaii) or purchased from the American Type Culture Collection. The mouse cell cultures were then infected with SV40 virus and incubated 2–3 mo until foci of transformed cells were observed, as described previously.45
Karyotyping human and mouse cells
One to 2 days before karyotyping, cells were seeded at about 50% confluence in 3 ml RPMI 1640 medium containing 5% fetal calf serum in a 5-cm culture dish. After reaching ~75% confluence, Colcemid (KaryoMax Colcemid solution; Gibco Invitrogen) was added to 50 ng per ml medium. The culture was then incubated at 37°C for 4–12 h. Subsequently, cells were dissociated with trypsin, washed in 3 ml of physiological saline (depending on purity) and incubated in 0.075 molar KCl at 37°C for 15 min. Subsequently the solution was cooled in ice-water mixed with 0.1 volume of freshly mixed glacial acetic acid-methanol (1:3, vol. per vol.) and centrifuged at 800 g. The cell pellet was then gradually suspended in the methanol-acetic a solution and incubated at room temperature for 15 min. The cells were then pipetted onto a microscope slide to give a suitable density of metaphases for microscopic analysis. If necessary, residual cellular debris was removed by resuspending the cells again in methanol-acetic acid and recentrifuging at 800 g. Slides with suitable metaphase chromosomes were hybridized with chromosome-specific, color-coded DNA probes (MetaSystems) as described by the manufacturer and by us previously.45,84
Acknowledgments
We specifically thank Chiara Mondello (Istituto di Genetica Molecolare, University of Pavia) for providing the cen3tel system of human skin fibroblasts to study immortalization and for her reliable and generous advice. Further, we thank Douglas Brash (Yale Comprehensive Cancer Center, Yale School of Medicine), Josh Nicholson (Department of Biological Sciences, Virginia Tech) and Rainer Sachs (Dept. Mathematics and Physics, UC Berkeley) for critical reviews and helpful comments on the manuscript. In addition we gratefully acknowledge Alfred Boecking (Universitätsklinikum Duesseldorf), Michele Carbone (Cancer Research Center of Hawaii, University of Hawaii), Alice Fabarius (University of Heidelberg at Mannheim), Richard Harland (Department Molecular and Cell Biology, UC Berkeley), Ruediger Hehlmann (University of Heidelberg at Mannheim), Daniele Mandrioli (Cesare Maltoni Cancer Research Center, Ramazzini Insitute), David Rasnick (former visiting scholar at UC Berkeley) and Peter Walian (Lawrence Berkeley National Laboratory, Donner Lab) for valuable information, many timely alerts and critical discussions. Finally we thank our three summer interns of 2012, Elisabeth Berger (Berkeley and Brown University), Gregory Hui and Max Duesberg for catalyzing this project by their lively discussions and ideas. Bong-Gyoon Han is specifically thanked for drafting and shaping Figure 11 (Lawrence Berkeley National Laboratory, Donner Lab). Our research would not have been possible without the generous support from the philanthropists Dr. Christian Fiala, Rajeev and Christine Joshi, Robert Leppo, Peter Rozsa of the Taubert Memorial Foundation, Howard Urnovitz (Chronix Biomedical), a Foundation that prefers to remain anonymous and other private sources.
Suxbmitted
11/01/12
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23720
References
- 1.Huxley J. Cancer biology: comparative and genetic. Biol Rev Camb Philos Soc. 1956;31:474–514. doi: 10.1111/j.1469-185X.1956.tb01558.x. [DOI] [Google Scholar]
- 2.Hauschka TS. The chromosomes in ontogeny and oncogeny. Cancer Res. 1961;21:957–74. [PubMed] [Google Scholar]
- 3.Koller PC. The role of chromosomes in cancer biology. Recent Results Cancer Res. 1972;38:1–122. doi: 10.1007/978-3-642-80680-3_1. [DOI] [PubMed] [Google Scholar]
- 4.Dermer G. The immortal cells: why cancer research fails. Garden City Park, New York: Avery Publishing Group Inc., 1993. [Google Scholar]
- 5.Weinberg R. The biology of cancer. Garland Science, 2007. [Google Scholar]
- 6.Blagosklonny MV. Cell immortality and hallmarks of cancer. Cell Cycle. 2003;2:296–9. doi: 10.4161/cc.2.4.470. [DOI] [PubMed] [Google Scholar]
- 7.Lewin B, Cassimeris L, Lingappa VR, Plopper B, eds. Cells. Sudbury, Massachusetts (Boston, Toronto, London, Singapore): Jones & Bartlett publishers, 2007. [Google Scholar]
- 8.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York: Garland Publishing, Inc., 2008. [Google Scholar]
- 9.Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, et al. Molecular Cell Biology. New York and Basingstoke UK: W. H. Freeman, 2008. [Google Scholar]
- 10.Vincent MD. The animal within: carcinogenesis and the clonal evolution of cancer cells are speciation events sensu stricto. Evolution. 2010;64:1173–83. doi: 10.1111/j.1558-5646.2009.00942.x. [DOI] [PubMed] [Google Scholar]
- 11.Duesberg P, Mandrioli D, McCormack A, Nicholson JM. Is carcinogenesis a form of speciation? Cell Cycle. 2011;10:2100–14. doi: 10.4161/cc.10.13.16352. [DOI] [PubMed] [Google Scholar]
- 12.Heng HH, Stevens JB, Bremer SW, Liu G, Abdallah BY, Ye CJ. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res. 2011;112:217–53. doi: 10.1016/B978-0-12-387688-1.00008-9. [DOI] [PubMed] [Google Scholar]
- 13.Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283–350. doi: 10.1016/B978-0-12-387688-1.00010-7. [DOI] [PubMed] [Google Scholar]
- 14.Knauss S, Klein A. From aneuploidy to cancer: the evolution of a new species? J Biosci. 2012;37:211–20. doi: 10.1007/s12038-012-9199-1. [DOI] [PubMed] [Google Scholar]
- 15.Rasnick D. The chromosomal imbalance theory of cancer. Boca Raton, FL, USA: CRC Press, Taylor and Francis Group, 2012. [Google Scholar]
- 16.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 17.Cheung PY, Deng W, Man C, Tse WW, Srivastava G, Law S, et al. Genetic alterations in a telomerase-immortalized human esophageal epithelial cell line: implications for carcinogenesis. Cancer Lett. 2010;293:41–51. doi: 10.1016/j.canlet.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Harris H. A long view of fashions in cancer research. Bioessays. 2005;27:833–8. doi: 10.1002/bies.20263. [DOI] [PubMed] [Google Scholar]
- 19.Nicholson JM, Cimini D. How mitotic errors contribute to karyotypic diversity in cancer. Adv Cancer Res. 2011;112:43–75. doi: 10.1016/B978-0-12-387688-1.00003-X. [DOI] [PubMed] [Google Scholar]
- 20.Muller H. The relation of recombination to mutation advance. Mutat Res. 1964;1:2e9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 21.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–56. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parris G. The cell clone ecology hypothesis and the cell fusion model of cancer progression and metastasis (II): three pathways for spontaneous cell-cell fusion and escape from the intercellular matrix. Med Hypotheses. 2006;67:172–6. doi: 10.1016/j.mehy.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 23.Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126:477–87. doi: 10.1016/j.cell.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bignold LP. Aneuploidy as a counter to “Muller’s ratchet” in cancer cells. Cancer Genet Cytogenet. 2007;178:173–4. doi: 10.1016/j.cancergencyto.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Bignold LP. Variation, “evolution”, immortality and genetic instabilities in tumour cells. Cancer Lett. 2007;253:155–69. doi: 10.1016/j.canlet.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Naugler CT. Population genetics of cancer cell clones: possible implications of cancer stem cells. Theor Biol Med Model. 2010;7:42. doi: 10.1186/1742-4682-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 28.Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA. 1998;95:14723–8. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–86. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 31.Milyavsky M, Shats I, Erez N, Tang X, Senderovich S, Meerson A, et al. Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res. 2003;63:7147–57. [PubMed] [Google Scholar]
- 32.Mondello C, Chiesa M, Rebuzzini P, Zongaro S, Verri A, Colombo T, et al. Karyotype instability and anchorage-independent growth in telomerase-immortalized fibroblasts from two centenarian individuals. Biochem Biophys Res Commun. 2003;308:914–21. doi: 10.1016/S0006-291X(03)01484-0. [DOI] [PubMed] [Google Scholar]
- 33.Counter CM, Meyerson M, Eaton EN, Ellisen LW, Caddle SD, Haber DA, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998;16:1217–22. doi: 10.1038/sj.onc.1201882. [DOI] [PubMed] [Google Scholar]
- 34.Kendall SD, Linardic CM, Adam SJ, Counter CM. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005;65:9824–8. doi: 10.1158/0008-5472.CAN-05-1543. [DOI] [PubMed] [Google Scholar]
- 35.Vaziri H, Squire JA, Pandita TK, Bradley G, Kuba RM, Zhang H, et al. Analysis of genomic integrity and p53-dependent G1 checkpoint in telomerase-induced extended-life-span human fibroblasts. Mol Cell Biol. 1999;19:2373–9. doi: 10.1128/mcb.19.3.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol. 2002;34:1382–94. doi: 10.1016/S1357-2725(02)00047-X. [DOI] [PubMed] [Google Scholar]
- 37.Steinert S, White DM, Zou Y, Shay JW, Wright WE. Telomere biology and cellular aging in nonhuman primate cells. Exp Cell Res. 2002;272:146–52. doi: 10.1006/excr.2001.5409. [DOI] [PubMed] [Google Scholar]
- 38.Toouli CD, Huschtscha LI, Neumann AA, Noble JR, Colgin LM, Hukku B, et al. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene. 2002;21:128–39. doi: 10.1038/sj.onc.1205014. [DOI] [PubMed] [Google Scholar]
- 39.Fauth C, O’Hare MJ, Lederer G, Jat PS, Speicher MR. Order of genetic events is critical determinant of aberrations in chromosome count and structure. Genes Chromosomes Cancer. 2004;40:298–306. doi: 10.1002/gcc.20051. [DOI] [PubMed] [Google Scholar]
- 40.Zongaro S, de Stanchina E, Colombo T, D’Incalci M, Giulotto E, Mondello C. Stepwise neoplastic transformation of a telomerase immortalized fibroblast cell line. Cancer Res. 2005;65:11411–8. doi: 10.1158/0008-5472.CAN-05-1140. [DOI] [PubMed] [Google Scholar]
- 41.Piao CQ, Liu L, Zhao YL, Balajee AS, Suzuki M, Hei TK. Immortalization of human small airway epithelial cells by ectopic expression of telomerase. Carcinogenesis. 2005;26:725–31. doi: 10.1093/carcin/bgi016. [DOI] [PubMed] [Google Scholar]
- 42.Mahale AM, Khan ZA, Igarashi M, Nanjangud GJ, Qiao RF, Yao S, et al. Clonal selection in malignant transformation of human fibroblasts transduced with defined cellular oncogenes. Cancer Res. 2008;68:1417–26. doi: 10.1158/0008-5472.CAN-07-3021. [DOI] [PubMed] [Google Scholar]
- 43.Tabach Y, Kogan-Sakin I, Buganim Y, Solomon H, Goldfinger N, Hovland R, et al. Amplification of the 20q chromosomal arm occurs early in tumorigenic transformation and may initiate cancer. PLoS ONE. 2011;6:e14632. doi: 10.1371/journal.pone.0014632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franco S, MacKenzie KL, Dias S, Alvarez S, Rafii S, Moore MA. Clonal variation in phenotype and life span of human embryonic fibroblasts (MRC-5) transduced with the catalytic component of telomerase (hTERT) Exp Cell Res. 2001;268:14–25. doi: 10.1006/excr.2001.5264. [DOI] [PubMed] [Google Scholar]
- 45.Li L, McCormack AA, Nicholson JM, Fabarius A, Hehlmann R, Sachs RK, et al. Cancer-causing karyotypes: chromosomal equilibria between destabilizing aneuploidy and stabilizing selection for oncogenic function. Cancer Genet Cytogenet. 2009;188:1–25. doi: 10.1016/j.cancergencyto.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 46.Argilla D, Chin K, Singh M, Hodgson JG, Bosenberg M, de Solórzano CO, et al. Absence of telomerase and shortened telomeres have minimal effects on skin and pancreatic carcinogenesis elicited by viral oncogenes. Cancer Cell. 2004;6:373–85. doi: 10.1016/j.ccr.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 47.Shay JW, Wright WE. Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen. Exp Cell Res. 1989;184:109–18. doi: 10.1016/0014-4827(89)90369-8. [DOI] [PubMed] [Google Scholar]
- 48.Huschtscha LI, Holliday R. Limited and unlimited growth of SV40-transformed cells from human diploid MRC-5 fibroblasts. J Cell Sci. 1983;63:77–99. doi: 10.1242/jcs.63.1.77. [DOI] [PubMed] [Google Scholar]
- 49.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/S0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 50.King M. Species evolution: the role of chromosome change. Cambridge: Cambridge University Press, 1993. [Google Scholar]
- 51.O’Brien SJ, Menotti-Raymond M, Murphy WJ, Nash WG, Wienberg J, Stanyon R, et al. The promise of comparative genomics in mammals. Science. 1999;286:458–62, 479-81. doi: 10.1126/science.286.5439.458. [DOI] [PubMed] [Google Scholar]
- 52.Matzuk MM. Germ-line immortality. Proc Natl Acad Sci USA. 2004;101:16395–6. doi: 10.1073/pnas.0407344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones DL. Aging and the germ line: where mortality and immortality meet. Stem Cell Rev. 2007;3:192–200. doi: 10.1007/s12015-007-0009-3. [DOI] [PubMed] [Google Scholar]
- 54.Cairns-Smith AG. Chp 2, The immortal germline In: Marsh J, Goode J, eds. Ciba Foundation Symposium 182 - Germline Development. Cambridge university press: Norvatis, 2007. [Google Scholar]
- 55.Brown JD, O’Neill RJ. Chromosomes, conflict, and epigenetics: chromosomal speciation revisited. Annu Rev Genomics Hum Genet. 2010;11:291–316. doi: 10.1146/annurev-genom-082509-141554. [DOI] [PubMed] [Google Scholar]
- 56.Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–65. [PubMed] [Google Scholar]
- 57.Wolman SR. Cytogenetic heterogeneity: its role in tumor evolution. Cancer Genet Cytogenet. 1986;19:129–40. doi: 10.1016/0165-4608(86)90380-8. [DOI] [PubMed] [Google Scholar]
- 58.Wolman SR, Camuto PM, Perle MA. Cytogenetic diversity in primary human tumors. J Cell Biochem. 1988;36:147–56. doi: 10.1002/jcb.240360206. [DOI] [PubMed] [Google Scholar]
- 59.Li R, Sonik A, Stindl R, Rasnick D, Duesberg P. Aneuploidy vs. gene mutation hypothesis of cancer: recent study claims mutation but is found to support aneuploidy. Proc Natl Acad Sci USA. 2000;97:3236–41. doi: 10.1073/pnas.97.7.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li R, Rasnick D, Duesberg P. Correspondence re: D. Zimonjic et al., Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res., 61: 8838-8844, 2001. Cancer Res. 2002;62:6345–8, author reply 6348-9. [PubMed] [Google Scholar]
- 61.Nicholson JM, Duesberg P. On the karyotypic origin and evolution of cancer cells. Cancer Genet Cytogenet. 2009;194:96–110. doi: 10.1016/j.cancergencyto.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Fabarius A, Li R, Yerganian G, Hehlmann R, Duesberg P. Specific clones of spontaneously evolving karyotypes generate individuality of cancers. Cancer Genet Cytogenet. 2008;180:89–99. doi: 10.1016/j.cancergencyto.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 63.Levan A, Biesele JJ. Role of chromosomes in cancerogenesis, as studied in serial tissue culture of mammalian cells. Ann N Y Acad Sci. 1958;71:1022–53. doi: 10.1111/j.1749-6632.1958.tb46820.x. [DOI] [PubMed] [Google Scholar]
- 64.Yerganian G, Shein HM, Enders JF. Chromosomal disturbances observed in human fetal renal cells transformed in vitro by simian virus 40 and carried in culture. Cytogenetics. 1962;1:314–24. doi: 10.1159/000129742. [DOI] [PubMed] [Google Scholar]
- 65.Vogt M, Dulbecco R. Steps in the neoplastic transformation of hamster embryo cells by polyoma virus. Proc Natl Acad Sci USA. 1963;49:171–9. doi: 10.1073/pnas.49.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oshimura M, Barrett JC. Chemically induced aneuploidy in mammalian cells: mechanisms and biological significance in cancer. Environ Mutagen. 1986;8:129–59. doi: 10.1002/em.2860080112. [DOI] [PubMed] [Google Scholar]
- 67.Duesberg P, Li R, Rasnick D, Rausch C, Willer A, Kraemer A, et al. Aneuploidy precedes and segregates with chemical carcinogenesis. Cancer Genet Cytogenet. 2000;119:83–93. doi: 10.1016/S0165-4608(99)00236-8. [DOI] [PubMed] [Google Scholar]
- 68.Klein A, Li N, Nicholson JM, McCormack AA, Graessmann A, Duesberg P. Transgenic oncogenes induce oncogene-independent cancers with individual karyotypes and phenotypes. Cancer Genet Cytogenet. 2010;200:79–99. doi: 10.1016/j.cancergencyto.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 69.Heng HH, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy PV, et al. Stochastic cancer progression driven by non-clonal chromosome aberrations. J Cell Physiol. 2006;208:461–72. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- 70.Berman J. Evolutionary genomics: When abnormality is beneficial. Nature. 2010;468:183–4. doi: 10.1038/468183a. [DOI] [PubMed] [Google Scholar]
- 71.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–5. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mellors RC, Keane JF, Jr., Papanicolaou GN. Nucleic acid content of the squamous cancer cell. Science. 1952;116:265–9. doi: 10.1126/science.116.3011.265. [DOI] [PubMed] [Google Scholar]
- 73.Spriggs AI. Cytogenetics of cancer and precancerous states of the cervix uteri. In: German J, ed. Chromosomes and cancer. New York: John Wiley, 1974:423. [Google Scholar]
- 74.Cairns J. Cancer. Science and Society. San Francisco: W. H. Freeman, 1978. [Google Scholar]
- 75.Ostano P, Bione S, Belgiovine C, Chiodi I, Ghimenti C, Scovassi AI, et al. Cross-analysis of gene and miRNA genome-wide expression profiles in human fibroblasts at different stages of transformation. OMICS. 2012;16:24–36. doi: 10.1089/omi.2011.0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 77.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 78.Wang TL, Rago C, Silliman N, Ptak J, Markowitz S, Willson JK, et al. Prevalence of somatic alterations in the colorectal cancer cell genome. Proc Natl Acad Sci USA. 2002;99:3076–80. doi: 10.1073/pnas.261714699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shibata D, Lieber MR. Is there any genetic instability in human cancer? DNA Repair (Amst) 2010;9:858–, discussion 859-60. doi: 10.1016/j.dnarep.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li R, Hehlman R, Sachs R, Duesberg P. Chromosomal alterations cause the high rates and wide ranges of drug resistance in cancer cells. Cancer Genet Cytogenet. 2005;163:44–56. doi: 10.1016/j.cancergencyto.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 81.Tooze J. The Molecular Biology of Tumour Viruses. New York: Cold Spring Harbor,1973. [Google Scholar]
- 82.Ahuja D, Sáenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–45. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 83.Ewald D, Li M, Efrat S, Auer G, Wall RJ, Furth PA, et al. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science. 1996;273:1384–6. doi: 10.1126/science.273.5280.1384. [DOI] [PubMed] [Google Scholar]
- 84.Duesberg P, Iacobuzio-Donahue C, Brosnan JA, McCormack A, Mandrioli D, Chen L. Origin of metastases: subspecies of cancers generated by intrinsic karyotypic variations. Cell Cycle. 2012;11:1151–66. doi: 10.4161/cc.11.6.19580. [DOI] [PubMed] [Google Scholar]
- 85.Duesberg P, Li R, Sachs R, Fabarius A, Upender MB, Hehlmann R. Cancer drug resistance: the central role of the karyotype. Drug Resist Updat. 2007;10:51–8. doi: 10.1016/j.drup.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 86.Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations in Cancer, <http://cgap.nci.nih.gov/Chromosomes/Mitelman>. NIH, 2012.
- 87.McAllister RM, Corriell LL. Tumorigenicity of tissue culture cells. Cancer Res. 1959;19:1040–4. [Google Scholar]
- 88.Schmitt H, Blin N, Zankl H, Scherthan H. Telomere length variation in normal and malignant human tissues. Genes Chromosomes Cancer. 1994;11:171–7. doi: 10.1002/gcc.2870110306. [DOI] [PubMed] [Google Scholar]
- 89.Holliday R. Endless quest. Bioessays. 1996;18:3–5. doi: 10.1002/bies.950180103. [DOI] [PubMed] [Google Scholar]
- 90.Bryan TM, Reddel RR. Telomere dynamics and telomerase activity in in vitro immortalised human cells. Eur J Cancer. 1997;33:767–73. doi: 10.1016/S0959-8049(97)00065-8. [DOI] [PubMed] [Google Scholar]
- 91.Pitot HC. Fundamentals of Oncology. New York: Marcel Dekker, Inc., 2002. [Google Scholar]
- 92.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 93.Zhai G, Aviv A, Hunter DJ, Hart DJ, Gardner JP, Kimura M, et al. Reduction of leucocyte telomere length in radiographic hand osteoarthritis: a population-based study. Ann Rheum Dis. 2006;65:1444–8. doi: 10.1136/ard.2006.056903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.MacIntyre A, Brouilette SW, Lamb K, Radhakrishnan K, McGlynn L, Chee MM, et al. Association of increased telomere lengths in limited scleroderma, with a lack of age-related telomere erosion. Ann Rheum Dis. 2008;67:1780–2. doi: 10.1136/ard.2007.086652. [DOI] [PubMed] [Google Scholar]
- 95.Xing J, Ajani JA, Chen M, Izzo J, Lin J, Chen Z, et al. Constitutive short telomere length of chromosome 17p and 12q but not 11q and 2p is associated with an increased risk for esophageal cancer. Cancer Prev Res (Phila) 2009;2:459–65. doi: 10.1158/1940-6207.CAPR-08-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chiang YJ, Calado RT, Hathcock KS, Lansdorp PM, Young NS, Hodes RJ. Telomere length is inherited with resetting of the telomere set-point. Proc Natl Acad Sci USA. 2010;107:10148–53. doi: 10.1073/pnas.0913125107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duesberg P, Stindl R, Hehlmann R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc Natl Acad Sci USA. 2000;97:14295–300. doi: 10.1073/pnas.97.26.14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duesberg P, Stindl R, Hehlmann R. Origin of multidrug resistance in cells with and without multidrug resistance genes: chromosome reassortments catalyzed by aneuploidy. Proc Natl Acad Sci USA. 2001;98:11283–8. doi: 10.1073/pnas.201398998. [DOI] [PMC free article] [PubMed] [Google Scholar]