

Abstract
Objective
Pioglitazone (PIO), an antidiabetic drug of the thiazolidinedione family, improves glucose and lipid metabolism in muscle, adipose, and liver tissues via peroxisome proliferator-activated receptor gamma (PPARγ) activation. We hypothesize that PIO therapy will improve the metabolic status of offspring exposed to maternal obesity in a mouse model developmentally programmed for metabolic syndrome (MetS).
Study Design
CD-1 female mice were fed a high-fat diet for 3 months prior to breeding and throughout pregnancy and lactation. The pups were weaned to a standard-fat diet. Offspring were randomly assigned to receive 40 mg/kg of PIO in 0.5% of methyl cellulose or 0.5% methyl cellulose by daily oral gavage for 2 weeks. The pre- and post-treatment total body weights of the pups were recorded. Visceral (VAT) and subcutaneous (SAT) adipose tissue were evaluated using micro-computed tomography. Serum analytes were measured. Post-treatment, minimally invasive microendoscopic fluorescence confocal imaging and intraperitoneal glucose tolerance tests were performed. The data were analyzed using appropriate statistical tests (significance, P<0.05).
Results
PIO therapy resulted in lower total body weight and lower VAT gain, and increased SAT. PIO significantly lowered triglyceride, insulin levels, and HOMA-IR in males, and fasting glucose in females. There was a trend toward larger adipocyte size.
Conclusion
Short-term PIO therapy in the offspring of obese mothers attenuates metabolic changes associated with the developmental programming of metabolic syndrome. These novel data suggest a potential role for drugs that activate PPARγ receptors to prevent MetS in the adult offspring at risk to develop metabolic alterations.
Keywords: fetal programming, in vivo adipose tissue imaging, maternal obesity, mice, offspring, pioglitazone
INTRODUCTION
According to the International Diabetes Federation, metabolic syndrome (MetS) is defined by the presence of central obesity and at least two of the following components: hypertension, glucose intolerance, and dyslipidemia. The MetS has reached epidemic proportions in many developed countries. Consequently, understanding the detrimental potential of obesity and its unfavorable consequences is vital to the development of preventative measures.1 Traditionally, metabolic syndrome and obesity research has focused on lifestyle factors in childhood and adulthood. More recently, the role of pregnancy in the obesity epidemic is beginning to be recognized. The rate of obesity in pregnant women worldwide is between 18.5% and 38.3%, and these rates continue to rise.2 Epidemiological and experimental studies demonstrate that offspring from obese mothers are at an increased risk of obesity as well as other features and complications of MetS.3–7 For example, the relative risk of childhood obesity associated with maternal obesity in the first trimester of pregnancy was found to be increased by 2-fold at age 2 to 3 years, and 2.3-fold at 4 years of age.3 MetS was found to be more common in adolescents who were born to obese mothers.5 Others have shown that the mother’s body mass index is one of the strongest predictors of overweight/obesity and percentage of body fat in her children.4;6 Adults born to obese mothers were more insulin resistant than those born to non obese mothers.8 Moreover, maternal prenatal fat intake was one of the strongest predictors of fat mass in the offspring at 10 years of age.9 Thus, there is strong accumulating evidence that offspring born of pregnancies complicated by obesity are at increased risk of obesity and other features of MetS.
Visceral adipose tissue dysfunction is emerging as the primary defect leading to MetS.10;11 Pathologic lipid accumulation and adipocyte hypertrophy, in addition to hypoxia and a variety of other processes lead to adipose tissue dysfunction and the development of metabolic and/or cardiovascular disorders. Previous studies, including our own, have demonstrated that adipose tissue dysfunction is present in fetal programming models of MetS. The most notable examples of adipose dysfunction in fetal programming models include elevated serum leptin levels, increased adipose tissue lipid accumulation, augmented expression of angiotensin, HIF-1α and PPARγ receptor, amplified activities of lipoprotein lipase and glycerol-3-phosphate dehydrogenase and decreased levels of antioxidant enzymes.7;12–15
Peroxisome proliferator-activated receptor gamma (PPARγ) is highly expressed in white and brown adipose tissue and plays a critical role in adipocyte differentiation and mature adipocyte maintenance. PPARγ regulates systemic insulin signaling via ligand-dependent transcriptional activation of target genes. In animal models, mRNA expression of the PPARγ is significantly increased in the adipose tissue of the offspring of obese dams.7;12 When administered to pregnant animals, thiazolidinediones, PPARγ agonists (TZDs,) improve maternal and fetal metabolic markers.16–20. However, none of these studies followed offspring development into adulthood nor there are studies which investigated the effects of TZDs if administered postnatally to offspring at risk to develop MetS, such as born to obese mothers.
In this study we tested the hypothesis that postnatal administration of the pioglitazone – a TZD class drug – will improve the metabolic status of adult offspring at risk to develop MetS due to exposure to a maternal high-fat diet during the prenatal and early postnatal periods.
MATERIAL AND METHODS
All procedures were approved by the Animal Care and Use Committee of the University of Texas Medical Branch. The animals were housed separately in temperature- and humidity-controlled quarters with constant 12:12-h light-dark cycles and were provided with food and water ad libitum.
Study design
In our lab we have established a mouse model of developmental MetS by feeding female mice a high-fat diet for 3 months before pregnancy, during pregnancy and until weaning. At 6 months of age offspring of these dams develop significant adiposity, hypertension, elevated fasting glycemia, and dyslipidemia when compared to pups born to mothers fed standard chow.21;22
For the current study female and male CD-1 mice were obtained from Charles Rivers Laboratories (Wilmington, MA). Female mice were approximately 4–5 weeks of age upon arrival. Females were fed high-fat (34.9% fat; D12492; Research diets Inc, New Brunswick, NJ) rodent chow for 3 months prior to breeding, during pregnancy, and until weaning (3 weeks postpartum), when all animals (mothers and offspring) were placed on the standard chow diet (5.6% fat; Teklad 7012: Harlan Teklad LM-485 Mouse/Rat Sterilizable diet; Harlan Teklad, Madison, WI). The fat source differed between diets: the high-fat diet used lard, and the standard-fat diet used soybean oil.
At 8 weeks of age, the offspring were subjected to fasting blood glucose measurements and blood collection for later analysis of triglyceride and insulin levels. Mice also underwent in vivo whole-body micro-computed tomography (CT) imaging. At 10 weeks of age, the pups were divided into control and treatment groups to include animals born to the same mother in both groups. The treatment group (PIO group, males n=6, females n=4) received 40 mg/kg pioglitazone (PIO, Sigma-Aldrich, ST. Louis, MO) dissolved in 0.5% of methyl cellulose by oral gavage (Fisher Scientific, Pittsburgh, PA).23 The control group (CTR group, males n=6, females n=4) received a similar amount of vehicle (0.5% methyl cellulose) only. Treatment was given daily (except on weekends) for 2 weeks, until 12 weeks of age. The selection of dosage and timing of the treatment was based on evidence from literature, where db/db mice, which are at risk to develop MetS (same as our animals), showed improvements in their metabolic parameters after 2–3 week PIO treatment.23;24 Following the treatment, intraperitoneal glucose tolerance tests (IGTT), fasting glucose measurements, in vivo micro-CT and confocal imaging were performed. One male per group died during post-treatment manipulations. Blood was collected for insulin and triglyceride analysis from the remaining animals, and then the offspring were euthanized. Visceral adipose tissue, including mesenteric, epididymal and perirenal fat, was excised and weighed.
In vivo imaging
Previously validated imaging methods for the quantitative assessment of visceral (VAT) and subcutaneous (SAT) adipose tissue and liver fat infiltration in rodents were used.25–27 Experimental protocols for non-invasive CT and minimally invasive confocal imaging were optimized in our lab over several months prior to the study. After administering anesthesia intraperitoneally with ketamine/xylazine (80–100 mg/kg and 5–10 mg/kg, respectively), mice were imaged with a small animal micro-CT scanner (Inveon™, Siemens Preclinical Solutions, Knoxville, Tennessee). Imaging parameters were set as follows: voltage 70 kV, current 500 μA, resolution 0.107 mm, exposure time 1000 ms, 520 steps and 360 degrees of rotations. Scan time was approximately 15 minutes per mouse. Mice were given supplemental oxygen via nose cone during scanning. The transverse views of CT images (one per animal) at the level of the 5th lumbar vertebra were selected for analysis of VAT, while transverse images at the level of 6th lumbar vertebra were used for analysis of SAT.25;26 The cross-sectional total body area and adipose tissue area were measured using Inveon Research Workplace software. The percentage of VAT (%VAT) and SAT (%SAT) was calculated from the cross-sectional total body area. The correlations between weight of VAT determined after extraction and determined by CT were significant (Pearson r=0.82, P=0.003 for males and r=0.82, P=0.01 for females). Liver and spleen radiodensity values in a transverse section between the 13th thoracic and 1st lumbar vertebrae were expressed as liver-to-spleen density ratio and compared between the groups. The radiodensity of the liver normalized to the radiodensity of the spleen (liver-to-spleen radiodensity ratio) indicated fat infiltration in the liver.25;26;28;29
To quantitatively assess PIO treatment-induced changes in adipocyte size, offspring underwent high-resolution minimally invasive microendoscopic fluorescence confocal imaging using a Cell-Vizio-488 confocal microendoscope (Mauna Kea Technologies, France). A fluorescent 1% Fluorescein Sodium solution (0.2–0.3 ml/animal) was injected via the tail vein or intraperitoneally. A 1.8 mm probe was inserted through an abdominal incision. Confocal images of mesenteric adipocytes in VAT were exported into Image J software (NIH) and the cell area was measured by manually tracing the cell borders.30 Every cell within five imaging fields was measured and the mean cell area was calculated for each animal. The mean area values were used to compare the two groups.
Individuals interpreting VAT, SAT liver/spleen radiodensity and adipocyte size were blinded to the treatment/control group allocation.
Intraperitoneal Glucose Tolerance Test (IGTT)
IGTTs were performed following 2 weeks of PIO or vehicle administration. Mice were fasted overnight (16–18 hrs) prior to a 20 % glucose bolus (1 g/kg per mouse) by intraperitoneal injection. Blood glucose levels were recorded prior to glucose injection and at 15, 30, 60, 90, and 120 min post injection. Overnight fasting and the glucose dose was selected based on our pilot studies in CD-1 mice. Sixty μl of blood was collected via tail vein prior to glucose injection for fasting insulin measurements. The blood was immediately centrifuged, and the serum was stored at −80°C until the insulin assay was performed.
Serologic Analysis
Blood glucose levels were measured using an OneTouch Ultra glucometer (LifeScan, Milpitas, CA) after an overnight (16–18 hrs) fast. Blood was collected from the offspring before and after treatment. Commercially available kits were used according to the manufacturer’s instructions to determine serum levels of triglycerides, cholesterol (BioAssay Systems, Hayward, CA), and insulin (CrystalChem, Downers Grove, IL).
Data Analysis
Data were analyzed as the difference from baseline (post treatment value - pretreatment value) and expressed as the mean ± SEM. The area under the IGTT curve for glucose was calculated using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). Insulin sensitivity was determined using the Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) and calculated by (fasting glucose level (mg/dL) × fasting insulin level (mU/L))/405.31;32 Statistical significance was determined using an unpaired Student’s t-test or Mann-Whitney test if the data were not normally distributed. The IGTT results were analyzed using a Two-way Repeated Measures ANOVA with Bonferonni correction. A probability (P) value of ≤0.05 was considered statistically significant.
RESULTS
There were no differences in total body weight, VAT and SAT area, fasting blood glucose and insulin levels between the two groups of mice at the start of the study (Tables 1, 2). Triglycerides levels were significantly higher in male mice assigned to PIO group (Table 2, P=0.01)
Table 1.
Baseline values before treatment for males in non-treated and Pioglitazone-treated groups after prenatal and early postnatal exposure to maternal high fat diet.
| Non-treated | Pioglitazone treated | P value | |
|---|---|---|---|
| Body weight (gm) | 38.4 ± 3.5 | 39.8 ± 4.7 | 0.6 |
| VAT as determined by CT (% from total area) | 28.7 ± 5.7 | 34.5 ± 2.9 | 0.4 |
| SAT as determined by CT % from total area) | 8.3 ± 1.3 | 8.9 ± 1.8 | 0.7 |
| Liver/spleen radiodensity (HU) | 0.7 ± 0.04 | 0.6 ± 0.02 | 0.6 |
| Triglyceride (mmol/L) | 2.3 ± 0.2 | 3.1 ± 0.2* | 0.01 |
| Fasting glucose (mg/dL) | 178 ± 11.5 | 164 ± 11.0 | 0.4 |
| Fasting insulin(ng/mL) | 0.8 ± .02 | 1.3 ± 0.2 | 0.1 |
| HOMA-IR | 5.8 ± 2 | 12 ± 3.2 | 0.2 |
Data are means ± SEM. VAT – visceral adipose tissue, SAT – subcutaneous adipose tissue, HOMA-IR – homeostasis model assessment of insulin resistance.
indicate statistically significant difference between the groups (P<0.05.)
Table 2.
Baseline values before treatment for females in non-treated and Pioglitazone-treated groups after prenatal and early postnatal exposure to maternal high fat diet.
| Non-treated | Pioglitazone treated | P value | |
|---|---|---|---|
| Body weight (gm) | 29.6 ± 2.4 | 32.2 ± 2.3 | 0.2 |
| VAT as determined by CT (% from total area) | 32.8 ± 3.5 | 37.5 ± 5.5 | 0.7 |
| SAT as determined by CT % from total area) | 10.8 ± 2.5 | 10.9 ± 2.4 | 1.0 |
| Liver/spleen radiodensity (HU) | 0.7 ± 0.02 | 0.7 ± 0.03 | 0.9 |
| Triglyceride (mmol/L) | 1.9 ± 0.3 | 2.4 ± 0.4 | 0.5 |
| Fasting glucose (mg/dL) | 70 ± 5.7 | 79 ± 7.4 | 0.6 |
| Fasting insulin(ng/mL) | 0.4 ± 0.07 | 0.5 ±0.1 | 0.7 |
| HOMA-IR | 2.5 ± 0.5 | 2.2 ± 0.6 | 0.9 |
Data are means ± SEM. VAT – visceral adipose tissue, SAT – subcutaneous adipose tissue, HOMA-IR – homeostasis model assessment of insulin resistance.
Effects of PIO treatment on outcome measures are presented in Tables 3 and 4: The PIO-treated males and females group gained less total body weight. Treated males acquired less VAT than animals in the CTR group. Females in both groups lost VAT with the PIO group losing less than CTR mice. The area of SAT increased in PIO-treated animals. The liver-to-spleen radiodensity ratio increased in males and females treated with PIO, suggesting that fat infiltration in the liver decreased as a result of PIO exposure (Tables 3–4, respectively). None of these changes/parameters were statistically significant between the groups.
Table 3.
Effects of Pioglitazone (change from baseline) on outcome measures for males in non-treated and Pioglitazone-treated groups after prenatal and early postnatal exposure to maternal high fat diet
| Non-treated | Pioglitazone treated | P value | |
|---|---|---|---|
| Body weight (gm) | 4.2 ± 1.0 | 0.3 ± 2.0 | 0.3 |
| VAT as determined by CT (% from total area) | 10.5 ± 5.0 | 6.2 ± 3.1 | 0.4 |
| SAT as determined by CT % from total area) | 1.2 ± 1.4 | 5.2± 1.8 | 0.2 |
| Liver/spleen radiodensity (HU) | 0.01 ± 0.04 | 0.08 ± 0.04 | 0.2 |
| Triglyceride (mmol/L) | 0.8 ± 0.2 | −0.9 ± 0.04* | 0.02 |
| Fasting glucose (mg/dL) | −32.4 ± 11.9 | −55.8 ± 7.6 | 0.1 |
| Fasting insulin(ng/mL) | 0.58 ± 0.2 | −0.6 ± 0.3* | 0.03 |
| HOMA-IR | 3.7 ± 1.8 | −7.3 ± 2.6* | 0.03 |
Data are means ± SEM. VAT – visceral adipose tissue, SAT – subcutaneous adipose tissue, HOMA-IR – homeostasis model assessment of insulin resistance.
indicate statistically significant difference between the groups (P<0.05.)
Table 4.
Effects of Pioglitazone (change from baseline) on outcome measures for females in non-treated and Pioglitazone-treated groups after prenatal and early postnatal exposure to maternal high fat diet
| Non-treated | Pioglitazone treated | P value | |
|---|---|---|---|
| Body weight (gm) | 2.3 ± 0.2 | 0.3 ± 0.9 | 0.34 |
| VAT as determined by CT (% from total area) | −10.5 ± 5.1 | −4.8 ± 2.7 | 0.5 |
| SAT as determined by CT (% from total area) | −0.7 ± 2.9 | 1.2 ± 1.5 | 0.8 |
| Liver/spleen radiodensity (HU) | 0.0006 ± 0.04 | 0.08 ± 0.02 | 0.3 |
| Triglyceride (mmol/L) | −0.3 ± 0.1 | −0.7 ± 0.6 | 0.7 |
| Fasting glucose (mg/dL) | 41.0 ± 8.1 | 5.5 ± 4.3* | 0.04 |
| Fasting insulin(ng/mL) | −0.15 ± 0.07 | 0.2 ± 0.3 | 0.4 |
| HOMA-IR | −0.7 ± 0.4 | 1.4 ± 1.3 | 0.4 |
Data are means ± SEM. VAT – visceral adipose tissue, SAT – subcutaneous adipose tissue, HOMA-IR – homeostasis model assessment of insulin resistance.
indicate statistically significant difference between the groups (P<0.05.)
Serum triglyceride levels significantly decreased in PIO-treated males, while increasing in control animals (Table 3, P=0.02). This change is especially significant, since PIO-assigned males had higher triglyceride levels before the treatment. In females, a decrease in triglyceride levels was also observed, though not statistically significant (Table 4).
There was no change in serum HDL, LDL/VLDL, and total cholesterol levels following treatment with PIO or vehicle (data not shown).
Both groups of males had reduced, but statistically not significant, fasting blood glucose levels, though the change in fasting glycemia of PIO-treated mice was larger (Table 3). In females fasting glucose levels increased, however, the change from baseline was significantly lower in mice which were administered PIO (Table 4, P=0.04). There were no significant differences in the IGTT blood glucose levels between the two male groups after treatment, although the PIO group blood glucose trended lower (Figures 1A and B). In females, there was a significant improvement in glucose IGTT as evidenced in Figure 2 – with glucose levels being prominently lower at 30 min post glucose challenge (Figure 2A, P<0.05) and with significantly lower area under the IGTT curve (Figure 2B, P=0.04). The area under the curve calculated from IGTT is one index of insulin sensitivity, thus insulin sensitivity was improved in mice treated with PIO reaching statistical significance in females.
Figure 1.
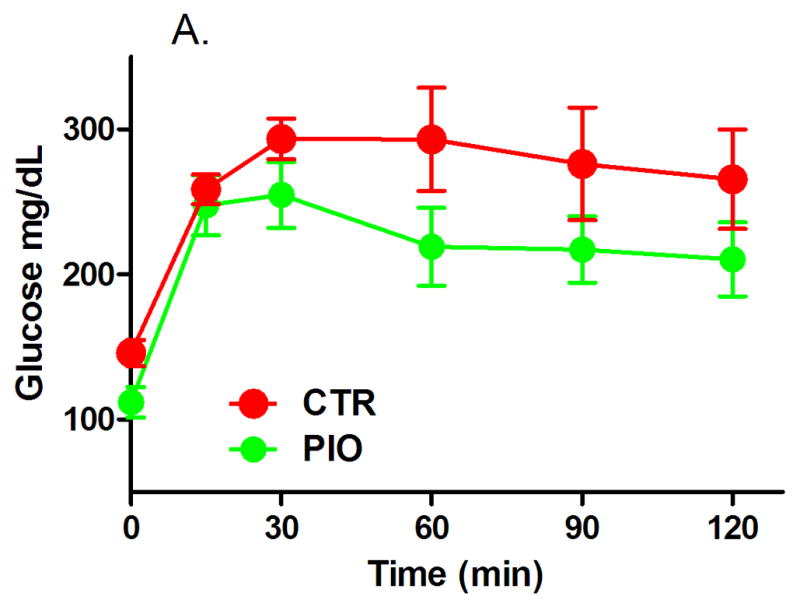

Results of intraperitoneal glucose tolerance test (A – IGTT, B – area under the curve) in male offspring with prenatal and early postnatal exposure to maternal high fat diet after treatment with vehicle (CTR) or pioglitazone (PIO). Data expressed as mean ± SEM.
Figure 2.
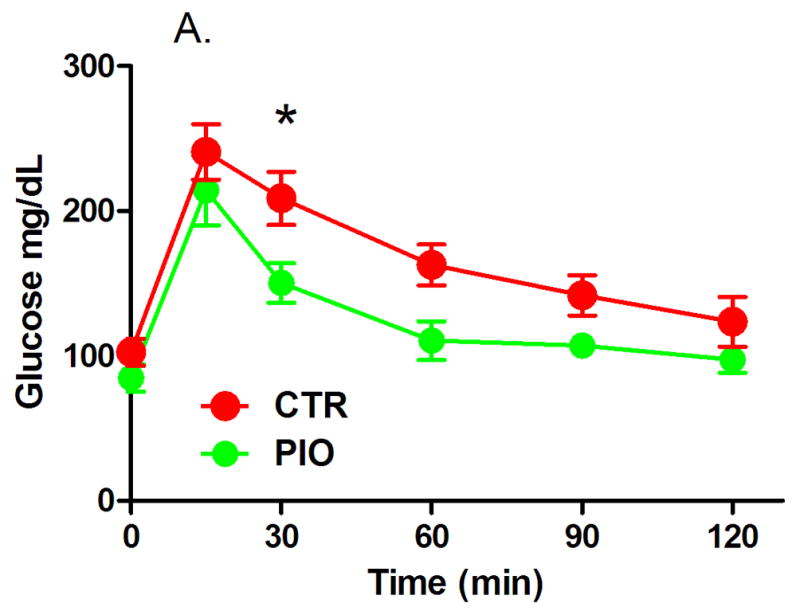
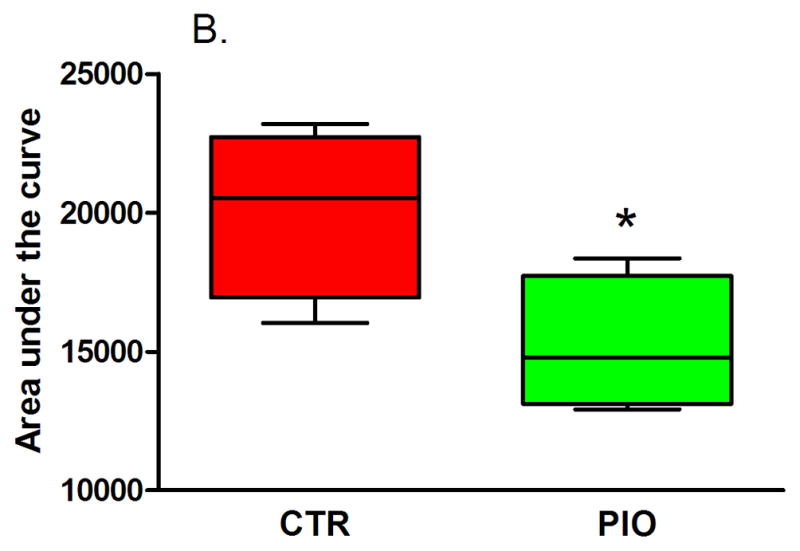
Results of intraperitoneal glucose tolerance test (A – IGTT, B – area under the curve) in female offspring with prenatal and early postnatal exposure to maternal high fat diet after treatment with vehicle (CTR) or pioglitazone (PIO). Data expressed as mean ± SEM. *indicates statistical significance (P<0.05).
Fasting insulin decreased after the treatment with PIO in males, while increasing in control animals (Table 3, P=0.03). In females, the opposite occurred – insulin increased in treated and somewhat decreased in control mice, though changes were not significantly different (Table 4). Similarly, insulin resistance assessed by HOMA-IR was improved in PIO-treated males (Table 3, P=0.03). In females, again, the opposite was observed – HOMA-IR was elevated in PIO-treated mice (Table 4).
Confocal imaging revealed that there were fewer adipocytes and they were larger in the PIO-treated mice compared to the non-treated controls (Table 5), though did not differ significantly between the groups.
Table 5.
Adipocyte size and numbers in male and females offspring after admisntration of pioglitazone or vehicle.
| Non-treated | Pioglitazone-treated | P value | |
|---|---|---|---|
| Average adipocyte size (μm2) | |||
| Males | 5740 ± 546.5 | 6460 ± 575.7 | 0.4 |
| Females | 3737 ± 412.9 | 4901 ± 711.4 | 0.2 |
| Average number of adipocytes | |||
| Males | 105.5 ± 11.9 | 102.2 ± 9.5 | 0.9 |
| Females | 169.5 ± 22.1 | 113.8 ± 15.6 | 0.08 |
COMMENT
In this study, we examined the metabolic effects of a 2-week PIO treatment in offspring exposed to a maternal prenatal and early postnatal high-fat diet. PIO therapy significantly reduced serum triglyceride, fasting glucose and insulin levels. There was a trend toward a smaller increase in VAT and body weight, a larger increase in SAT, improved glucose tolerance and insulin sensitivity, and larger adipocytes after exposure to PIO. Some of these changes were different in males and females. In addition being the first to investigate postnatal treatment in offspring of obese mothers, we are the first to demonstrate that PIO could be used to treat MetS overall.
A monthly quantitative noninvasive micro-CT scan of mice between 1 and 6 months of age determined that visceral adiposity and fat deposition in the liver appears in 3 month old animals if the mice were exposed to a maternal prenatal and early postnatal high-fat diet, therefore, the mice in our study were at risk to develop metabolic syndrome.22 PIO was first administered at 2.5 months of age, before a significant increase in the amount of VAT and hepatic fat infiltration was observed. Our demonstration that PIO can be administered to mice at-risk to develop MetS and that this treatment can improve their metabolic status compared to their non-treated counterparts is significant.
Some of the PIO effects (fasting glucose, triglyceride levels) were different in males and females. To our knowledge there are no definitive data on gender differences on the effects of TZDs, though some studies report a favorable effects of PIO on blood glucose control especially in female patients.33 We are inclined to attribute gender differences observed in our model to a short duration of PIO treatment.
TZDs are high-affinity ligands for PPARγ, and TZD activation of PPARγ in adipose tissue alters glucose and lipid metabolism to improve insulin sensitivity. Activated PPARγ promotes adipogenesis and enhances the ability of white fat to store dietary fatty acids. As a result, fatty acids are not stored in skeletal muscle or liver where their accumulation could impair insulin action; instead, fatty acids are retained in adipose tissue, leading to increases in body weight and adiposity. Consequently, TZDs reduce circulating free fatty acid levels and improve glycemic control as a result of reduced insulin resistance.34;35 In our study, we determined that a short, 2-week treatment with PIO was sufficient to lower triglyceride, fasting glucose and insulin levels in offspring that were born to mothers on high-fat diets. Similar results have been shown in humans with type 2 diabetes who received PIO for 24 weeks, in mice fed a high-fat diet for 6 months and then treated with PIO for 2–3 months, and in db/db mice after 4-week of PIO treatment.34;36–38 TZD therapy is associated with weight gain in some patients and in mouse models.34;36;38;39 PIO treatment did not significantly increase total body weight in our mice. On the contrary, the PIO group gained less weight than the CTR animals. There are several explanations for our findings. First, an increase in the treatment duration from two weeks to several months may have resulted in similar weight gain as that observed in the aforementioned studies. Second, when TZDs are given to pregnant animals on standard chow for a few days to a few weeks, fetal weight does not increase. In contrary, embryonic and neonatal weight reductions have been reported.16–19 Our findings confirm that short-duration treatment has no effect on weight gain and raises the possibility that TZDs do not affect body weight in fetal programming models.
Weight gain associated with TZD therapy is attributable to an increase in fat mass and fluid retention.39 TZD-related increase in fat mass is depot specific: CT scans indicate PIO treatment in humans increase SAT, but not VAT.37 Furthermore, when administered to pregnant animals, TZD does not lead to increased fetal visceral adiposity.16 Our CT imaging results of VAT and SAT are consistent with these findings.
Clinical studies have reported reduced hepatic fat following administration of TZDs.40 Similarly, the liver-to-spleen radiodensity ratio increased in our PIO-treated mice, which reflects a decrease in liver fat infiltration.
Previous studies regarding the effect of TZDs on adipocyte size were inconclusive. 34;38;41 In our study, although not statistically significant, we found larger adipocytes after the PIO treatment, which are in line with several earlier reports.38
TZDs, especially PIO, are the only antidiabetic drug class that affects dyslipidemia, one of the markers of MetS. PIO decreases triglyceride levels and increases HDL cholesterol levels without affecting LDL and total cholesterol levels.36 While we observed a significant decrease in triglyceride levels after PIO treatment, no other lipid-lowering effects were determined. We attribute this discrepancy to short-duration treatment, which was sufficient to significantly change fasting blood glucose levels, but not long enough to improve dyslipidemia other than circulating triglyceride levels. A longer treatment regimen may be required to observe changes in total cholesterol, HDL cholesterol and LDL/VLDL cholesterol levels.
The main limitation of our study is the short duration of treatment with PIO. We hypothesize that administering treatment for a longer period of time, up to several months, may lead to a significant improvement in the metabolic status of offspring born to obese mothers. In addition, post-treatment monitoring would determine if improvements in metabolic parameters in the offspring continue or diminish after the treatment is discontinued.
In summary, postnatal pioglitazone therapy attenuates metabolic changes associated with developmental programming in the offspring of obese mothers. Our data are novel and we propose a potential role for PPARγ ligands not only in the prevention of MetS in adult offspring of obese mothers but also as a novel treatment for MetS.
Acknowledgments
We would like to thank Ms. Fernanda Vergara for her assistance with imaging studies.
Dr. Egle Bytautiene, is supported by a research career development award (K12HD052023: Building Interdisciplinary Research Careers in Women’s Health Program -BIRCWH) from the National Institute of Allergy and Infectious Diseases (NIAID), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the Office of the Director (OD), National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID, NICHD, OD, or the National Institutes of Health.
This study was conducted with the support of the Institute for Translational Sciences at the University of Texas Medical Branch, supported in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, National Institutes of Health
Footnotes
DISCLOSURE: The authors report no conflict of interest.
Paper was presented at the 32nd Annual Meeting, Society of Maternal-Fetal Medicine, Dallas, TX, February 6–11, 2012
Reprints will not be available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl Health Stat Report. 2009;(13):1–7. [PubMed] [Google Scholar]
- 2.Galtier-Dereure F, Boegner C, Bringer J. Obesity and pregnancy: complications and cost. Am J Clin Nutr. 2000;71(5):1242S–1248. doi: 10.1093/ajcn/71.5.1242s. [DOI] [PubMed] [Google Scholar]
- 3.Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114(1):e29–e36. doi: 10.1542/peds.114.1.e29. [DOI] [PubMed] [Google Scholar]
- 4.Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113(10):1126–1133. doi: 10.1111/j.1471-0528.2006.00989.x. [DOI] [PubMed] [Google Scholar]
- 5.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 6.Koupil I, Toivanen P. Social and early-life determinants of overweight and obesity in 18-year-old Swedish men. Int J Obes. 2008;32(1):73–81. doi: 10.1038/sj.ijo.0803681. [DOI] [PubMed] [Google Scholar]
- 7.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EHJ, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51(2):383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 8.Mingrone G, Manco M, Valera Mora ME, Guidone C, Iaconelli A, Gniuli D, et al. Influence of Maternal Obesity on Insulin Sensitivity and Secretion in Offspring. Diabetes Care. 2008;31(9):1872–1876. doi: 10.2337/dc08-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brion MJ, Ness AR, Rogers I, Emmett P, Cribb V, vey Smith G, et al. Maternal macronutrient and energy intakes in pregnancy and offspring intake at 10 y: exploring parental comparisons and prenatal effects. The American Journal of Clinical Nutrition. 2010;91(3):748–756. doi: 10.3945/ajcn.2009.28623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bluher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117(6):241–250. doi: 10.1055/s-0029-1192044. [DOI] [PubMed] [Google Scholar]
- 11.Hajer GR, van Haeften TW, Visseren FLJ. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J. 2008;29(24):2959–2971. doi: 10.1093/eurheartj/ehn387. [DOI] [PubMed] [Google Scholar]
- 12.Muhlhausler BS, Duffield JA, McMillen IC. Increased maternal nutrition stimulates peroxisome proliferator activated receptor γ, adiponectin, and leptin messenger ribonucleic acid expression in adipose tissue before birth. Endocrinology. 2007;148(2):878–885. doi: 10.1210/en.2006-1115. [DOI] [PubMed] [Google Scholar]
- 13.Benkalfat NB, Merzouk H, Bouanane S, Merzouk SA, Bellenger J, Gresti J, et al. Altered adipose tissue metabolism in offspring of dietary obese rat dams. Clin Sci (Lond ) 2011;121(1):19–28. doi: 10.1042/CS20100534. [DOI] [PubMed] [Google Scholar]
- 14.Bytautiene E, Banerjee D, Kechichian T, Yin H, Sbrana E, Tamayo E, et al. Adipose tissue dysfunction in a model of developmental programming of metabolic syndrome. Am J Obstet Gynecol. 2012;206(1):S97–S98. [Google Scholar]
- 15.Bytautiene E, Kechichian T, Gamble P, Longo M, Saade GR. Antioxidant capacity of adipose tissue in offspring exposed to prenatal high fat diet. Reprod Sci. 2012;19(3):154A. [Google Scholar]
- 16.Muhlhausler BS, Morrison JL, McMillen IC. Rosiglitazone increases the expression of peroxisome proliferator-activated receptor γ target genes in adipose tissue, liver, and skeletal muscle in the sheep fetus in late gestation. Endocrinology. 2009;150(9):4287–4294. doi: 10.1210/en.2009-0462. [DOI] [PubMed] [Google Scholar]
- 17.Sevillano J, Lopez-Perez IC, Herrera E, Del Pilar RM, Bocos C. Englitazone administration to late pregnant rats produces delayed body growth and insulin resistance in their fetuses and neonates. Biochem J. 2005;389(Pt 3):913–918. doi: 10.1042/BJ20041837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaiff WT, Knapp FF, Barak Y, Biron-Shental T, Nelson DM, Sadovsky Y. Ligand-activated peroxisome proliferator activated receptor γ alters placental morphology and placental fatty acid uptake in mice. Endocrinology. 2007;148(8):3625–3634. doi: 10.1210/en.2007-0211. [DOI] [PubMed] [Google Scholar]
- 19.Klinkner DB, Lim HJ, Strawn J, Oldham KT, Sander TL. An in vivo murine model of rosiglitazone use in pregnancy. Fertil Steril. 2006;86(4, Supplement):1074–1079. doi: 10.1016/j.fertnstert.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 20.Sevillano J, de Castro J, Bocos C, Herrera E, Ramos MP. Role of insulin receptor substrate-1 serine 307 phosphorylation and adiponectin in adipose tissue insulin resistance in late pregnancy. Endocrinology. 2007;148(12):5933–5942. doi: 10.1210/en.2007-0352. [DOI] [PubMed] [Google Scholar]
- 21.Bytautiene E, Patrikeev I, Vergara F, Wei J, Vincent K, Motamedi M, et al. The relation between adiposity and metabolic profile: an In Vivo imaging study in a mouse model of fetal programming of obesity. Reprod Sci. 2011;18(3 S):369A. [Google Scholar]
- 22.Bytautiene E, Abate N, Vincent K, Patrikeev I, Wei J, Motamedi M, et al. Changes in visceral adiposity and liver fat in the offspring in a mouse model of pre-pregnancy obesity: a longitudinal study using computed tomography. Am J Obstet Gynecol. 2012;206(1, Supplement):S97. [Google Scholar]
- 23.Yamanaka M, Itakura Y, Tsuchida A, Nakagawa T, Noguchi H, Taiji M. Comparison of the antidiabetic effects of brain-derived neurotrophic factor and thiazolidinediones in obese diabetic mice. Diabetes Obes Metab. 2007;9(6):879–888. doi: 10.1111/j.1463-1326.2006.00675.x. [DOI] [PubMed] [Google Scholar]
- 24.Kanda Y, Shimoda M, Hamamoto S, Tawaramoto K, Kawasaki F, Hashiramoto M, et al. Molecular mechanism by which pioglitazone preserves pancreatic β-cells in obese diabetic mice: evidence for acute and chronic actions as a PPARγ agonist. Am J Physiol Endocrinol Metab. 2010;298(2):E278–E286. doi: 10.1152/ajpendo.00388.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lublinsky S, Luu Y, Rubin C, Judex S. Automated separation of visceral and subcutaneous adiposity in in vivo microcomputed tomographies of mice. J Digit Imaging. 2009;22(3):222–231. doi: 10.1007/s10278-008-9152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Judex S, Luu YK, Ozcivici E, Adler B, Lublinsky S, Rubin CT. Quantification of adiposity in small rodents using micro-CT. Methods. 2010;50(1):14–19. doi: 10.1016/j.ymeth.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lubura M, Hesse D, Neumann N, Scherneck S, Wiedmer P, Schümann A. Non-Invasive Quantification of White and Brown Adipose Tissues and Liver Fat Content by Computed Tomography in Mice. PLoS ONE. 2012;7(5):e37026. doi: 10.1371/journal.pone.0037026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodama Y, Ng CS, Wu TT, Ayers GD, Curley SA, Abdalla EK, et al. Comparison of CT methods for determining the fat content of the liver. AJR Am J Roentgenol. 2007;188(5):1307–1312. doi: 10.2214/AJR.06.0992. [DOI] [PubMed] [Google Scholar]
- 29.Speliotes EK, Massaro JM, Hoffmann U, Vasan RS, Meigs JB, Sahani DV, et al. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The Framingham heart study. Hepatology. 2010;51(6):1979–1987. doi: 10.1002/hep.23593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimura S, Manabe I, Nagasaki M, Seo K, Yamashita H, Hosoya Y, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. 2008;118(2):710–721. doi: 10.1172/JCI33328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Homeostatic model assessment. 2012 Available from: URL: http://en.wikipedia.org/wiki/Homeostatic_model_assessment.
- 32.Akagiri S, Naito Y, Ichikawa H, Mizushima K, Takagi T, Handa O, et al. A mouse model of metabolic syndrome; increase in visceral adipose tissue precedes the development of fatty liver and insulin resistance in high-fat diet-fed male KK/Ta mice. J Clin Biochem Nutr. 2008;42(2):150–157. doi: 10.3164/jcbn.2008022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tajiri Y. Indicators for the efficacy of pioglitazone before and during treatment in Japanese patients with type 2 diabetes. Diabetes technology & therapeutics. 2007;9(5):429–437. doi: 10.1089/dia.2007.0204. [DOI] [PubMed] [Google Scholar]
- 34.Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, et al. PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. PNAS. 2009;106(52):22504–22509. doi: 10.1073/pnas.0912487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tontonoz P, Spiegelman BM. Fat and Beyond: The Diverse Biology of PPARγ. Annu Rev Biochem. 2008;77(1):289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 36.Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A Comparison of Lipid and Glycemic Effects of Pioglitazone and Rosiglitazone in Patients With Type 2 Diabetes and Dyslipidemia. Diabetes Care. 2005;28(7):1547–1554. doi: 10.2337/diacare.28.7.1547. [DOI] [PubMed] [Google Scholar]
- 37.Smith SR, de Jonge L, Volaufova J, Li Y, Xie H, Bray GA. Effect of pioglitazone on body composition and energy expenditure: a randomized controlled trial. Metabolism. 2005;54(1):24–32. doi: 10.1016/j.metabol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Yang KJ, Noh JR, Kim YH, Gang GT, Hwang JH, Yang SJ, et al. Differential modulatory effects of rosiglitazone and pioglitazone on white adipose tissue in db/db mice. Life Sci. 2010;87(13–14):405–410. doi: 10.1016/j.lfs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Wilding J. Thiazolidinediones, insulin resistance and obesity: finding a balance. Int J Clin Pract. 2006;60(10):1272–1280. doi: 10.1111/j.1742-1241.2006.01128.x. [DOI] [PubMed] [Google Scholar]
- 40.Bajaj M, Suraamornkul S, Pratipanawatr T, Hardies LJ, Pratipanawatr W, Glass L, et al. Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes. 2003;52(6):1364–1370. doi: 10.2337/diabetes.52.6.1364. [DOI] [PubMed] [Google Scholar]
- 41.Okuno A, Tamemoto H, Tobe K, Ueki K, Mori Y, Iwamoto K, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest. 1998;101(6):1354–1361. doi: 10.1172/JCI1235. [DOI] [PMC free article] [PubMed] [Google Scholar]