

Abstract
Context
Patients with pseudohypoparathyroidism type 1a (PHP-1a) develop early-onset obesity. The abnormality in energy expenditure and/or energy intake responsible for this weight gain is unknown.
Objective
The aim of this study was to evaluate energy expenditure in children with PHP-1a compared with obese controls.
Patients
We studied 6 obese females with PHP-1a and 17 obese female controls. Patients were recruited from a single academic center.
Measurements
Resting energy expenditure and thermogenic effect of a high fat meal were measured using whole room indirect calorimetry. Body composition was assessed using whole body dual energy x-ray absorptiometry. Fasting glucose, insulin and hemoglobin A1C were measured.
Results
Children with PHP-1a had decreased resting energy expenditure compared with obese controls (P <0.01). After adjustment for fat free mass, the PHP-1a group’s resting energy expenditure was 346.4 kcals/day less than obese controls (95% CI [−585.5 to −106.9], P <0.01). The thermogenic effect of food, expressed as percent increase in postprandial energy expenditure over resting energy expenditure, was lower in PHP-1a patients than obese controls but did not reach statistical significance (absolute reduction of 5.9%, 95% CI [−12.2% to 0.3%], P = 0.06).
Conclusions
Our data indicate that children with PHP-1a have decreased resting energy expenditure compared with obese controls and that may contribute to the development of obesity in these children. These patients may also have abnormal diet-induced thermogenesis in response to a high fat meal. Understanding the causes of obesity in PHP-1a may allow for targeted nutritional or pharmacologic treatments in the future.
Keywords: Pseudohypoparathyroidism, Obesity, Pediatrics, Energy Expenditure
Introduction
Pseudohypoparathyroidism is a rare disorder caused by mutations in the gene GNAS which encodes for the alpha subunit of the stimulatory G-protein (Gsα). The phenotype varies based on whether the mutant allele is inherited from the paternal or maternal chromosome, as the paternal allele is imprinted and silenced in some tissues. Patients with a maternally inherited mutation, termed pseudohypoparathyroidism type 1a (PHP-1a), demonstrate the classic Albright Hereditary Osteodystrophy (AHO) phenotype, including short stature, brachydactyly, subcutaneous ossifications and cognitive impairment. These patients also have multi-hormone resistance, most commonly parathyroid (PTH) resistance and thyroid stimulating hormone (TSH) resistance. In contrast, patients with a paternally inherited mutation, termed pseudopseudohypoparathyroidism (PPHP), have a mild AHO phenotype and no hormone resistance.
A recently recognized feature of PHP-1a is early-onset obesity. In one study, children with PHP-1a had an average BMI z-score of 2.58 ± 0.23 compared with an average BMI z-score of −0.41 ± 0.87 in children with PPHP.(1) Over 89% of the children with PHP-1a were classified as obese compared with 17% of children in the general population.(1) Adults with PHP-1a were more obese than both adults with PPHP and the general population.(1) The gnas exon 1 knockout mouse model also demonstrates increased obesity in mice with a maternally inherited mutant allele and paternally inherited mutant alleles, although to different degrees of severity.(2)
The etiology of early-onset obesity in PHP-1a is unknown. Hypothyroidism and growth hormone deficiency may contribute to obesity in these patients.(1) Abnormal function of the melanocortin-4 receptor (MC4R) is another possibility. MC4R is a Gsα-coupled receptor (GPCR) that plays a critical role in energy homeostasis. Heterozygous and homozygous mutations in MC4R are well documented in humans to lead to a syndrome of early-onset obesity, hyperinsulinemia, increased bone mineral density, and accelerated linear growth.(3, 4) Similarities between the obesity syndromes in mc4r and gnas exon 1 knockout mice, along with the coupling of the MC4R through Gsα support the possibility of abnormal MC4R function in PHP-1a. Mice have paternal imprinting of gnas in the paraventricular nucleus of the hypothalamus, an important site of MC4R action.(5) Like the mc4r knockout, the gnas exon 1 knockout mouse has abnormalities in energy expenditure and sympathetic nervous system activity.(2, 5) Both the MC4R and Gsα murine models demonstrate insulin resistance and impaired glucose tolerance that may develop prior to the onset of obesity.(5-7) Previous work in our lab demonstrated absence of appropriate diet-induced thermogenesis in the mc4r knockout mouse in response to a high fat diet.(8)
We postulated that children with PHP-1a have abnormal hypothalamic MC4R signaling. Based on data from the MC4R and Gsα murine model, we hypothesized that children with PHP-1a would have abnormal energy expenditure compared with obese controls. We also hypothesized that children with PHP-1a have increased insulin resistance compared with obese controls. We measured energy expenditure and insulin resistance in children with PHP-1a compared with a group of obese control patients to test this hypothesis.
Subjects and Methods
Participants
Children between the ages of 7 and 18 years old were recruited from the Pediatric Endocrinology Clinic and Pediatric Weight Management Clinic at Vanderbilt University from November, 2010 through December, 2011. Through review of clinic records, 8 eligible patients with a clinical diagnosis of PHP-1a were identified and 6 female patients enrolled in the study. Obese control patients were identified through clinic visits and advertisements in clinic waiting rooms. Inclusion criteria included a current BMI >95th percentile for age and gender and onset of obesity prior to 10 years old. We enrolled 21 female obese controls in the study. Exclusion criteria included diabetes, Cushing syndrome, Prader-Willi syndrome, growth hormone deficiency and use of metformin or other appetite altering drug in the past 3 months. Patients with treated hypothyroidism were eligible to participate. Study visits were held at the Clinical Research Center (CRC) at Vanderbilt University (Nashville, TN, USA). All studies were approved by the Institutional Review Board of Vanderbilt University. Informed consent was obtained from all participants or a parent of the participant and assent was obtained from participants under 18 years old.
Experimental procedure
Participants were asked to maintain their usual diet and abstain from vigorous exercise in the two days prior to the study visit. Participants arrived to the CRC in the fasting state. A physician performed a physical exam, including assessment of pubertal development using Tanner stages. Patients were defined as pubertal if Tanner stage 2 or above for breast development (females) or testicular size (males). Standing height was measured using a wall-mounted stadiometer. Weight was measured using a digital scale, lightly clothed and without shoes. Body mass index (BMI) was calculated using the equation BMI= weight (kg)/height (m)2. BMI, height and weight z-scores were also calculated as standard deviations from the mean using gender and age specific Centers for Disease Control growth charts. Fat mass, fat free mass, and bone mineral density (BMD) were measured by whole body dual energy x-ray absorptiometry using pediatric software (DXA, Lunar Prodigy, GE Medical Systems, Madison, WI, USA).
A fasting blood sample was obtained for measurement of glucose (mg/dL), insulin (μU/mL) and hemoglobin A1C. Insulin resistance was calculated using the homeostasis model assessment index equation HOMA-IR = insulin * glucose/405.(9)
Energy expenditure was measured using whole-room indirect calorimetry. The room has strictly controlled temperature and humidity and is equipped with a window to the outside, window to the adjacent room, food pass window, sink, toilet, chair, and multimedia panel that includes TV. Participants were allowed to view movies, read, or use an iPad during the study. Participants rested quietly with minimal movement in a reclining chair for 30 minutes prior to determining the resting energy expenditure (REE). Measurements were recorded in 1 minute intervals and REEmeasured was calculated over a 30 minute period from rates of oxygen consumption and carbon dioxide production using the Weir equation.(10) The predicted REE was calculated using the female specific equation REEpredicted = 51.2 * weight (kg) + 24.5 * height (cm) −207.5 * age (years) + 1 629.8 (11, 12) and converted to kcal/day by multiplying by 0.239. The percentage of expected REE was calculated using the formula REE% predicted= 100 * REEmeasured/REEpredicted.
Participants were given a high fat test meal through the window pass through. The meal consisted of a milkshake of chocolate Carnation® instant breakfast and whipping cream (81% fat, 17% carbohydrate, 2% protein). Energy content of the meal was standardized individually to provide 35% of the REEmeasured. The meal was consumed in less than 30 minutes and the remaining shake was weighed. The thermogenic effect of food (TEF) was calculated as the postprandial increase in energy expenditure above the REE over 180 minutes. Participants were observed throughout the study. Only data from when participants were seated quietly with minimal movement as recorded minute-by-minute was included in the analysis.
Mutation analysis
Genetic testing for PHP-1a was not part of the study protocol. Three PHP-1a patients had previous DNA sequencing of GNAS1 during routine clinical care by the Johns Hopkins University School of Medicine DNA Diagnostic Laboratory (Baltimore, MD, USA). Genetic testing for GNAS mutations detects approximately 80% of mutations associated with PHP-1a.
All obese controls were genotyped for mutations in MC4R. MC4R primers were used to PCR amplify genomic DNA isolated from whole blood. The Vanderbilt University Center for Human Genetics Research DNA Resources Core provided technical assistance for this work. The entire DNA coding sequence as well as ~400 bp upstream and ~300 bp downstream of the MC4R gene was amplified using two PCR primers 1921 bp apart (Fwd: gtgtcaccctgcaatttgtg, and Rev: tgttacgaaagcacgcaaag). Amplified samples were treated with exosap-IT® (USB Corp., Cleveland, OH, USA) and sequenced using 3 forward primers to ensure signal overlap and accurate sequencing (Fwd 1: tctcacagactcccattgca; Fwd 2: cacagcaatgccagtgagtc; Fwd 3: cccggcactggtgccatccg) using BigDye Terminator chemistry resolved on the ABI 3730xl DNA Analyzer. DNA sequences of patients were compared with the NCBI MC4R Reference Sequence: NC_000018.9 (software Seqscape V2.5, Applied Biosystems).
Data Collection
Study data were collected and managed using REDCap electronic data capture tools hosted at Vanderbilt University.(13) REDCap (Research Electronic Data Capture) is a secure, web-based application designed to support data capture for research studies.
Statistical Analysis
We enrolled 3 controls per case. Total sample size was limited by the number of patients with PHP-1a identified in our endocrinology clinic. Unless specified otherwise, data are expressed as median (lower quartile, upper quartile) and nonparametric tests were used. General characteristics were compared using the Mann-Whitney U test and Fisher’s exact test. The nonparametric 95% confidence interval estimates the median of the difference between a sample from the PHP-1a patients and a sample from obese controls (this does not estimate the difference in medians but rather the median of the difference between a sample from ‘x’ and a sample from ‘y’). REE was assessed using the Mann-Whitney U test and linear regression. Each case was matched with a control based on fat free mass and REE was assessed using a Wilcoxon signed rank test. If there was no control with a fat free mass within 3 kg of a case, that case was excluded from the paired analysis. The thermogenic effect of food was assessed over multiple time points using a linear regression model with time and patient group as predictors. To allow for the potential non-linearity in the trend of TEF over time, restricted cubic splines with 3 knots were used for the time predictor. Huber’s robust sandwich estimator was used to account for repeated observations.(14) Statistical analysis was conducted using SPSS version 19 and R version 2.13.1.
Results
Study Population
We evaluated 6 patients with PHP-1a. Since all PHP-1a patients were female, we evaluated 21 female obese controls. Two control patients were withdrawn from the study, one due to inability to obtain a blood sample and one due to protocol noncompliance. Two control patients were found to have MC4R mutations; they are not included in this analysis. Thus, a total of 17 control patients were included in the analysis. One PHP-1a participant and 5 obese controls ate <80% of the test meal; the data for those participants are not included in the analysis of TEF. A total of 5 PHP-1a participants and 12 obese controls were included in the TEF analysis.
Baseline Characteristics
Table 1 summarizes the general characteristics and metabolic profiles of the participants. There was a difference in race between the two groups; the PHP-1a group was Caucasian while the obese control group was 59% Caucasian, 35% African American and 6% Asian. Age, weight and body fat percentage were not significantly different between groups. The average BMI z-score was very similar between the PHP-1a group and the control group (2.6 (2.2, 2.8) vs. 2.5 (2.2, 2.6)). The age range was 7 to 18 years old in the PHP-1a group and 8 to 18 years old in the control group. In the PHP-1a group, 50% of the participants were pubertal as compared to 82% of the control group. As expected, height z-score was significantly lower in the PHP-1a group than the obese controls (−0.4 (−2.3, 0.7) vs. 1.3 (0.7, 2.0), P <0.01). Height z-score differed between the pubertal and pre-pubertal PHP-1a patients (−2.8 (−2.9, −1.8, n=3) vs. 0.9 (0.4, 0.9, n=3)), no difference was present between pubertal and pre-pubertal controls. The total body BMD z-scores were above average for all participants and there was no difference between groups. Systolic blood pressure and heart rate were not significantly different between groups, although these tests may be underpowered due to small sample size. Four obese controls had treated hypothyroidism; all were adequately controlled with normal free T4 and TSH. There was no significant difference between euthyroid controls and those treated for hypothyroidism in any variable.
Table 1.
General Characteristics and Metabolic Profile
| PHP-1a (n = 6) |
Obese Controls (n= 17) |
Median of differences (Nonparametric CI*) |
|
|---|---|---|---|
| Age (years) | 11.8 (8.5, 15.0) | 12.6 (10.8, 15.3) | −0.8 (−5.4 to 3.4) |
| Race | |||
| Caucasian | 100% (n=6) | 58.8% (n= 10) | |
| African-American | 0% | 35.3% (n= 6) | |
| Asian | 0% | 5.9% (n= 1) | |
| Weight Z-score | 2.7 (1.6, 3.0) | 2.8 (2.3, 3.0) | −0.1 (−1.6 to 0.5) |
| Height Z-score | −0.4 (−2.3, 0.7) | 1.3 (0.7, 2.0) | −1.9 (−4.0 to -0.6) |
| BMI Z-score | 2.6 (2.2, 2.8) | 2.5 (2.2, 2.6) | 0.1 (−0.4 to 0.4) |
| Body fat percentage | 48 (46, 50) | 47 (46,52) | −0.7 (−7.8 to 3.8) |
| BMD Z-score | 1.6 (0.8, 3.2) | 2.0 (1.4, 2.4) | −0.2 (−1.4 to 1.4) |
| Heart Rate (bpm) | 74 (70, 80) | 86 (79, 91) | −9 (−19 to 2) |
| Systolic BP (mmHg) | 116 (107,125) | 123 (110,128) | −5 (−19 to 7) |
| Hemoglobin A1C (%) | 5.2 (5.1, 5.4) | 5.5 (5.4, 5.6) | −0.3 (−0.4 to 0.1) |
| Fasting glucose (mg/dL) | 80 (78, 87) | 91 (83, 94) | −7 (−16 to 4) |
| Fasting insulin (mcU/mL) | 16 (5, 23) | 23 (12, 35) | −9.1 (−22.3 to 6.2) |
| HOMA-IR | 3.2 (1.1, 3.9) | 4.2 (2.8, 7.1) | −2.2 (−5.1 to 0.7) |
PHP-1a, pseudohypoparathyroidism type 1a; BMI, body mass index; BMD, bone mineral density; HOMA-IR, homeostasis model assessment index of insulin resistance. Data expressed as median (lower quartile, upper quartile). BMI, height and weight z-scores were calculated as standard deviations from the mean using gender and age specific Centers for Disease Control growth charts.
The nonparametric 95% confidence interval estimates the median of the difference between a sample from the PHP-1a patients and a sample from obese patients; it does not estimate the difference in medians.
Table 2 summarizes the GNAS mutation analysis and diagnostic criteria of the PHP-1a patients. None of the patients were treated with growth hormone (GH) at the time of the study. Patient #6 had been treated with GH from 12 to 14 years of age; the other patients all had normal growth rates during childhood. Five of six PHP-1a patients had TSH resistance and were treated with levothyroxine. All patients had normal free T4 levels. Three patients had elevated TSH levels though the free T4 levels were at the upper limit of normal.
Table 2.
GNAS mutation analysis and clinical phenotype of patients with PHP-1a
| Patient | Phenotype | TSH | Free T4 | Age | Height | Weight | BMI |
|---|---|---|---|---|---|---|---|
| 1 | PTH and TSH resistance, obesity, subcutaneous ossifications, cognitive impairment, brachydactyly. Mutation: Exon 13: c.1107-1108 deletion TG |
3.82 | 8.4 | −.11 | 2.99 | 2.83 | |
| 2* | PTH resistance, obesity, short stature, subcutaneous ossifications, cognitive impairment. |
3.67 | 0.60 | 14.9 | −2.81 | .45 | 1.56 |
| 3* | PTH and TSH resistance, obesity, short stature, subcutaneous ossifications, cognitive impairment, brachydactyly |
10.70 | 1.20 | 18.3 | −.73 | 2.61 | 2.47 |
| 4 | PTH and TSH resistance, obesity, cognitive impairment, brachydactyly. Mutation: Exon 10: c.728C>T (A243V)a |
9.17 | 1.20 | 8.6 | .94 | 3.39 | 2.91 |
| 5 | PTH and TSH resistance, obesity, subcutaneous ossifications, cognitive impairment. Mutations: Exon 12: c.1024C>T (R342X) |
4.15 | 0.97 | 7.2 | .94 | 2.87 | 2.65 |
| 6 | PTH, GHRH and TSH resistance, obesity, short stature, cognitive impairment, brachydactyly |
7.03 | 0.92 | 15.0 | −3.09 | 1.27 | 2.16 |
PHP-1a, pseudohypoparathyroidism type 1a; PTH, parathyroid hormone; GHRH, growth hormone releasing hormone; TSH, thyroid stimulating hormone (normal 0.30-5.00 mcU/mL); free T4 (normal 0.50-1.20 ng/dL); age (years), height (SD), weight (SD), BMI (SD). BMI, height and weight z-scores were calculated as standard deviations from the mean using gender and age specific Centers for Disease Control growth charts. Most recent laboratory values are shown. All patients with TSH resistance were treated with levothyroxine. Results of genetic testing included when known.
Siblings
Laboratory tests
One obese control had an abnormal fasting glucose (103 mg/dL) and one obese control had a hemoglobin A1C in the prediabetes range (5.8%). No patients were diagnosed with diabetes. All obese controls and 4 of 6 PHP-1a patients had evidence of insulin resistance by HOMA-IR. There were no statistically significant differences in insulin, glucose, HOMA-IR or hemoglobin A1C between groups, although these tests may be underpowered due to small sample size (Table 1). There was also no statistically significant difference in insulin, glucose, HOMA-IR or hemoglobin A1when the PHP-1a group was compared with only the Caucasian controls.
Energy Expenditure
Patients with PHP-1a had decreased resting energy expenditure (Table 3 and Figure 1). The PHP-1a group had lower REE%predicted than obese controls (107.9% (105.9, 111.8) vs. 115.5% (112.1, 123.1), P <0.01). The same result was found when comparing the PHP-1a group and only the Caucasian controls (107.9% (105.9, 111.8) vs. 115.6% (112.1, 123.1), P= 0.03). In the obese control group there was no statistically significant difference between REE%predicted in Caucasians versus African Americans (median of the difference 0.033 (−0.78, 0.171), P= 0.6) or in euthyroid patients versus treated hypothyroid patients (median of the difference 0.107 (−0.017, 0.204), P= 0.1). Figure 1 shows REE as a function of fat free mass. Patients with PHP-1a had decreased REE after adjusting for fat free mass (−346.4 kcals/day, 95% CI [−585.5, −106.9], P <0.01 by linear regression). Due to the differences in height, the control patients had a higher average fat free mass (50.4 kg (39.1, 58.1) vs. 36.2 kg (30.9, 37.1), P= 0.09). We therefore further analyzed REE in five PHP-1a patients closely matched with controls based on fat free mass (Table 3). There was one PHP-1a patient for whom an appropriate control was not identified. In the paired analysis, PHP-1a patients again had a lower REE than matched obese controls (P= 0.04 by Wilcoxon signed rank test).
Table 3.
Resting energy expenditure of cases and controls matched based on fat free mass
| PHP-1a | Matched Obese Control | |||
|---|---|---|---|---|
|
| ||||
| Patient | Fat Free Mass (kg) |
REEmeasured (kcals/day) |
Fat Free Mass (kg) |
REEmeasured (kcals/day) |
| 1 | 30.9 | 1540 | Not available | Not available |
| 2* | 36.2 | 1 252 | 35.7 | 2 176 |
| 3* | 61.5 | 2 145 | 61.4 | 2 355 |
| 4 | 36.2 | 1 853 | 35.5 | 2 088 |
| 5 | 27.8 | 1 322 | 25.6 | 1461 |
| 6 | 37.1 | 1432 | 37.0 | 2 112 |
PHP-1a, pseudohypoparathyroidism type 1a. REEmeasured, resting energy expenditure as determined by whole-room indirect calorimetry.
Siblings
Figure 1.
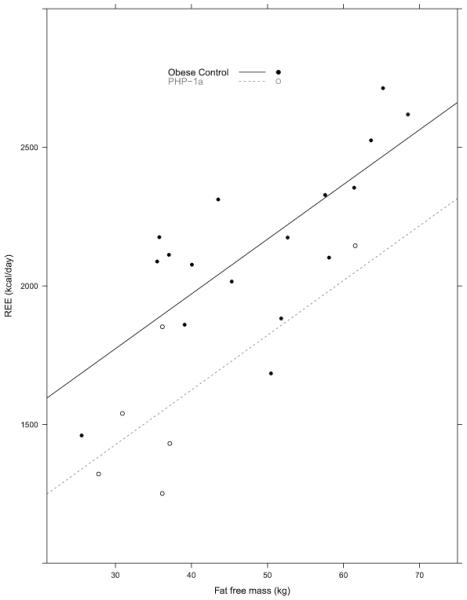
Patients with pseudohypoparathyroidism 1a (PHP-1a) have decreased resting energy expenditure (REE) after adjusting for fat free mass (−346.4 kcals/day, 95% CI [−585.5, −106.9], p= 0.007, linear regression). The linear regression equation is represented on the graph as a solid line for obese controls and a dotted line for PHP-1a patients. Patients with PHP-1a are represented by open circles and obese control patients are represented by black dots. REE was measured using whole-room indirect calorimetry. Fat free mass was determined using whole-body dual energy x-ray absorptiometry.
Data was available for calculation of TEF from 12 obese controls and 5 PHP-1a participants. In a linear regression model of TEF (expressed as percent increase in postprandial energy expenditure over REE) over 180 minutes, the PHP-1a group’s TEF was more than 50% lower than obese controls though this did not reach statistical significance (absolute reduction of 5.9%, 95% CI [−12.2%, 0.3%], P = 0.06, Figure 2). There was no statistically significant difference between groups in the percent of the meal eaten or the time it took for the participant to finish the meal. Two of the six (33%) PHP-1a patients had self-reported hyperphagia on review of systems which was similar to the obese controls (5 of 17, 29%). In contrast, the two patients with MC4R mutations had marked hyperphagia and finished the meal in less than 5 minutes versus a median of 17 minutes (15, 22) for obese controls and 19 minutes (15, 29) for the PHP-1a group (P= 0.4).
Figure 2.

Estimated means of the percent increase in thermogenic effect of food over time stratified by mutation type. Due to the sample size, the model was constrained to have a constant mean difference between groups. Patients with PHP-1a are represented by the dotted line and obese control patients are represented by the solid black line. The light gray shading represents 95% CI; areas where the 95% CI overlap are represented by dark gray shading. Patients stood up to receive the meal which resulted in an increase in energy expenditure at time 0; they were instructed to remain seated for the remainder of the study.
Discussion
Our results show that patients with PHP-1a have decreased REE when compared with obese controls with similar body composition. The differences in REE are clinically significant, patients with PHP-1a had a REE that was 346.4 kcals/day lower (95% CI −585.5 to −106.9) than obese controls with comparable fat free mass. To our knowledge, this is the first study to measure energy expenditure in patients with PHP-1a. The dramatically decreased REE, in the absence of apparent hyperphagia, may explain a component of the rapid weight gain seen in children with PHP-1a.(15-17) Our findings are supported by previous studies in the murine model.(2, 5)
Since a defining characteristic with PHP-1a is hormone resistance on the basis of defective Gsα signaling, hormonal abnormalities have been suspected as a cause of the early-onset obesity. GH deficiency due to growth hormone releasing hormone resistance may contribute to the obese phenotype in some patients.(18, 19) In our cohort, however, only one PHP-1a patient had evidence of GH deficiency. This patient was treated with GH for two years but her BMI continued to increase from 30.2 kg/m2 to 34.0 kg/m2 during that time period. While GH deficiency alters body composition, adults with growth hormone deficiency have normal REE both pre and post-treatment with recombinant GH, particularly after adjustment for fat free mass.(20, 21)
TSH resistance is very common in PHP-1a but it is not clear that hypothyroidism causes weight gain in children or adults.(22, 23) Hypothyroidism is unlikely to contribute to the obesity phenotype in PHP-1a as most patients are treated with adequate thyroid hormone replacement from an early age, particularly in the era of newborn screening. In addition, patients with PHP-1a may not be truly hypothyroid as they are often able to maintain normal thyroxine levels by increasing their TSH levels. In our cohort, all five PHP-1a patients with TSH resistance were diagnosed as infants. While three PHP-1a patients had mildly elevated TSH levels, all had free T4 levels at the upper end of the reference range. It is possible that these patients had only recently become compliant with levothyroxine, or that they are chronically non-compliant but able to overcome the TSH resistance and maintain normal thyroxine levels by increasing their TSH level. None of our participants had overt hypothyroidism and changes in REE and body composition have not been seen in subclinical hypothyroidism.(24)
We postulate that abnormal function of the MC4R in the hypothalamus contributes to the obesity phenotype in PHP-1a. The MC4R is a Gsα-coupled GPCR and a critical regulator of energy homeostasis. In tissues where GNAS is biallelically expressed, there should be no difference in phenotype between PHP-1a and PPHP. Since early-onset obesity is present only in PHP-1a patients,(1) imprinting must play a role. Unlike adipose and liver tissue, there is evidence of paternal imprinting of gnas in the mouse hypothalamus.(5) In a brain specific gnas knockout, disruption of the maternal allele led to decreased REE and a more than two-fold increase in feed efficiency (weight gain/kcal intake) compared with disruption of the paternal allele.(5) The mice also failed to increase energy expenditure in response to MTII, a melanocortin agonist, though the anorexic effects of MTII were maintained.(5) Our data agree with the animal data as our PHP-1a patients appear to gain weight due to decreased REE without patient report of hyperphagia. In at least one study, patients heterozygous for MC4R mutations were also found to have decreased REE compared with obese controls. (25)
Another similarity between the obesity phenotype of MC4R mutations and Gsα mutations is a defect in thermogenesis. Diet induced thermogenesis accounts for approximately 10% of daily energy expenditure.(26) Individual variability in TEF may be related to obesity and insulin resistance.(27, 28) Both the MC4R and brain-specific Gsα mouse models have impaired diet-induced thermogenesis and do not increase their energy expenditure while on a high fat diet,(5, 8) likely due to decreased sympathetic nervous system (SNS) activity.(5, 29) A decrease in TEF means that food is digested more efficiently with less energy lost as heat. This increase in feeding efficiency could lead to greater than expected weight gain and may also contribute to the obesity phenotype of patients with PHP-1a. In our study, we found that PHP-1a patients had more than a 50% decrease in TEF in response to a high fat meal compared with controls, though this did not meet statistical significance due to the small sample size. The subjects also had difficulty remaining sedentary for the entire study which may account for the increasing TEF seen during the last 60 minutes in both groups. We measured TEF for 3 hours but it is possible that the effect continues for up to 6 hours. Additional studies are needed to confirm these findings and to explore whether the defect in TEF is nutrient specific. The effect of MC4R mutations on TEF in humans is unknown.
MC4R mutations are associated with increased insulin resistance and type 2 diabetes in childhood.(3, 4) Both the MC4R and Gsα murine models demonstrate insulin resistance and impaired glucose tolerance that may develop prior to the onset of obesity.(5-7) In our study, we found that 4 of 6 children with PHP-1a had already developed hyperinsulinemia and insulin resistance, however, we did not find evidence that that the insulin resistance in PHP-1a is more severe or earlier in onset than in obese controls. Surprisingly, patients with PHP-1a had a lower mean HOMA than obese controls (3.2 (1.1, 3.9) vs. 4.2 (2.8, 7.1) P= 0.14) though this was not statistically significant and may have been influenced by racial differences between groups.
While CNS changes may be sufficient to cause obesity in PHP-1a, there are differences between the phenotype of MC4R and Gsα mutations. Humans and mice with abnormal MC4R demonstrate hyperphagia, changes in autonomic tone, and subsequent obesity (3, 8, 29) while the obesity due to Gsα mutations appears to be due primarily to decreased energy expenditure, with no apparent hyperphagia in the murine model(5). Food intake and hunger have not been systematically investigated in patients with PHP-1a and there are conflicting case reports in the literature.(15, 17) The possible lack of hyperphagia in PHP-1a may be explained by Gsα being biallelically expressed in brain regions involved in food intake. There is conflicting evidence on REE in humans with MC4R mutations; this may be due to the heterogeneity of the mutations and resulting variability in receptor function.(3, 25, 30) The effect on REE may be more pronounced in PHP-1a as these patients are functionally homozygous for GNAS mutations in imprinted tissues, while most patients identified with MC4R mutations are heterozygous. Alternatively, the pronounced effects on REE in PHP-1a may result from defects in both MC4R and other Gsα-dependent GPCRs.
The main limitation of our study is the small sample size, implying low power and wide confidence intervals for many of the parameters of interest. While the two groups were similar in degree of adiposity, the PHP-1a group was shorter and therefore had a lower fat free mass. We attempted to control for the difference in fat free mass in our analysis of REE but this is a limitation of the study. We have only studied female patients and do not know if our results are generalizable to male patients. All PHP-1a patients were Caucasian while the obese control group included African American and Asian patients. Previous studies have shown decreased REE in African Americans compared with white subjects whereas we found an increased REE in our obese control patients compared to PHP-1a patients. When we compared REE in the PHP-1a group to only the Caucasian controls, our results remained statistically significant. Unfortunately, our small sample size limits the ability to adjust for race as a potential confounder in the linear regression models. TEF is a difficult parameter to measure and there is no universally accepted protocol. Our study monitored TEF for 180 minutes; it is possible that PHP-1a patients have a delayed TEF when compared to obese controls that was not captured in this study. TEF may also be affected by the ambient temperature. While the study was not conducted at thermoneutrality, the metabolic chamber used in this study has strictly controlled temperature and humidity so the environment was similar for each subject and participants had access to blankets if cold. Finally, it is possible that menstrual cycle in pubertal females contributes to variability in energy expenditure. The timing of menses was not controlled for in our study, but we found similar results for our pubertal and prepubertal participants.(31, 32)
Understanding the etiology of weight gain is critical to design targeted nutritional and/or pharmacological interventions for patients with PHP-1a. Our data provides evidence that decreased REE occurs in children with PHP-1a. We did not find evidence of increased insulin resistance in children with PHP-1a compared to obese controls. Further studies are needed to confirm these findings and to better understand the risk of insulin resistance and diabetes in patients with PHP-1a.
Acknowledgements
We thank Rachel Lippert for her assistance with the mutation analysis and Lauren Whitaker for her assistance with the indirect calorimetry. We would like to acknowledge all the patients and their families who made this research possible.
Grant support: This study was supported in part by NIH grants 5T32HD060554-02 (AHS), the Vanderbilt CTSA grant URL1 RR024975-01 from NCRR/NIH and 5 M01 RR-000095 from the NCRR/NIH (AHS), RO1DK070332 (RDC), and by a Fellows Development Research Grant in Diabetes, Obesity and Fat Cell Biology from the Endocrine Fellows Foundation (AHS).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Galpha(s) in the development of human obesity. J Clin Endocrinol Metab. 2007 Mar;92(3):1073–9. doi: 10.1210/jc.2006-1497. [DOI] [PubMed] [Google Scholar]
- 2.Chen M, Gavrilova O, Liu J, Xie T, Deng C, Nguyen AT, et al. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci U S A. 2005 May 17;102(20):7386–91. doi: 10.1073/pnas.0408268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003 Mar 20;348(12):1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 4.Thearle MS, Muller YL, Hanson RL, Mullins M, Abdussamad M, Tran J, et al. Greater Impact of Melanocortin-4 Receptor Deficiency on Rates of Growth and Risk of Type 2 Diabetes Mellitus During Childhood Compared With Adulthood in Pima Indians. Diabetes. 2011 Nov 21; doi: 10.2337/db11-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen M, Wang J, Dickerson KE, Kelleher J, Xie T, Gupta D, et al. Central nervous system imprinting of the G protein G(s)alpha and its role in metabolic regulation. Cell Metab. 2009 Jun;9(6):548–55. doi: 10.1016/j.cmet.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The Central Melanocortin System Can Directly Regulate Serum Insulin Levels. Endocrinology. 2000 Sep 1;141(9):3072–9. doi: 10.1210/endo.141.9.7665. 2000. [DOI] [PubMed] [Google Scholar]
- 7.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. The Journal of Clinical Investigation. 2001;108(7):1079–85. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci. 2001 Jun;4(6):605–11. doi: 10.1038/88423. [DOI] [PubMed] [Google Scholar]
- 9.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985 Jul;28(7):412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 10.Weir JB. New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol. 1949 Aug;109(1-2):1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molnar D, Jeges S, Erhardt E, Schutz Y. Measured and predicted resting metabolic rate in obese and nonobese adolescents. J Pediatr. 1995 Oct;127(4):571–7. doi: 10.1016/s0022-3476(95)70114-1. [DOI] [PubMed] [Google Scholar]
- 12.Hofsteenge GH, Chinapaw MJ, Delemarre-van de Waal HA, Weijs PJ. Validation of predictive equations for resting energy expenditure in obese adolescents. Am J Clin Nutr. 2010 May;91(5):1244–54. doi: 10.3945/ajcn.2009.28330. [DOI] [PubMed] [Google Scholar]
- 13.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009 Apr;42(2):377–81. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber P. The Behavior of Maximum Likelihood Estimates under Nonstandard Conditions. Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability. 1967;I:221–33. [Google Scholar]
- 15.Ong KK, Amin R, Dunger DB. Pseudohypoparathyroidism--another monogenic obesity syndrome. Clin Endocrinol (Oxf) 2000 Mar;52(3):389–91. doi: 10.1046/j.1365-2265.2000.00911.x. [DOI] [PubMed] [Google Scholar]
- 16.Nwosu BU, Lee MM. Pseudohypoparathyroidism type 1a and insulin resistance in a child. Nat Rev Endocrinol. 2009 Jun;5(6):345–50. doi: 10.1038/nrendo.2009.81. [DOI] [PubMed] [Google Scholar]
- 17.Dekelbab BH, Aughton DJ, Levine MA. Pseudohypoparathyroidism type 1A and morbid obesity in infancy. Endocr Pract. 2009 Apr;15(3):249–53. doi: 10.4158/EP.15.3.249. [DOI] [PubMed] [Google Scholar]
- 18.Germain-Lee EL, Groman J, Crane JL, Jan de Beur SM, Levine MA. Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. J Clin Endocrinol Metab. 2003 Sep;88(9):4059–69. doi: 10.1210/jc.2003-030028. [DOI] [PubMed] [Google Scholar]
- 19.Mantovani G, Maghnie M, Weber G, De Menis E, Brunelli V, Cappa M, et al. Growth hormone-releasing hormone resistance in pseudohypoparathyroidism type ia: new evidence for imprinting of the Gs alpha gene. J Clin Endocrinol Metab. 2003 Sep;88(9):4070–4. doi: 10.1210/jc.2002-022028. [DOI] [PubMed] [Google Scholar]
- 20.Stenlof K, Sjostrom L, Lonn L, Bosaeus I, Kvist H, Tolli J, et al. Effects of recombinant human growth hormone on basal metabolic rate in adults with pituitary deficiency. Metabolism. 1995 Jan;44(1):67–74. doi: 10.1016/0026-0495(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 21.Chong PK, Jung RT, Scrimgeour CM, Rennie MJ, Paterson CR. Energy expenditure and body composition in growth hormone deficient adults on exogenous growth hormone. Clin Endocrinol (Oxf) 1994 Jan;40(1):103–10. doi: 10.1111/j.1365-2265.1994.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 22.Lomenick JP, El-Sayyid M, Smith WJ. Effect of levo-thyroxine treatment on weight and body mass index in children with acquired hypothyroidism. J Pediatr. 2008 Jan;152(1):96–100. doi: 10.1016/j.jpeds.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Karmisholt J, Andersen S, Laurberg P. Weight loss after therapy of hypothyroidism is mainly caused by excretion of excess body water associated with myxoedema. J Clin Endocrinol Metab. 2011 Jan;96(1):E99–103. doi: 10.1210/jc.2010-1521. [DOI] [PubMed] [Google Scholar]
- 24.Kong WM, Sheikh MH, Lumb PJ, Naoumova RP, Freedman DB, Crook M, et al. A 6-month randomized trial of thyroxine treatment in women with mild subclinical hypothyroidism. Am J Med. 2002 Apr 1;112(5):348–54. doi: 10.1016/s0002-9343(02)01022-7. [DOI] [PubMed] [Google Scholar]
- 25.Krakoff J, Ma L, Kobes S, Knowler WC, Hanson RL, Bogardus C, et al. Lower metabolic rate in individuals heterozygous for either a frameshift or a functional missense MC4R variant. Diabetes. 2008 Dec;57(12):3267–72. doi: 10.2337/db08-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schutz Y, Bessard T, Jequier E. Diet-induced thermogenesis measured over a whole day in obese and nonobese women. Am J Clin Nutr. 1984 Sep;40(3):542–52. doi: 10.1093/ajcn/40.3.542. [DOI] [PubMed] [Google Scholar]
- 27.Camastra S, Bonora E, Del Prato S, Rett K, Weck M, Ferrannini E, EGIR (European Group for the Study of Insulin Resistance) Effect of obesity and insulin resistance on resting and glucose-induced thermogenesis in man. Int J Obes Relat Metab Disord. 1999 Dec;23(12):1307–13. doi: 10.1038/sj.ijo.0801072. [DOI] [PubMed] [Google Scholar]
- 28.Segal KR, Albu J, Chun A, Edano A, Legaspi B, Pi-Sunyer FX. Independent effects of obesity and insulin resistance on postprandial thermogenesis in men. J Clin Invest. 1992 Mar;89(3):824–33. doi: 10.1172/JCI115661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009 Jan 1;360(1):44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 30.Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000 Jul;106(2):271–9. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry CJ, Lightowler HJ, Marchini J. Intra-individual variation in resting metabolic rate during the menstrual cycle. Br J Nutr. 2003 Jun;89(6):811–7. doi: 10.1079/BJN2003839. [DOI] [PubMed] [Google Scholar]
- 32.Melanson KJ, Saltzman E, Russell R, Roberts SB. Postabsorptive and postprandial energy expenditure and substrate oxidation do not change during the menstrual cycle in young women. J Nutr. 1996 Oct;126(10):2531–8. doi: 10.1093/jn/126.10.2531. [DOI] [PubMed] [Google Scholar]