

Background: N-Linked glycosylation is catalyzed by oligosaccharyltransferase (OST).
Results: Specific amino acids in enzyme and acceptor substrate are identified as key determinants for substrate binding and turnover.
Conclusion: Quantification of substrate binding and turnover reveal a delicate interplay between acceptor substrate, enzyme, and metal ion.
Significance: The study represents the first quantitative analysis of substrate binding and turnover in N-linked glycosylation.
Keywords: Enzyme Kinetics, Glycosylation, Glycosyltransferases, Membrane Proteins, Oligosaccharide, Fluorescence Anisotropy, PglB, Campylobacter lari, Oligosaccharyltransferase, Protein N-Glycosylation
Abstract
N-Linked glycosylation is an essential post-translational protein modification in the eukaryotic cell. The initial transfer of an oligosaccharide from a lipid carrier onto asparagine residues within a consensus sequon is catalyzed by oligosaccharyltransferase (OST). The first X-ray structure of a complete bacterial OST enzyme, Campylobacter lari PglB, was recently determined. To understand the mechanism of PglB, we have quantified sequon binding and glycosylation turnover in vitro using purified enzyme and fluorescently labeled, synthetic peptide substrates. Using fluorescence anisotropy, we determined a dissociation constant of 1.0 μm and a strict requirement for divalent metal ions for consensus (DQNAT) sequon binding. Using in-gel fluorescence detection, we quantified exceedingly low glycosylation rates that remained undetected using in vivo assays. We found that an alanine in the −2 sequon position, converting the bacterial sequon to a eukaryotic one, resulted in strongly lowered sequon binding, with in vitro turnover reduced 50,000-fold. A threonine is preferred over serine in the +2 sequon position, reflected by a 4-fold higher affinity and a 1.2-fold higher glycosylation rate. The interaction of the +2 sequon position with PglB is modulated by isoleucine 572. Our study demonstrates an intricate interplay of peptide and metal binding as the first step of protein N-glycosylation.
Introduction
N-Linked glycosylation is a post-translational protein modification that takes place in the secretory pathway of eukaryotic cells. N-Glycans have a central role in diverse biological processes such as protein folding, intracellular trafficking, regulation of protein turnover, or cell-cell recognition (1). Protein N-glycosylation is common in Archaea (2–4) and is also found in defined taxa of Bacteria (5–7). The molecular mechanism of N-linked protein glycosylation is similar in all domains of life. An oligosaccharide is assembled on a lipid carrier, resulting in a lipid-linked oligosaccharide (LLO),3 and transferred en bloc to the amide nitrogen of an acceptor asparagine, resulting in an N-glycosidic linkage. The process is catalyzed by oligosaccharyltransferase (OST), an integral membrane enzyme that modifies asparagines that are present in a consensus sequence N-X-(S/T), where X must not be proline (8). The N-X-(S/T) motif is found in eukaryotes, Archaea, and, in extended form, in Bacteria (8–12).
Although the presence of an N-X-(S/T) sequon is essential, it is not sufficient to define a substrate for N-linked glycosylation. In fact, only two-thirds of all existing sequons in potential substrate proteins are modified with a glycan (13). The underlying principles that govern N-glycosylation site occupancy are poorly understood. A statistical analysis of eukaryotic glycoproteins with an entry in the Protein Data Bank revealed a surprisingly large number of glycosylated asparagines of low accessibility (13) suggesting that glycosylation occurs before protein folding (14). Therefore the fate of an individual sequon might depend on its structural environment and the competition between glycosylation and protein folding (15). The OST enzyme of higher eukaryotes is a multiprotein complex, with the catalytic Stt3 subunit surrounded by seven other subunits, some of which have chaperone or oxidoreductase activities, suggesting that OST directly modulates glycoprotein folding (16–20). However, another factor governing site occupancy is the primary sequence of the glycosylation site. Statistical analyses have shown that sequons with a Thr in the +2 position are more frequently glycosylated than those containing Ser (10, 13, 21). This is consistent with biochemical studies revealing that N-X-T sites in acceptor peptides and proteins are better substrates than N-X-S sites (22–25). On the other hand, a comprehensive analysis of the mouse glycoproteome revealed the existence of non-canonical sequons, among which N-X-C, N-X-V, and N-G-X sites appeared with statistical significance (21). In bacteria, a single-subunit OST termed PglB, which is homologous to the catalytic Stt3 subunit of eukaryotic OST, catalyzes the transfer of glycans onto acceptor proteins.
The X-ray structure of the PglB protein from Campylobacter lari has helped identify the structural basis of sequon recognition (26). It was shown that the +2 Thr/Ser of the sequon is optimally positioned to allow specific hydrogen bonds to be formed with the WWD motif of PglB, which is strictly conserved in Stt3 homologues (18). The structure suggested van der Waals interactions as a possible explanation for the preference of Thr over Ser at the +2 position of the sequon. It also revealed a possible salt bridge between an arginine side chain on the surface of the enzyme and the negatively charged aspartate in the −2 position of the sequon, which may explain the observation that bacterial glycosylation sequons are extended ((D/E)-Z-N-X-(S/T), with X, Z ≠ P (27)) compared with their eukaryotic or archaeal counterparts. Nevertheless, glycosylation of non-canonical sites lacking either the interaction in the −2 position or the +2 position have been reported for the PglB enzyme in vivo (28, 29).
To understand the catalytic mechanism of OST, quantitative in vitro assays and detailed structure-function analyses are essential. We exploited the stability and activity of detergent-solubilized C. lari PglB to determine sequon binding affinities and glycosylation rates. We chemically synthesized fluorescently labeled peptide substrates and purified milligram amounts of PglB enzyme, where we introduced mutations guided by the crystal structure. After developing highly sensitive assays, we quantified sequon binding using fluorescence anisotropy and determined glycosylation turnover rates even for highly disfavored mutant/peptide combinations. We were thus able to gain insight into (i) the influence of active site residues and divalent metal ions for sequon binding, (ii) the specificity of the Ser/Thr binding pocket, and (iii) bacteria-specific requirements of sequon recognition. Our quantitative assessment of peptide binding and catalysis provides insight into the natural selection of the N-X-(S/T) sequon in eukaryotes and the extended sequon in Bacteria.
EXPERIMENTAL PROCEDURES
Reagents
All reagents were purchased from Sigma, Fluka, Acros Organics, or Alfa Aesar. 5-Carboxyfluorescein was purchased either from Novabiochem (Switzerland) or OChem Incorporation (USA). The amino acid building blocks and Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate were purchased from Advanced ChemTech (USA) or Novabiochem (Switzerland). Fmoc-protected TentaGel S RAM resin was purchased from Rapp Polymere (Germany).
Construction of Plasmids
Mutations in the pglB gene of C. lari were generated by the QuikChange method on plasmid pSF2 (26). For the in vivo glycosylation assay pglB variants were subcloned into a pMLBAD plasmid (30). For in vitro glycosylation studies the two endogenous glycosylation sites were removed (N535Q and N556Q) to prevent autoglycosylation of PglB. The resulting construct was referred to as wild type construct and all subsequent mutations were based on this construct.
Mutation of the DQNAT sequon within the 3D5 acceptor protein was performed by ligation of phosphorylated, double-stranded DNA of oligonucleotides 5′-CTAGCGGTGGTGGTGGTTCTGGTGGTGGTGCCCAGAACGCCA-3′ and 5′-CCGGTGGCGTTCTGGGCACCACCACCAGAACCACCACCACCG-3′ into the plasmid pCL21 (31) digested with restriction enzymes NheI and AgeI. This resulted in plasmid pCL64 carrying an AQNAT acceptor site. All plasmids were validated by DNA sequencing.
In Vivo Glycosylation Assay
In vivo complementation analysis of PglB mutants was performed as described before (26). Briefly, Escherichia coli SCM6 cells were transformed with three separate plasmids carrying: 1) the C. jejuni pglBmut cluster (containing an inactivated pglB gene) to generate LLO; 2) the glycosylation acceptor protein 3D5 containing a DQNAT or an AQNAT site; 3) C. lari PglB, wild type or mutants. Expression and glycosylation of 3D5 was monitored by SDS-PAGE of periplasmic cell extracts and visualized by mobility shift due to increased size in an immunoblot using anti-c-Myc antibody, or the reactivity of the glycoprotein in an anti-glycan immunoblot using hR6 antiserum.
Expression and Purification of PglB Mutants
Overexpression and purification of PglB mutants was performed as previously described (26). Shortly, proteins were overexpressed in E. coli BL21(DE3) Gold cells in a 30-liter fermenter or in 5-liter baffled flasks using Terrific Broth medium supplemented with 1% (w/v) glycerol. Cells were grown to A600 of 10.0 (fermenter) or 3.0 (flasks) at 37 °C and induced with 0.1% (w/v) arabinose for 2 or 4 h, respectively. Cells were harvested by centrifugation and pellets were stored at −80 °C before membrane preparation. All subsequent steps were carried out at 4 °C. PglB was solubilized in 25 mm Tris-HCl, pH 8.0, 250 mm NaCl, 10% (v/v) glycerol, and 1% (w/v) N-dodecyl-β-d-maltopyranoside (Anatrace) for 1 h. All purification buffers contained N-dodecyl-β-d-maltopyranoside. PglB was purified on a nickel-nitrilotriacetic acid Superflow affinity column (Qiagen) and desalted into 10 mm MES, pH 6.5, 100 mm NaCl, 0.5 mm EDTA, and 3% (v/v) glycerol. Desalted protein was analyzed by size exclusion chromatography (Superdex 200, GE Healthcare) and, if needed, concentrated up to 26 mg/ml in a 100-kDa molecular mass cutoff Ultra-15 concentrator (Amicon, Millipore). The concentrated sample was re-analyzed by gel filtration and protein concentrations were determined by absorption at 280 nm of diluted samples using spectrophotometry.
Synthesis of Acceptor Peptides Labeled with 5-Carboxyfluorescein
Peptide synthesis was initiated by loading TentaGel S RAM resin (500 mg, loading: 0.24 mmol g−1) in a 10-ml polypropylene syringe fitted with a polypropylene frit, a Teflon stopcock, and a stopper. The resin was swollen in DCM (6 ml, 20 min). After removal of the DCM, the Fmoc protecting group of the resin was removed by using a solution of 20% piperidine in NMP. Stirring of the reaction mixture at any given step was performed by attaching the closed syringes to a rotating axis. The completion of the reaction was checked using the 2,4,6-trinitrobenzenesulphonic acid test. Removal of the Fmoc protecting group was performed by using a solution of 20% piperidine in NMP (6 ml, 2 × 10 min). After filtration, the resin was washed with NMP (2 × 6 ml), MeOH (2 × 6 ml), and DCM (2 × 6 ml). Coupling of amino acids was performed using Fmoc-protected amino acids (3 eq), Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (3 eq), and N,N-Diisopropylethylamine (6 eq) in NMP (6 ml). The resin was stirred for 2 h before it was washed with NMP, MeOH, and DCM (2 × 6 ml each). Acetylation of the resin was performed after each amino acids coupling using a solution of acetic anhydride:DCM (1:1). Coupling of 5-carboxyfluorescein was performed by using 5-carboxyfluorescein (2 eq), HOBt (5 eq), and N,N′-Diisopropylcarbodiimide (5 eq) in NMP (6 ml). The resin was stirred overnight and protected from light using aluminum foil. The resin was washed with NMP, MeOH, and DCM (2 × 6 ml each). A solution of 20% piperidine in NMP (6 ml, 6 × 5 min) was added to the resin to remove the excess of free 5-carboxyfluorescein before the resin was finally washed with DCM (6 × 6 ml). After coupling of the fluorophore, the compounds were protected from light with aluminum foil at all times. TFA cleavage was performed by adding a solution of TFA/TIS/H2O (94:5:1, v/v/v) to the resin for 1 h (if the linear peptide contained a Cys, TFA/TIS/H2O/EDT (94/2.5/1.0/2.5, v/v/v/v) was used instead). The peptide was precipitated with tert-butyl methyl ether and dissolved in H2O/MeCN with 0.1% TFA mixture. Labeled acceptor peptides were purified by preparative RP-HPLC and obtained as TFA salts after lyophilization. Peptides were dissolved to 2–5 mm in dimethyl sulfoxide and the concentration of peptides was determined from a dilution in 100 mm Tris-HCl, pH 9.0, by absorbance at 495 nm, with an extinction coefficient of 79,000 m−1 cm−1. A detailed description for the synthesis of individual peptides can be found in the supplemental Methods.
Fluorescence Anisotropy Measurements
70 μl of desalt buffer supplemented with 1 μm peptide substrate and 10 mm MnCl2 was titrated with concentrated PglB containing equal concentrations of peptide substrate and MnCl2. Anisotropy was measured with a Quantamaster QM-7/2003 spectrofluorometer (Photon Technology International) in a T-format at 4 °C. Each concentration was measured 20 times for 1 s at 494 nm excitation and 521 nm emission wavelengths. Anisotropy was calculated using the equation,
 |
and plotted against total protein concentration. Data were fitted in Prism (GraphPad Software) using the following equation for the 1:1 stoichiometry,
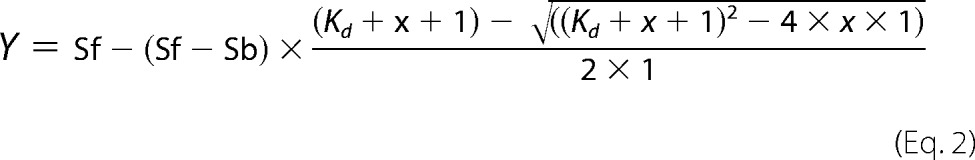 |
or with
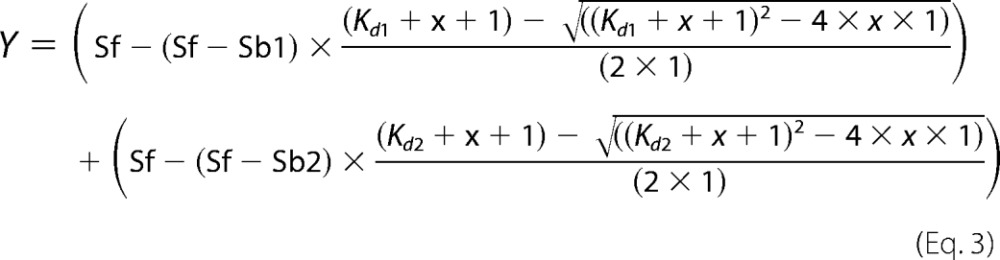 |
for the mutant R375A, where a 2:1 stoichiometry yielded a higher R2 value. The protein solution from the cuvette was analyzed again by size exclusion chromatography after each anisotropy measurement to prove stability of the protein over the course of the experiment.
Extraction of LLOs
Isolation of LLOs was performed as described before (32). Briefly, LLOs were extracted from E. coli SCM6 cells carrying the Campylobacter jejuni pglBmut cluster (containing an inactivated pglB gene) by a mixture of chloroform:MeOH:H2O, 10:20:3. Extracts were dried in a rotavap and reconstituted in a buffer containing 10 mm MES, pH 6.5, 100 mm NaCl, and 1% Triton X-100 (w/v). The concentration of reconstituted LLOs was determined by titrating various amounts of LLO against a constant amount of acceptor peptide in an in vitro glycosylation assay.
In Vitro Glycosylation Assay
In a reaction containing 10 mm MES, pH 6.5, 100 mm NaCl, 10 mm MnCl2, 3% glycerol (v/v), and 1% Triton X-100 (w/v), 10% LLO extract (v/v, corresponding to 35 μm) and various amounts of PglB (1 nm to 25 μm) were mixed. The mixture was preincubated at 30 °C in a water bath for 5 min before the reaction was started by addition of fluorescently labeled acceptor peptide. Reactions were incubated in a water bath at 30 °C. Samples were taken at different time points and reactions were stopped by the addition of 4× SDS sample buffer. Samples were diluted 200-fold prior to analysis by Tricine SDS-PAGE in minigels (8 × 8 cm) consisting of a 16% resolving gel with 6 m urea, a 10% spacer gel, and a 4% stacking gel (33). Fluorescent bands for peptide and glycopeptide were visualized by using a Typhoon Trio Plus imager (GE Healthcare) with excitation at 488 nm and a 526-nm SP emission filter. The amount of formed glycopeptide was determined from band intensities of fluorescence gel scans (ImageJ) where the sum of signals for glycosylated and non-glycosylated peptide for each lane was defined as 100%.
For Michaelis-Menten kinetics, 1 nm PglB was incubated with various amounts of acceptor peptide (0.25–30 μm) for 10 min. Data were fitted in Prism (GraphPad Software) by nonlinear regression according to the Michaelis-Menten formula.
For turnover rate determination, various amounts of PglB enzyme were incubated with 10 μm acceptor peptide. A total of 10 samples were taken in 3-min intervals and data were fitted by linear regression.
For M2+ titration experiments, the nickel-nitrilotriacetic acid eluate of purified PglB was supplemented with 0.5 mm EDTA, but no EDTA was added to the desalt buffer. 4 nm PglB enzyme was incubated with 10 μm acceptor peptide and various amounts of MnCl2 and MgCl2, respectively. Four samples were taken at the appropriate time intervals (3 to 25 min) so that the reaction was in the linear range. Data were fitted by linear regression to determine turnover rates. These rates were plotted against the M2+ concentration and data were fitted with a model for one metal binding site using the following equation.
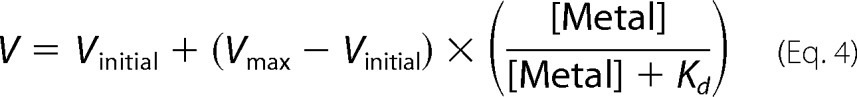 |
RESULTS
Novel in Vitro Assays Allow Accurate Determination of Sequon Binding Affinity and Glycosylation Turnover
Fig. 1 illustrates a hypothetical reaction mechanism based on previous biochemical studies and on the X-ray structure of C. lari PglB. The physical separation of acceptor peptide binding and catalytic sites suggested that sequon binding could be studied as a discrete, initial step of the reaction. We synthetically attached a fluorophore and a Gly-Ser linker to the N terminus, and a phenylalanine to the C terminus, of a consensus acceptor sequon DQNAT (the underlined N representing the glycosylated asparagine and the zero position of the sequon). The resulting peptide, 5-carboxyfluorescein-GSDQNATF-NH2 (the underlined region representing the sequon, Fig. 2A) was used both for binding and glycosylation studies. C. lari PglB was overexpressed and purified in detergent (n-dodecyl-β-d-maltoside) as described before (26). The purity of all PglB variants was analyzed by SDS-PAGE, and the monodispersity of the concentrated (>15 mg/ml) protein preparations was ascertained using analytical size exclusion chromatography (supplemental Fig. S1).
FIGURE 1.
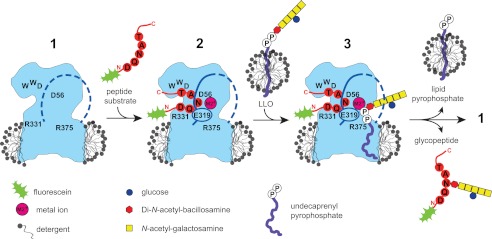
Schematic of a working model of the PglB reaction cycle. The contour of C. lari PglB in a detergent micelle is shown in blue, with positions of functionally relevant and conserved residues indicated in black. In the ground state (1), the external loop 5 (EL5) is assumed to be disordered (dashed blue line). Binding of a fluorescein-labeled substrate peptide and divalent metal cation (M2+, pink) causes EL5 (including residue Glu-319) to become ordered (continuous line) (2). Addition of extracted, LLO from a separate micelle triggers glycosylation, with state 3 likely reflecting a transient state. Following the glycosylation reaction, glycopeptide and undecaprenyl pyrophosphate are released.
FIGURE 2.

Quantitative in vitro determination of sequon binding and catalysis. A, structure of synthesized, fluorescein-labeled substrate peptide containing the pentapeptide sequon DQNAT. Three-letter codes are shown for all amino acids except the acceptor asparagine. The −2 and +2 positions of the sequon are indicated. B, quantification of substrate peptide binding using fluorescence anisotropy. Purified PglB was titrated into a solution containing 1 μm fluorescently labeled peptide containing the DQNAT sequon. Data points reflect the mean of 20 measurements of the same sample, error bars indicate standard deviations. Curve fitting was performed assuming a single binding site (R2 = 0.9940). C, analysis of peptide glycosylation determined by quantification of fluorescently labeled substrate and product following Tricine SDS-PAGE analysis. Glycosylated peptide (g1) is separated from non-glycosylated peptide (g0), bands were visualized by a fluorescence gel scan at 488 nm excitation and 526 nm emission. The concentration of substrate peptide used in the reaction is indicated above the lanes, the PglB concentration was 1 nm. Before gel loading, samples were diluted such that the concentration of total peptide in each lane was identical. D, determination of Michaelis-Menten kinetics for the in vitro glycosylation reaction shown in C. The amount of glycopeptide was determined from band intensities of fluorescence gel scans. The sum of the signals for glycosylated and non-glycosylated peptide for each lane was defined as 100%. Data were fitted by nonlinear regression according to the Michaelis-Menten formula (R2 = 0.9676). E, time course of in vitro glycosylation using 10 μm fluorescently labeled peptide and 1 nm PglB. Separation of glycopeptides and gel scanning was performed as in C. F, determination of turnover rate from the reaction shown in E. Quantification of glycopeptide was done as in D. Data were fitted by linear regression and the turnover rate was calculated from the slope of the fit (R2 = 0.9787). D and F, each data point represents the average of three individual reactions and error bars indicate S.D.
Peptide substrate binding was quantified by fluorescence anisotropy and yielded a dissociation constant, Kd, of 1.02 ± 0.06 μm for wild type PglB and the DQNAT consensus sequon (Fig. 2B), determined in the presence of 10 mm Mn2+ (see also below). Control experiments using an unrelated membrane protein in a similar detergent solution (Fig. 4A) indicated that binding of the fluorescently labeled peptide to PglB was specific.
FIGURE 4.
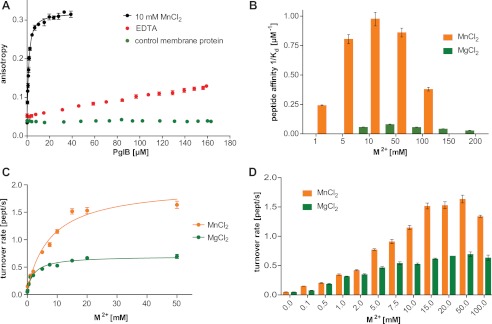
Metal ion dependence of in vitro peptide binding and glycosylation. A, fluorescence anisotropy experiments using fluorescently labeled peptide containing the DQNAT sequon, performed as in Fig. 2B but testing the role of MnCl2. A control measurement with an unrelated membrane protein prepared in the same detergent is shown. The WT data are identical to that shown in Fig. 2B. B, peptide binding affinities (1/Kd) as a function of MnCl2 and MgCl2 concentrations, respectively. Each bar indicates a single fluorescence anisotropy curve (supplemental Fig. S3), error bars indicate the S.E. of the obtained fit for each dataset. C, turnover rates of WT PglB (4 nm) and fluorescently labeled peptide (10 μm) containing the DQNAT sequon as a function of Mn2+ and Mg2+ concentrations. Apparent dissociation constants for metal ions were obtained by fitting the data using a single binding site model (R2(MnCl2) = 0.981; R2(MgCl2) = 0.981). D, overview of glycosylation turnover rates at different concentrations of MnCl2 and MgCl2 (C and D). Each data point reflects a turnover rate determination as shown in Fig. 2F, error bars indicate the S.E. of each fit.
To determine turnover rates, PglB was prepared in the same conditions as for peptide binding analysis, and LLO was added as a glycan donor. The glycosylated product was separated from the unmodified substrate by gel electrophoresis, and the educt/product ratio was determined by measuring the in-gel fluorescence intensities of both educt and product bands. Michaelis-Menten kinetics could thus be determined for wild type PglB using increasing concentrations of peptide substrate (Fig. 2C). We determined Km and Vmax values of 2.60 ± 0.28 μm and 2.20 ± 0.07 peptide/s, respectively (Fig. 2D). The turnover rate for wild type PglB and the DQNAT sequon was determined for the initial, linear range of the reaction and resulted in 1.5 ± 0.04 peptide/s (Fig. 2, E and F). Because our assay system limited the use of substrate peptide to a concentration of 30 μm and many mutations introduced in PglB or the acceptor peptide reduced the enzymatic performance drastically, we decided to determine and compare initial turnover rates instead of Michaelis-Menten kinetics, because impractically high peptide concentrations would be required to reach Vmax. After optimizing the system using wild type PglB and the consensus sequon, we concluded that the two methods allowed us to perform quantitative comparisons of sequon binding and turnover rates using any combination of PglB mutant or sequon variant.
The Role of Active Site Residues for Sequon Binding
The PglB structure revealed a cavity that contains the active site of the enzyme and is believed to bind LLO (Fig. 3A). Based on a tentative modeling of LLO into the crystal structure it was hypothesized that Arg-375 might interact with the pyrophosphate moiety of the LLO substrate (Fig. 3A). To test this hypothesis, we mutated Arg-375 to alanine and lysine and analyzed the in vivo activity of PglB using glycosylation-competent E. coli cells (18) expressing an scFv fragment as acceptor protein (31). We found that although the activity of the R375K mutant was only slightly reduced, the R375A mutant had no detectable activity (Fig. 3B). We then analyzed these mutants in vitro. Anisotropy measurements using the DQNAT sequon yielded dissociation constants of 0.35 ± 0.02 μm for R375K and 2.10 ± 0.16 μm for R375A (Fig. 3C and Table 1). A slightly improved data fit was obtained for the R375A mutant using two distinct binding sites, yielding Kd1 of 4.58 ± 0.68 μm and Kd2 of 0.02 ± 0.036 μm (Table 1). We concluded that the positive charge of Arg-375 was not required for peptide binding, but was essential for later steps in catalysis. Activity measurements at high enzyme concentrations revealed that the glycosylation rate of the R375A mutant was reduced 45,000-fold, and the R375K mutant 40-fold relative to WT PglB (Table 1 and supplemental Fig. S2). These results were consistent with the in vivo activity measurements (Fig. 3B). In addition, the data demonstrated that extremely low turnover rates could be determined in vitro even when no in vivo activity could be observed.
FIGURE 3.

Uncoupling peptide binding from glycosylation. A, surface representation of the active site of C. lari PglB (PDB code 3RCE). Catalytically important residues (cyan) and acceptor peptide (yellow) are shown in stick representation and are labeled. The bound divalent metal ion (M2+) is shown as a purple sphere. A hydrophobic groove possibly accommodating the isoprenoid part of LLO is indicated. B, immunoblots of in vivo glycosylation reactions detecting acceptor protein 3D5 (top) or bacterial N-glycans (bottom). Glycosylation results in a mobility shift from the non-glycosylated (g0) to the glycosylated form of the acceptor protein (g1). PglB mutants are indicated above the lanes. The lanes originate from the same gel and exposure time. C, fluorescence anisotropy experiments using fluorescently labeled peptide containing the DQNAT sequon, performed as described in the legend to Fig. 2B. The WT data are identical to that shown in Fig. 2B. Curve fitting was performed assuming a single binding site for all curves (R2(wt) = 0.9940; R2(R375K) = 0.9934; R2(R375A) = 0.9881). For the R375A mutant, a two-site model yielded a slightly better fit (R2 = 0.9951).
TABLE 1.
Glycosylation turnover rates and dissociation constants of PglB mutants D56A, E319A, R375A, and R375K
Turnover rates and dissociation constants were derived from curves shown in supplemental Figs. S2 and S4. The errors represent the S.E. of the fit for each turnover rate and dissociation constant. Values for relative turnover and relative binding refer to the ratio of an individual mutant and wt PglB.
| Mutant | Turnover rate | Relative turnover/fold reduction | Dissociation constant (Kd) | Relative binding |
|---|---|---|---|---|
| peptide/s | μm | |||
| WT | 1.50 ± 0.04 | 1 | 1.02 ± 0.06 | 1 |
| R375A | (3.35 ± 0.12) × 10−5 | 2.23 × 10−5/45,000-fold | One site: | 0.49 |
| 2.10 ± 0.17 | ||||
| Two sites: | ||||
| Kd1: 4.58 ± 0.68a | ||||
| Kd2: 0.02 ± 0.04 | ||||
| R375K | (3.93 ± 0.04) × 10−2 | 2.62 × 10−2/38-fold | 0.35 ± 0.02 | 2.91 |
| D56A | (3.56 ± 0.13) × 10−5 | 2.37 × 10−5/42,000-fold | –b | |
| E319A | (1.70 ± 0.04) × 10−3 | 1.13 × 10−3/900-fold | –b |
a Curve fitting yields R2 of 0.9881 for a model with one binding site and R2 of 0.9951 for two binding sites.
b Peptide binding was observed, but no Kd could be determined because the fluorescence anisotropy signal reached only 30–50% saturation at the limiting PglB concentration of 100–150 μm.
We next investigated the catalytically essential residues Asn-56 and Glu-319. The structure suggested that the side chains of these residues are in contact with the acceptor asparagine and simultaneously contribute to metal binding (26). Individual mutations of Asp-56 and Glu-319 to alanine strongly reduced C. lari PglB activity in vivo (26), and mutations of the equivalent residues in C. jejuni PglB yielded similarly reduced in vitro glycosylation activities (34). We performed peptide binding experiments with PglB D56A and E319A mutants and found substantially lowered affinities in both cases (Fig. 3C). Dissociation constants could not be accurately determined because at the maximum enzyme concentration (110–120 μm), the anisotropy signal reached only about 30% of the signal expected for saturation. Analysis of the corresponding glycosylation rates showed a 900-fold reduction for mutant E319A (Table 1 and supplemental Fig. S2). For the mutant D56A, we observed differences in activity in distinct batches of purified protein. Compared with wild type, the activity of D56A was reduced between 11,000- and 140,000-fold, with an average reduction of 42,000-fold (Table 1 and supplemental Fig. S2). Irrespective of this fluctuation, mutant D56A had a much more pronounced effect on turnover than mutant E319A, which is in agreement with the observed in vivo activities. We conclude that the side chains of Asp-56 and Glu-319 have a role both in peptide binding and turnover.
Dual Role of the Divalent Metal Ion in Sequon Binding and Catalysis
Divalent metal ions are essential cofactors of most GT-A glycosyltransferases (35) and several studies have described the requirement of Mn2+ or Mg2+ for the activity of OSTs from various organisms (14, 23, 36–38). To understand the role of the metal ion for PglB function (Fig. 3A), we measured peptide binding in the presence of EDTA. The results revealed very weak binding for the DQNAT sequon, too low for accurate Kd determination (Fig. 4A). We subsequently performed a series of fluorescence anisotropy measurements at various Mn2+ and Mg2+ concentrations (Fig. 4B, supplemental Fig. S3, and supplemental Table S2). The data revealed a concentration-dependent increase of acceptor sequon affinity, with the highest affinity observed in the presence of 10 mm Mn2+ and 50 mm Mg2+, respectively. At higher concentrations of the metal ions, the observed binding affinity was reduced.
To quantify the effect of increasing Mn2+ and Mg2+ concentrations on catalysis, glycosylation rates were determined (Fig. 4D). The resulting turnover rates increased and decreased analogously to the observed sequon affinities, but with a slight shift of the maxima (Fig. 4, B and D). Whereas the strongest peptide binding was observed at 10 mm Mn2+ and 50 mm Mg2+, the highest turnover was observed at ∼50 mm Mn2+ and between 15 and 50 mm Mg2+. Our data allowed us to determine the apparent metal ion binding affinities (Fig. 4C). We used data points at concentrations of up to 50 mm for both metal ions because of the reduced activity at higher concentrations, as described above (Fig. 4, B and D). The data revealed an apparent Kd for Mg2+ binding of 2.32 ± 0.51 mm, which was ∼3-fold lower than that of Mn2+ (apparent Kd 7.88 ± 1.76 mm). However, the maximum turnover rate in the presence of Mn2+ was almost 3-fold higher than when Mg2+ is present (Vmax of 2.00 ± 0.14 and 0.70 ± 0.03 peptide/s, respectively). This is consistent with a ∼3-fold higher activity of eukaryotic OST enzymes in the presence of manganese compared with magnesium, determined at 10 mm metal ion concentrations for yeast OST (37) and 3 mm metal ion concentrations for hen oviduct OST (39).
Specific Interactions of the +2 Position for Sequon Binding
The PglB structure suggested specific interactions between the sequon and the conserved WWD-motif. The hydroxyl group of the +2 Thr side chain can form hydrogen bonds to Trp-463, Trp-464, and Asp-465. An additional hydrogen bond is possible between Asp-465 and the backbone of the acceptor peptide (Fig. 5A). In addition, a van der Waals (vdW) contact between Ile-572 and the methyl group of the +2 Thr was proposed. Ile-572 is not only conserved in bacterial homologues, but can also be found in eukaryotes and some archaeal homologues (Fig. 5B). A mutation of the corresponding isoleucine to alanine in C. jejuni PglB reduced its in vitro activity (40). To test the role of the postulated vdW interaction with Ile-572, we performed binding studies with different sequon and PglB variants. We observed that peptide binding affinity was reduced 4-fold when the +2 Thr was replaced by a serine (Fig. 5C, Table 2, and supplemental Fig. S4). Intriguingly, a similar reduction in peptide affinity was observed when the DQNAT peptide was bound to the I572V mutant (Fig. 5C, Table 2, and supplemental Fig. S4). This suggested that the removal of a methyl group either from the +2 Thr of the sequon or from Ile-572 of the enzyme leads to a comparable reduction in sequon binding. The I572V mutant revealed a ∼2-fold higher affinity for a +2 Thr compared with a +2 Ser, suggesting that the methyl group of Thr can still benefit from vdW interactions with a Val-572. A further truncation of Ile-572 to an alanine reduced binding of the DQNAT sequon 57-fold and that of the DQNAS sequon 165-fold compared with the WT enzyme (Fig. 5C, Table 2, and supplemental Fig. S4). To compare these effects with catalytic activities, we determined turnover rates of Ile-572 mutants. We found a stepwise reduction in activity for PglB mutants I572V and I572A. In both cases, turnover was 1.2-fold faster for the DQNAT sequon compared with DQNAS (Fig. 5C, Table 2, and supplemental Fig. S2).
FIGURE 5.

Interactions between the +2 sequon position and PglB. A, ball and stick representation of acceptor peptide (yellow) and interacting PglB residues (cyan), as observed in the X-ray structure (PDB code 3RCE). Sequon and PglB residues are labeled in black and cyan, respectively. Yellow lines indicate N and C termini of the sequon. Proposed hydrogen bonds are shown as dashed lines. B, sequence alignment of different bacterial (B), archaeal (A), and eukaryotic (E) Stt3 homologues, highlighting the conservation of Ile-572. The alignment was performed using M-Coffee (56). C, peptide binding affinities (1/Kd; left panel) and turnover rates (right panel) for DQNAT and DQNAS-containing peptides and WT, I572A, and I572V PglB, respectively. Error bars indicate the S.E. of each fit.
TABLE 2.
Glycosylation turnover rates and dissociation constants for PglP and peptide variants affecting interactions at the +2 position
Turnover rates and dissociation constants were derived from curves shown in supplemental Figs. S2 and S4. The errors represent the S.E. of the fit for each turnover rate and dissociation constant. Values for relative turnover and relative binding refer to the ratio of an individual mutant/sequon and WT PglB/DQNAT sequon.
| Mutant | Sequon | Turnover rate | Relative turnover/fold reduction | Dissociation constant (Kd) | Relative binding |
|---|---|---|---|---|---|
| peptides/s | μm | ||||
| WT | DQNAT | 1.50 ± 0.04 | 1 | 1.02 ± 0.06 | 1 |
| DQNAS | 1.25 ± 0.04 | 0.83/1.2-fold | 4.02 ± 0.20 | 0.25 | |
| DQNAC | (3.65 ± 0.12) × 10−3 | 2.43 × 10−3/400-fold | –a | ||
| DQNAV | (2.21 ± 0.07) × 10−4 | 1.47 × 10−4/7,000-fold | –a | ||
| DQNAA | (3.60 ± 0.12) × 10−4 | 2.40 × 10−4/4,000-fold | –a | ||
| W464F | DQNAT | (1.55 ± 0.05) × 10−3 | 1.03 × 10−3/1,000-fold | –a | |
| I572V | DQNAT | 1.08 ± 0.02 | 0.72/1.4-fold | 3.75 ± 0.15 | 0.27 |
| DQNAS | 0.61 ± 0.02 | 0.41/2.5-fold | 7.71 ± 0.11 | 0.13 | |
| DQNAA | (3.60 ± 0.06) × 10−4 | 2.40 × 10−4/4,000-fold | NDb | ||
| I572A | DQNAT | 0.33 ± 0.08 | 0.22/4.5-fold | 57.31 ± 0.93 | 0.02 |
| DQNAS | 0.13 ± 0.04 | 8.67 × 10−2/12-fold | 165.60 ± 5.50 | 0.01 | |
| DQNAA | (1.14 ± 0.05) × 10−4 | 7.60 × 10−5/13,000-fold | ND |
a Peptide binding was observed, but no Kd could be determined because the fluorescence anisotropy signal reached only 30–50% saturation at the limiting PglB concentration of 100–150 μm.
b ND, data not determined.
In contrast to the I572A mutation, changes in the WWD motif affected peptide binding much more. Mutation of the second Trp residue of the motif (W464F, Fig. 5A) resulted in a 1000-fold slower turnover rate and strongly decreased peptide affinity, making it impossible to determine accurate Kd values (Table 2 and supplemental Figs. S2 and S4). Given the sensitivity of our assays, we were able to quantify binding and turnover of non-canonical sequons. We investigated binding and turnover for substrate peptides containing cysteine, valine, or alanine in the +2 position, because the PglB structure revealed that these amino acids could fit into the binding pocket. Binding was strongly reduced for the DQNAC, DQNAV, and DQNAA sequons (supplemental Fig. S4 and Table 2), and no accurate Kd values could be determined. The glycosylation turnover of the DQNAC sequon was reduced 400-fold compared with DQNAT (and 350-fold compared with DQNAS) (Table 2 and supplemental Fig. S2). Turnover of DQNAV or DQNAA was reduced 7000- and 4000-fold compared with DQNAT, respectively (Table 2 and supplemental Fig. S2).
Strong Contribution of −2 Position for Sequon Binding
In contrast to archaeal and eukaryotic Stt3 homologues, bacterial OSTs recognize and specifically interact with a negatively charged amino acid at the −2 position of the sequon (11, 13, 27). In vitro studies of C. jejuni PglB expressed in E. coli membranes revealed a preference for a −2 Asp over a −2 Glu (41). The X-ray structure of C. lari PglB revealed a possible salt bridge between the −2 Asp with the side chain of Arg-331 (Fig. 6A), a residue only conserved in bacterial homologues. We have quantified this interaction in C. lari PglB. The affinity of the R331A mutant for DQNAT, as well as that of WT PglB for AQNAT, was virtually abolished (supplemental Fig. S4, Table 3, and supplemental Table S1), and no Kd values could be determined. In vitro glycosylation of the R331A mutant revealed a 400-fold reduction in turnover for the DQNAT sequon, but a 15,000-fold reduction for AQNAT (Table 3 and supplemental Fig. S2). Even though the R331A mutant was unable to form a salt bridge, it nevertheless, revealed a preference for a −2 Asp over a −2 Ala in the sequon. Notably, the turnover of WT PglB enzyme with AQNAT was reduced 50,000-fold compared with the consensus DQNAT sequon (Table 3 and supplemental Fig. S2). We validated these findings using an in vivo glycosylation assay with an acceptor protein containing either a DQNAT or an AQNAT sequon. Wild type PglB glycosylated DQNAT-containing acceptor protein almost quantitatively, but was unable to process AQNAT. The R331A mutant showed reduced glycosylation efficiency for DQNAT but, intriguingly, a very low but non-negligible glycosylation of AQNAT, which was detectable in the anti-glycan immunoblot (Fig. 6B). We thus demonstrate that a mutant bacterial OST enzyme can glycosylate a eukaryotic sequon in vitro and in vivo. However, an approach to invert the interaction at the −2 position was not successful. Neither PglB mutants R331D nor R331E were able to glycosylate a RQNAT sequon in vivo (data not shown). Another strongly conserved the PglB residue in close vicinity of the −2 Asp of the sequon is Arg-147, which forms a hydrogen bond with the catalytic Glu-319 (Fig. 6A). A R147K mutant revealed reduced in vivo glycosylation of a DQNAT-containing acceptor protein. A double mutant R147K,R331A was completely inactive (Fig. 6B).
FIGURE 6.

Interactions between the −2 sequon position and PglB. A, ball and stick representation of acceptor peptide (yellow) and interacting PglB residues (cyan), as observed in the X-ray structure (PDB code 3RCE). Sequon and PglB residues are labeled in black and cyan, respectively. Yellow lines indicate N and C termini of the sequon. Proposed hydrogen bonds are shown as dashed lines. A potential interaction between Arg-147 and the −2 Asp is indicated. B, immunoblots of in vivo glycosylation reactions detecting acceptor protein 3D5 (top) or bacterial N-glycans (bottom). Glycosylation results in a mobility shift from the non-glycosylated (g0) to the glycosylated form of the acceptor protein (g1). PglB mutants are indicated above the lanes, the glycosylation sequons present in the acceptor proteins are indicated below the lanes.
TABLE 3.
Glycosylation turnover rates and dissociation constants for sequon and PglB variants affecting interactions at −2 position
Turnover rates and dissociation constants were derived from curves shown in supplemental Figs. S2 and S4. The errors represent the S.E. of the fit for each turnover rate and dissociation constant. Values for relative turnover and relative binding refer to the ratio of an individual mutant/sequon and WT PglB/DQNAT sequon.
| Mutant | Sequon | Turnover rate | Relative turnover/fold reduction | Dissociation constant (Kd) |
|---|---|---|---|---|
| peptide/s | μm | |||
| WT | DQNAT | 1.50 ± 0.04 | 1 | 1.02 ± 0.06 |
| WT | AQNAT | (3.08 ± 0.13) × 10−5 | 2.05 × 10−5/50,000-fold | –a |
| R331A | DQNAT | (3.71 ± 0.08) × 10−3 | 2.47 × 10−3/400-fold | –a |
| R331A | AQNAT | (1.03 ± 0.02) × 10−4 | 6.87 × 10−5/15,000-fold | NDb |
a Peptide binding was observed, but no Kd could be determined because the fluorescence anisotropy signal reached only 30–50% saturation at the limiting PglB concentration of 100–150 μm.
b ND, data not determined.
DISCUSSION
Despite its essential nature, the reaction mechanism of oligosaccharyltransferase is insufficiently understood. This is not only due to the scarcity of structural insight (only one structure of a full-length enzyme available), but also due to the challenges of studying dynamic and labile membrane proteins in vitro.
The crystal structure of C. lari PglB revealed bound acceptor substrate in the absence of the donor substrate, which distinguishes PglB from glycosyltransferases (35). Hence, this allowed us to study acceptor binding as a discrete step preceding catalysis. However, we are aware that the sequence of events of the glycosylation reaction might be different from the mechanistic model for the in vitro reaction as represented in Fig. 1.
Guided by our crystal structure, we have introduced specific mutations in PglB and acceptor peptides, and report the first quantification of sequon binding affinities and turnover rates obtained with purified enzyme. For this investigation, milligram amounts of monodisperse PglB retaining its stability at concentrations above 20 mg/ml were required, which was challenging due to the low expression levels and the inherent aggregation behavior of PglB in detergent micelles. PglB expression levels ranged from 0.08 to 0.12 mg/g of E. coli cells, and only protein variants that showed excellent stability and monodispersity at concentrations up to 300 μm were evaluated. To prevent aggregation, all anisotropy measurements were carried out at 4 °C. For wild type enzyme and consensus sequon DQNAT, binding was also measured at 23 °C and yielded a Kd value of 1.62 ± 0.02 μm (supplemental Fig. S5), indicating that the temperature increase only slightly reduces peptide binding affinity. The concentrated protein was analyzed by size exclusion chromatography before and after the measurements. The second experimental requirement was the preparation of reaction substrates. LLO could be prepared from E. coli cells transformed with the genes allowing it to biosynthetically produce C. jejuni LLO, which can serve as a substrate for the C. lari enzyme. Acceptor peptides with attached fluorescent dyes had previously been used for in vitro glycosylation (42). However, our synthetic fluorescently labeled peptides could also be used for independent measurements of peptide binding to PglB by fluorescence anisotropy, a highly sensitive technique.
For the consensus sequon (DQNAT), we performed both Michaelis-Menten kinetics and turnover rate analysis. The peptide concentration used in the time course (10 μm) was ∼4 times the observed Km value (2.6 μm), and the obtained turnover rate was close to the observed Vmax. For all other reactions reported here, initial turnover rates were measured instead of Michaelis-Menten kinetics. This was the consequence of a technical limitation. As illustrated in Fig. 2, C and E, accurate detection of the ratio of unmodified acceptor peptide to the glycosylated product is only possible when both are measurable from the same gel. In addition, for very unfavorable combinations of enzyme mutants or sequon variants, the solubility of the fluorescently labeled peptides prevented the use of concentrations that guaranteed maximum turnover (Vmax). We therefore determined turnover rates using constant concentrations of synthetic fluorescently labeled peptides and variable amounts of PglB mutants. For reactions with a low enzyme/substrate ratio, the obtained turnover rates can be directly compared with wild type because the peptide concentration was above the Km value and the determined turnover rates were consequently a good approximation of Vmax. For the reactions of highly disfavored enzyme mutants or poor substrate peptide analogues, turnover rates were quantified at high enzyme/substrate ratios. Under these conditions, the determined turnover rates did not reach Vmax. However, not only were comparisons largely reliable, but studying the enzymatic reaction at a fixed peptide concentration also reflects the natural scenario inside a cell, where a constant flow of peptide segments are presented to OST. In addition to the caveat above, the absolute turnover rates cannot be transferred directly to the in vivo situation because (i) the concentration of divalent metal ions inside a cell (periplasm or lumen of the ER) is much lower than that used in in vitro assays, (ii) glycosylation sequons are embedded in larger polypeptide chains, and (iii) in vivo OST and LLO are surrounded by a lipid bilayer rather than detergent micelles. Nevertheless, the rates reported here are highly useful for understanding the reaction mechanism of PglB. Our study therefore provides the first direct determination of a dissociation constant of an isolated acceptor sequon peptide and a purified OST enzyme. Our data revealed an intricate mutual dependence of peptide and metal binding for successful catalysis. Several conclusions can be drawn.
1) Residues in the active site of PglB are essential for catalysis, but not necessarily for peptide binding. Mutations of the essential Asp-56 and Glu-319 to alanine not only decreased in vivo and in vitro rates, but also drastically lower in vitro peptide affinity. This suggests that, along with bound metal, these residues provide stabilizing interactions to the amide group of the acceptor asparagine. In contrast, the conserved Arg-375 was essential for catalysis, but not for binding. Whereas replacing Arg-375 with a lysine preserved peptide binding and catalytic activity, changing it to an alanine abolished catalysis, but without affecting peptide binding. This observation agrees with the hypothesis that Arg-375 provides critical interactions with the pyrophosphate moiety in the LLO substrate. It also validates the experimental approach of studying peptide binding and catalysis using distinct techniques to obtain mechanistic insight.
2) The bound metal ion in the active site of PglB is not only essential for catalysis, but also for peptide binding. The requirement of a divalent cation for OST activity is generally accepted (14, 23, 36–38). However, it was primarily assumed to act as a Lewis acid to stabilize the lipid-pyrophosphate leaving group, in analogy to metal-dependent glycosyltransferases (35). As the metal ion is not in direct contact with the acceptor peptide, its strict requirement for peptide binding to PglB was unexpected. However, it is consistent with the finding that active site residues Asp-56 and Glu-319, which contact bound metal ion, also contribute to peptide binding.
3) The dependence of PglB activity on relatively high concentrations of metal ions has important implications for in vivo OST function. At 10 mm, bound manganese caused a 17-fold stronger sequon binding than magnesium. This effect may be due to different coordination properties of the two metals (43), because protein stability was not affected after exposure to elevated concentrations of either ion (supplemental Fig. S1). The preference for Mn2+ over Mg2+ was much less pronounced in the in vitro glycosylation reaction, and we conclude that tighter peptide binding induced by Mn2+ might not necessarily correlate with more efficient catalysis. However, our assays cannot reveal whether metal ion binding precedes sequon binding or if these events happen simultaneously. At lower metal concentrations, we observed similar activities for Mn2+ and Mg2+, which has implications for in vivo situation. Even though Mn2+ has been suggested as the physiological cation (44), it is estimated that the concentration of free Mn2+ in the ER is 102-103 times lower than that of Mg2+ (45, 46). Similar estimates can be made for the periplasm of Campylobacter species, because in their natural environment, Mg2+ is 103-105 times more abundant than Mn2+ (47–50). The growth media used for in vivo glycosylation in E. coli (LB media) contains ∼0.23 mm Mg2+, suggesting that OST operates far below its maximal activity. Therefore, it is not surprising that supplementing growth media with MnCl2 is a successful strategy to increase the catalytic efficiency in CHO cells (51). However, additional studies are required to determine whether both magnesium and manganese are used in vivo.
4) Hydrogen bonds between the +2 Thr/Ser of the sequon and the surface of PglB provide strong contributions to peptide binding. Only amino acids containing a hydroxyl group (Ser and Thr) can provide the specific network of hydrogen bonds formed with the conserved WWD motif. In principle, the thiol group of cysteine can also form such hydrogen bonds, but they are much weaker than those of the hydroxyl group. We observed strongly reduced in vitro affinity and glycosylation rates of acceptor peptides containing a DQNAC sequon. Nevertheless, glycosylated N-X-C sequons have been found in vivo, for example, in bovine protein C or in human von Willebrand factor (10). Also Bause and Legler (23) showed that a peptide containing an N-G-C site can be glycosylated in vitro using calf liver microsomes, albeit with low efficiency. A mapping of the mouse N-glycoproteome revealed that 1.3% of all N-glycosylated sites are N-X-C sequons, followed by 0.4% N-X-V sequons (21).
5) Isoleucine 572 modulates sequon binding by providing van der Waals interactions. The binding affinities we determined not only confirmed the preference of Thr over Ser (DQNAT versus DQNAS sequon) but could quantify the role of Ile-572 in this phenomenon. Removal of the stabilizing vdW interaction between the side chain of Ile-572 and the +2 Thr was found to reduce both peptide binding and glycosylation rates. Different Ile-572 mutants, combined with either +2 Thr or +2 Ser sequons, showed a positive correlation between these two events (Fig. 7). The mutant I572A, in combination with a DQNAS sequon, revealed the lowest affinity and turnover, whereas WT enzyme and DQNAT yielded the highest binding affinity and glycosylation rates. This correlation illustrates the additive effect of the vdW interactions contributing to increased substrate affinity and hence to increased glycosylation efficiency. However, the effect was less pronounced for catalysis than for sequon binding. We conclude that the reduced vdW interactions could speed up the reaction, possibly by facilitating faster release of glycopeptide from the enzyme, thus counteracting the reduced binding affinity (Fig. 1). Ile-572 is conserved in ϵ-proteobacteria and a mutation to a Val in C. jejuni PglB (I571V) revealed a slightly reduced glycosylation efficiency in vivo (52). In δ-proteobacteria, Ile-572 is occasionally replaced by another hydrophobic residue. For example, Desulfovibrio alaskensis PglB has a Leu in the corresponding position (Leu-597, Fig. 5B). Given that a Leu provides similar vdW interactions with the distal methyl group as an Ile, we expect that N-glycosylation in D. alaskensis is efficient as well. Identifying the analogues residues of Ile-572 in eukaryotic Stt3 homologues is not straightforward. If biased sequence alignments are used, Ile-572 of PglB corresponds to Ile-593 in the Stt3 subunit of Saccharomyces cerevisiae OST. Intriguingly, the mutation I593A of S. cerevisiae Stt3 indeed resulted in a temperature-sensitive phenotype, whereas the mutation W517Y was lethal (20).
FIGURE 7.

Correlation of peptide binding and glycosylation turnover. Values for peptide binding and turnover rates presented in Fig. 4C are correlated in a two-dimensional plot. Peptide binding is presented as log (1/Kd). Error bars indicate the S.E. of each fit, but are smaller than the symbols in most cases.
6) An aspartate in the −2 position of the bacterial sequon contributes strongly to the binding affinity. N-Linked glycosylation in Bacteria is more specific and less frequent than in eukaryotes. Also, only strains representing the clade ϵ- and δ-proteobacteria have been shown to N-glycosylate proteins (6). We studied the effect of removing either partner of the proposed salt bridge between the −2 Asp of the DQNAT sequon and Arg-331 of PglB. We observed abolished binding affinity and strongly reduced catalysis. Because removing the negatively charged −2 Asp of the sequon resulted in a much stronger effect than removing the positively charged R331A of PglB, we conclude that the recognition of the −2 Asp is not exclusively accomplished by Arg-331, but requires additional interactions. One candidate for such an interaction is the side chain of Arg-147, which also forms hydrogen bonds with catalytic residues (Fig. 6A). Its mutation to a lysine indeed reduced sequon binding and catalysis, but it is unclear if the effect is specific.
The 400-fold reduction of glycosylation activity of the R331A mutant correlates with the observation that Km values of sequons of eukaryotic OST enzymes are roughly 2 orders of magnitude higher than those of bacterial sequons binding to PglB (23, 41, 53). With the extension of the glycosylation sequon, Bacteria probably increase their substrate specificity, achieving higher stringency. This might be required to recruit acceptor sites in exposed and flexible regions of folded proteins in the periplasm (32, 54). In contrast, eukaryotic OST enzymes are proposed to be localized next to the secretion machinery in the ER membrane, resulting in a high local concentration of sequons in the nascent polypeptide chains (15, 17, 19, 55). In summary, our data highlights the details of the complex network of interactions between the sites of peptide, metal, and LLO binding on the surface of PglB, allowing sequon recognition and glycosylation to be studied as distinct steps.
Acknowledgments
We thank E. Weber-Ban and R. Glockshuber for helpful discussions and granting us access to their fluorometer. We thank the Mass Spectrometry Service (Department of Chemistry and Biochemistry, University of Berne) for recording MS spectra of labeled acceptor peptides. We thank M. Niederer for providing purified control membrane protein and S. Numao and A. Faridmoayer for helpful discussions.
This work was supported by a grant from the NCCR Structural Biology Zurich (to K. P. L.) and Swiss National Science Foundation Grants SNF 200020_125020 (to J. L. R.) and SNF 31003A-131075/1 (to K. P. L.).

This article contains supplemental Methods, Tables S1 and S2, and Figs. S1–S5.
- LLO
- lipid-linked oligosaccharide
- OST
- oligosaccharyltransferase
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- vdW
- van der Waals
- DCM
- dichloromethane.
REFERENCES
- 1. Varki A. (1993) Biological roles of oligosaccharides. All of the theories are correct. Glycobiology 3, 97–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calo D., Kaminski L., Eichler J. (2010) Protein glycosylation in Archaea. Sweet and extreme. Glycobiology 20, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 3. Mescher M. F., Strominger J. L. (1976) Purification and characterization of a prokaryotic glucoprotein from the cell envelope of Halobacterium salinarium. J. Biol. Chem. 251, 2005–2014 [PubMed] [Google Scholar]
- 4. Lechner J., Wieland F. (1989) Structure and biosynthesis of prokaryotic glycoproteins. Annu. Rev. Biochem. 58, 173–194 [DOI] [PubMed] [Google Scholar]
- 5. Jervis A. J., Butler J. A., Lawson A. J., Langdon R., Wren B. W., Linton D. (2012) Characterization of the structurally diverse N-linked glycans of Campylobacter species. J. Bacteriol. 194, 2355–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nothaft H., Szymanski C. M. (2010) Protein glycosylation in bacteria. Sweeter than ever. Nat. Rev. Microbiol. 8, 765–778 [DOI] [PubMed] [Google Scholar]
- 7. Szymanski C. M., Yao R., Ewing C. P., Trust T. J., Guerry P. (1999) Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol. Microbiol. 32, 1022–1030 [DOI] [PubMed] [Google Scholar]
- 8. Marshall R. D. (1972) Glycoproteins. Annu. Rev. Biochem. 41, 673–702 [DOI] [PubMed] [Google Scholar]
- 9. Abu-Qarn M., Eichler J. (2007) An analysis of amino acid sequences surrounding archaeal glycoprotein sequons. Archaea 2, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gavel Y., von Heijne G. (1990) Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites. Implications for protein engineering. Protein Eng. 3, 433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Igura M., Kohda D. (2011) Quantitative assessment of the preferences for the amino acid residues flanking archaeal N-linked glycosylation sites. Glycobiology 21, 575–583 [DOI] [PubMed] [Google Scholar]
- 12. Nita-Lazar M., Wacker M., Schegg B., Amber S., Aebi M. (2005) The N-X-S/T consensus sequence is required but not sufficient for bacterial N-linked protein glycosylation. Glycobiology 15, 361–367 [DOI] [PubMed] [Google Scholar]
- 13. Petrescu A. J., Milac A. L., Petrescu S. M., Dwek R. A., Wormald M. R. (2004) Statistical analysis of the protein environment of N-glycosylation sites. Implications for occupancy, structure, and folding. Glycobiology 14, 103–114 [DOI] [PubMed] [Google Scholar]
- 14. Pless D. D., Lennarz W. J. (1977) Enzymatic conversion of proteins to glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 74, 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruiz-Canada C., Kelleher D. J., Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nilsson I., Kelleher D. J., Miao Y., Shao Y., Kreibich G., Gilmore R., von Heijne G., Johnson A. E. (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell. Biol. 161, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schulz B. L., Stirnimann C. U., Grimshaw J. P., Brozzo M. S., Fritsch F., Mohorko E., Capitani G., Glockshuber R., Grütter M. G., Aebi M. (2009) Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc. Natl. Acad. Sci. U.S.A. 106, 11061–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., Aebi M. (2002) N-Linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793 [DOI] [PubMed] [Google Scholar]
- 19. Wilson C. M., High S. (2007) Ribophorin I acts as a substrate-specific facilitator of N-glycosylation. J. Cell. Sci. 120, 648–657 [DOI] [PubMed] [Google Scholar]
- 20. Yan Q., Lennarz W. J. (2002) Studies on the function of oligosaccharyl transferase subunits. Stt3p is directly involved in the glycosylation process. J. Biol. Chem. 277, 47692–47700 [DOI] [PubMed] [Google Scholar]
- 21. Zielinska D. F., Gnad F., Wiśniewski J. R., Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 22. Bause E. (1984) Model studies on N-glycosylation of proteins. Biochem. Soc. Trans. 12, 514–517 [DOI] [PubMed] [Google Scholar]
- 23. Bause E., Legler G. (1981) The role of the hydroxy amino acid in the triplet sequence Asn-Xaa-Thr(Ser) for the N-glycosylation step during glycoprotein biosynthesis. Biochem. J. 195, 639–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasturi L., Chen H., Shakin-Eshleman S. H. (1997) Regulation of N-linked core glycosylation. Use of a site-directed mutagenesis approach to identify Asn-Xaa-Ser/Thr sequons that are poor oligosaccharide acceptors. Biochem. J. 323, 415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasturi L., Eshleman J. R., Wunner W. H., Shakin-Eshleman S. H. (1995) The hydroxy amino acid in an Asn-X-Ser/Thr sequon can influence N-linked core glycosylation efficiency and the level of expression of a cell surface glycoprotein. J. Biol. Chem. 270, 14756–14761 [DOI] [PubMed] [Google Scholar]
- 26. Lizak C., Gerber S., Numao S., Aebi M., Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 27. Kowarik M., Young N. M., Numao S., Schulz B. L., Hug I., Callewaert N., Mills D. C., Watson D. C., Hernandez M., Kelly J. F., Wacker M., Aebi M. (2006) Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 25, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ielmini M. V., Feldman M. F. (2011) Desulfovibrio desulfuricans PglB homolog possesses oligosaccharyltransferase activity with relaxed glycan specificity and distinct protein acceptor sequence requirements. Glycobiology 21, 734–742 [DOI] [PubMed] [Google Scholar]
- 29. Schwarz F., Lizak C., Fan Y. Y., Fleurkens S., Kowarik M., Aebi M. (2011) Relaxed acceptor site specificity of bacterial oligosaccharyltransferase in vivo. Glycobiology 21, 45–54 [DOI] [PubMed] [Google Scholar]
- 30. Lefebre M. D., Valvano M. A. (2002) Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl. Environ. Microbiol. 68, 5956–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lizak C., Fan Y. Y., Weber T. C., Aebi M. (2011) N-Linked glycosylation of antibody fragments in Escherichia coli. Bioconjug. Chem. 22, 488–496 [DOI] [PubMed] [Google Scholar]
- 32. Kowarik M., Numao S., Feldman M. F., Schulz B. L., Callewaert N., Kiermaier E., Catrein I., Aebi M. (2006) N-Linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science 314, 1148–1150 [DOI] [PubMed] [Google Scholar]
- 33. Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protoc. 1, 16–22 [DOI] [PubMed] [Google Scholar]
- 34. Jaffee M. B., Imperiali B. (2011) Exploiting topological constraints to reveal buried sequence motifs in the membrane-bound N-linked oligosaccharyl transferases. Biochemistry 50, 7557–7567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases. Structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 36. Igura M., Maita N., Kamishikiryo J., Yamada M., Obita T., Maenaka K., Kohda D. (2008) Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J. 27, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharma C. B., Lehle L., Tanner W. (1981) N-Glycosylation of yeast proteins. Characterization of the solubilized oligosaccharyltransferase. Eur. J. Biochem. 116, 101–108 [DOI] [PubMed] [Google Scholar]
- 38. Welply J. K., Shenbagamurthi P., Lennarz W. J., Naider F. (1983) Substrate recognition by oligosaccharyltransferase. Studies on glycosylation of modified Asn-X-Thr/Ser tripeptides. J. Biol. Chem. 258, 11856–11863 [PubMed] [Google Scholar]
- 39. Das R. C., Heath E. C. (1980) Dolichyldiphosphoryloligosaccharide–protein oligosaccharyltransferase. Solubilization, purification, and properties. Proc. Natl. Acad. Sci. U.S.A. 77, 3811–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maita N., Nyirenda J., Igura M., Kamishikiryo J., Kohda D. (2010) Comparative structural biology of eubacterial and archaeal oligosaccharyltransferases. J. Biol. Chem. 285, 4941–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen M. M., Glover K. J., Imperiali B. (2007) From peptide to protein. Comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46, 5579–5585 [DOI] [PubMed] [Google Scholar]
- 42. Kohda D., Yamada M., Igura M., Kamishikiryo J., Maenaka K. (2007) New oligosaccharyltransferase assay method. Glycobiology 17, 1175–1182 [DOI] [PubMed] [Google Scholar]
- 43. Bock C. W., Katz A. K., Markham G. D., Glusker J. P. (1999) Manganese as a replacement for magnesium and zinc. Functional comparison of the divalent ions. J. Am. Chem. Soc. 121, 7360–7372 [Google Scholar]
- 44. Kaufman R. J., Swaroop M., Murtha-Riel P. (1994) Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry 33, 9813–9819 [DOI] [PubMed] [Google Scholar]
- 45. Sugiyama T., Goldman W. F. (1995) Measurement of SR free Ca2+ and Mg2+ in permeabilized smooth muscle cells with use of furaptra. Am. J. Physiol. 269, C698–705 [DOI] [PubMed] [Google Scholar]
- 46. Williams R. J. P., da Silva J. J. (2000) The distribution of elements in cells. Coordin. Chem. Rev. 200, 247–348 [Google Scholar]
- 47. Rükgauer M., Klein J., Kruse-Jarres J. D. (1997) Reference values for the trace elements copper, manganese, selenium, and zinc in the serum/plasma of children, adolescents, and adults. J. Trace Elem. Med. Biol. 11, 92–98 [DOI] [PubMed] [Google Scholar]
- 48. Weisinger J. R., Bellorín-Font E. (1998) Magnesium and phosphorus. Lancet 352, 391–396 [DOI] [PubMed] [Google Scholar]
- 49. Davison W. (1993) Iron and manganese in lakes. Earth Sci. Rev. 34, 119–163 [Google Scholar]
- 50. Majzurawska M., Rouilly M., Morf W. E., Simon W. (1989) Determination of magnesium and calcium in water with ion-selective electrodes. Anal. Chim. Acta 218, 47–59 [Google Scholar]
- 51. Gawlitzek M., Estacio M., Fürch T., Kiss R. (2009) Identification of cell culture conditions to control N-glycosylation site-occupancy of recombinant glycoproteins expressed in CHO cells. Biotechnol. Bioeng. 103, 1164–1175 [DOI] [PubMed] [Google Scholar]
- 52. Ihssen J., Kowarik M., Wiesli L., Reiss R., Wacker M., Thöny-Meyer L. (2012) Structural insights from random mutagenesis of Campylobacter jejuni oligosaccharyltransferase PglB. BMC Biotechnol. 12, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bause E., Hettkamp H., Legler G. (1982) Conformational aspects of N-glycosylation of proteins. Studies with linear and cyclic peptides as probes. Biochem. J. 203, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Slynko V., Schubert M., Numao S., Kowarik M., Aebi M., Allain F. H. (2009) NMR structure determination of a segmentally labeled glycoprotein using in vitro glycosylation. J. Am. Chem. Soc. 131, 1274–1281 [DOI] [PubMed] [Google Scholar]
- 55. Harada Y., Li H., Li H., Lennarz W. J. (2009) Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U.S.A. 106, 6945–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moretti S., Armougom F., Wallace I. M., Higgins D. G., Jongeneel C. V., Notredame C. (2007) The M-Coffee web server. A meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res. 35, W645–648 [DOI] [PMC free article] [PubMed] [Google Scholar]