

Background: The Na+/K+-ATPase maintains Na+/K+ gradients across the plasma membrane, essential for cellular functions.
Results: Zebrafish deficient of α3Na+/K+-ATPase display abnormal motility and brain ventricle dilation.
Conclusion: Zebrafish α3Na+/K+-ATPase is CNS-specific and required for brain ventricle maintenance and embryonic motility.
Significance: This is the first study assessing α3Na+/K+-ATPase in brain development and embryonic motility.
Keywords: Animal Models; ATPases; Membrane Proteins; Neurological Diseases; Proteomics; Na,K-ATPase; Brain Ventricle Dilation; Motility; Resting Membrane Potential; Zebrafish
Abstract
Na+/K+-ATPases are transmembrane ion pumps that maintain ion gradients across the basolateral plasma membrane in all animal cells to facilitate essential biological functions. Mutations in the Na+/K+-ATPase α3 subunit gene (ATP1A3) cause rapid-onset dystonia-parkinsonism, a rare movement disorder characterized by sudden onset of dystonic spasms and slow movements. In the brain, ATP1A3 is principally expressed in neurons. In zebrafish, the transcripts of the two ATP1A3 orthologs, Atp1a3a and Atp1a3b, show distinct expression in the brain. Surprisingly, targeted knockdown of either Atp1a3a or Atp1a3b leads to brain ventricle dilation, a likely consequence of ion imbalances across the plasma membrane that cause accumulation of cerebrospinal fluid in the ventricle. The brain ventricle dilation is accompanied by a depolarization of spinal Rohon-Beard neurons in Atp1a3a knockdown embryos, suggesting impaired neuronal excitability. This is further supported by Atp1a3a or Atp1a3b knockdown results where altered responses to tactile stimuli as well as abnormal motility were observed. Finally, proteomic analysis identified several protein candidates highlighting proteome changes associated with the knockdown of Atp1a3a or Atp1a3b. Our data thus strongly support the role of α3Na+/K+-ATPase in zebrafish motility and brain development, associating for the first time the α3Na+/K+-ATPase deficiency with brain ventricle dilation.
Introduction
The Na+/K+-ATPase is essential for maintaining Na+ and K+ gradients across the plasma membrane, required for many cellular functions, e.g. regulation of cell volume, pH, Na+-coupled secondary transport of molecules and neurotransmitters, and the excitability of muscle and neuronal cells (1, 2). In mammals, the four α (α1, α2, α3, and α4) isoforms display distinct tissue-specific expression patterns (2, 3). The α3 isoform (ATP1α3) is expressed in brain, eye, ear, muscle, cartilage, uterus, placenta, and heart (4–7). In the brain, the ATP1α3 subunit is present exclusively in neurons (8). Mutations in the gene cause the neurological disorder rapid-onset dystonia-parkinsonism (RDP)2 (9, 10), a rare movement disorder with an abrupt onset and rapid (hours to weeks) development of dystonia parkinsonism, primarily bradykinesia and postural instability (10, 11).
Besides ion pump roles, Na+/K+-ATPases also serve as signal transducers, modulating synaptic plasticity, e.g. inducing dendritic growth in cortical neurons (12). Evidence for neuronal roles of ATP1α3 was previously found in both human and rat dorsal root ganglia and also observed in rat embryos (embryonic day 21) (13).
Currently, two different genetically modified mouse models targeting the Atp1a3 gene exist: α3+/−KII4 (14) and α3+/−KII810N (Myshkin) (15). These mice display learning/memory deficits (14), epilepsy/seizures (15), stress-induced motor symptoms (16), anxious phenotype, and depression-like behavior (17). However, they do not fully capture the human RDP symptoms, and the pathology of RDP remains unsolved.
Recently, it became more evident that Danio rerio (zebrafish) is a valuable model for investigating Na+/K+-ATPase functions (18, 19). We employed zebrafish, with its advantageous properties, e.g. external development with optical clarity, small size, short generation time (2–3 months), and high fecundity (20, 21) to further study the role of ATP1α3 in early neuronal functions. In a comparison of the zebrafish brain structure with human, the gross architecture of many zebrafish brain areas, e.g. retina, olfactory bulb, hypothalamus, cerebellum, and spinal cord, is similar to that of humans, although there exist some differences between teleosts and mammals (22). Moreover, zebrafish enables several behavioral and drug tests and hence is relevant for many disease-related studies.
Zebrafish have two ATP1A3 orthologs, Atp1a3a and Atp1a3b (23). Surprisingly, knockdown (KD) of Atp1a3a or Atp1a3b results in severe brain ventricle dilation in contrast to previous data, where KD of Atp1a1 caused reduced brain ventricle inflation (33), thus supporting the role of Na+/K+-ATPase in brain ventricle development. The brain ventricle dilation in Atp1a3a/b KD embryos was accompanied by an ∼20-mV depolarization of the spinal Rohon-Beard (RB) neuron resting membrane potential (RMP), suggesting compromised functions in neurons as a consequence of Atp1a3a KD-generated defects in ion homeostasis. In support of this, morphant embryos further displayed both abnormal touch response and spontaneous movements. To identify additional neuronal functions that may depend on correct ion homeostasis for normal performance, we used a proteomic approach. Interestingly, this revealed several proteins, including cytoskeletal, ion-binding, muscle-associated proteins, etc., with expression levels that were affected by Atp1a3a or Atp1a3b KD.
This is the first detailed study of α3Na+/K+-ATPases in zebrafish and on the basis of conservation of zebrafish and mammalian α3Na+/K+-ATPase expression and functions. Zebrafish can serve as an advantageous model for analysis of brain ventricular volume maintenance and embryonic motility, both related to ion homeostasis.
EXPERIMENTAL PROCEDURES
Animals
TU (Tübingen) zebrafish strain (Nüsslein-Volhard Laboratory, Max Planck Institute, Tübingen, Germany) and Tg(gfap:GFP) transgenic line (Zebrafish International Resource Center, University of Oregon) of either sex were used in the experiments. Embryo maintenance and staging were performed as described previously (24, 25). To conserve optical clarity, embryos were raised in the presence of 0.2 mm 1-phenyl-2-thiourea (24).
Reverse Transcription (RT) and qRT-PCR
qRT-PCR protocol was performed on cDNAs from embryos at corresponding stages (1000-cell, 50% epiboly, 75% epiboly, 6-somite, prim-6, prim-22, pectoral fin, and adult (one male + one female)) as described previously (26). Pearl Primer software (27) and Roche Applied Science assay design were used to design the primers (supplemental Table S2) to detect transcripts of Atp1a3a (accession number: NM_131684.2), Atp1a3b (accession number: NM_131685.2), Actb2 (accession number: NM_181601.3), and Bhmt, Ckma, Ckmb, Gamt, Gpib, Krt4, Krt5, Mhyz2, Ndpkz2, Pgam2, Pvalb9, Zgc:91930, and 113d7.4, accession numbers of which are provided in Table 1. Generated PCR product sizes and identities were verified by gel electrophoresis (data not shown) and DNA sequencing.
TABLE 1.
List of regulated proteins selected from proteomics assay
Proteins relevant for the Atp1a3a and Atp1a3b KD phenotypes were selected from supplemental Table S1. Proteins were detected by iTRAQ LC-MS/MS. Database accession numbers, gene identity, relative-fold change, and tissue associations are given. The Atp1a3 KD/control protein expression ratio indicates up-regulation when it is equal/above 2 and down-regulation when it is equal/below 0.5.
| Accession number | Description | Gene | Atp1a3a KD/control (116/114) | Tissue association |
|---|---|---|---|---|
| 39645432 | Krt5 protein | Krt5 | 0.5 | Epidermis, eye |
| 44890667 | Krt4 protein | Krt4 | 0.5 | Epidermis |
| 158253775 | Zgc:165344 protein | Zgc:165344 | 0.5 | Myotome |
| Accession number | Description | Gene | Atp1a3b KD/control (117/114) | Tissue association |
|---|---|---|---|---|
| 45387573 | Parvalbumin isoform 1d | Pvalb1 | 0.2 | Myotome |
| 123916361 | RecName: Full = betaine-homocysteine S-methyltransferase 1 | Bhmt | 0.3 | All |
| 50512294 | Myosin, heavy polypeptide 2, fast muscle-specific | Mhyz2 | 0.3 | Muscle |
| 41053595 | Nucleoside diphosphate kinase B | Ndpkz2 | 0.3 | Brain, eye, muscle |
| 41056123 | Phosphoglycerate mutase 2 | Pgam2 | 0.4 | Heart, myotome |
| 68366260 | Predicted: glutathione S- transferase θ 1a | Gstt1a | 0.4 | Retina, head mesenchyme |
| 33636707 | Parvalbumin 9 | Pvalb9 | 0.4 | |
| 157787181 | Muscle creatine kinase b | Ckmb | 0.4 | Eye, heart, myotome |
| 123229625 | Creatine kinase, muscle | Ckma | 0.5 | Eye, heart, myotome |
| 51571925 | Adenylate kinase isoenzyme 1 | Zgc:91930 | 0.5 | Myotome, somite |
| 157888726 | Glucose phosphate isomerase b | Gpib | 0.5 | Eye, heart, myotome |
| 157743330 | Guanidinoacetate N- methyltransferase | Gamt | 0.5 | Brain |
| 167234796 | si:dkeyp-113d7.4 | si:dkeyp-113d7.4 | 0.5 | Intermediate filament |
To investigate SP-MO-mediated KD, we initially performed RT-PCR using primers enclosing the presumably extruded exons. This clearly indicated that both SP-MOs caused a KD via nonsense-mediated mRNA decay (data not shown). The quantitation of KD levels was performed by qRT-PCR on first strand cDNA generated from ∼30 WT, std-MO- (5.5, 11, and 3 ng), α3a-SP-MO- (5.5 and 11 ng), and α3b-SP-MO- (1.5 and 3 ng) injected embryos.
Efficiency of each primer pair used in qRT-PCR was detected to be ∼100%. Transcription levels were quantified using the relative quantification method based on comparative threshold cycle values (Ct).
Cloning of Zebrafish Atp1a3a and Atp1a3b cDNA
Total RNA was isolated from a male and a female zebrafish by the TRIzol® method (Invitrogen) (28). Total RNA was used as template to perform RT-PCR by using the SuperScriptTM III RT-PCR system (Invitrogen) according to the manufacturer's instructions with Atp1a3a- and Atp1a3b-specific primer pairs (supplemental Table S2) tagged with restriction enzyme recognition sites. The program for RT-PCR was one cycle for 30 min at 55 °C and 2 min at 94 °C followed by 40 cycles of 15 s at 94 °C, 30 s at 66 °C, and 4 min at 68 °C and a final extension for 15 min at 68 °C. Gel-purified RT-PCR fragments were cloned into pTZ57R vector (InsTAcloneTM PCR cloning kit, Fermentas) following the manufacturer's protocol and subsequently subjected to DNA sequencing.
In Situ Hybridization and Sectioning
Antisense and sense RNA probe templates were generated by PCR on pTZ57R vector harboring Atp1a3a probe coding sequence, for Atp1a3a, and by RT-PCR on adult zebrafish RNA, for Atp1a3b, introducing a T7 priming site in both scenarios. Probes were digoxigenin-labeled during synthesis from purified PCR products by using the digoxigenin RNA labeling mix (Roche Applied Science) and T7 polymerase (Roche Applied Science) following the manufacturer's protocol. Embryos were fixed overnight in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) and kept in MetOH in −20 °C. To detect Atp1a3a and Atp1a3b transcripts, whole-mount in situ hybridization was performed on embryos at 60 hpf, as described previously (29) with minor modifications.
Embryos were embedded and oriented in 5% agar so that they could be cut transversely following dehydrating in ascending alcohol solutions (70% ×2, 96% ×2, and 99% ×2), infiltration, and embedding in glycolmethacrylate (Technovit 7100). Using a Microm 355, 20-μm-thick sections were cut, and every 10th section was taken by systematic sampling. Whole-mount embryos and sections were observed using an inverted microscope, Olympus IX71.
MO-mediated Knockdown of Atp1a3a or Atp1a3b and mRNA Rescue
MOs (Gene Tools, LLC) for microinjection were diluted in distilled water and microinjected into embryos at the 1–4 cell stage. Translation-blocking MOs were: α3a-MO (5′-CTTTCTTCAGTCTGTCAAACGGCGT-3′) (12 ng) and α3b-MO (5′-AGAGTGATGGAGAAAGTGACAGCCT-3′) (3 ng); the splice-blocking MOs were: α3a-SP-MO (5′-TCCACCTGAGCAATGACACCAAACA-3′) (11 ng), targeting the intron 6-exon 7 boundary, and α3b-SP-MO (5′-AGTGCCTGACAGAAACAAAGCATTT-3′) (3 ng), targeting the intron 7-exon 8 boundary. The standard control MO (std-MO: 5′-CCTCTTACCTCAGTTACAATTTATA-3′) (3 and 12 ng) and the p53-MO (5′-GCGCCATTGCTTTGCAAGAATTG-3′) (4 ng) were injected at the indicated amounts. Embryos were grown for 60 h in embryo medium with 0.003% 1-phenyl-2-thiourea and observed under an inverted microscope, Olympus IX71. To visualize zebrafish brain ventricles, rhodamine-conjugated dextran was injected into the brain ventricle at 48 hpf as described previously (30), and embryos were subsequently imaged under bright field and fluorescent light (Olympus IX71).
The template for Atp1a3a and Atp1a3b mRNA was generated by PCR on pTZ57R vector harboring Atp1a3a (3069 bp) and Atp1a3b (3069 bp) coding sequences. In vitro synthesis of Atp1a3a and Atp1a3b mRNA was performed on gel-purified Atp1a3a and Atp1a3b PCR products by using the mMESSAGE mMACHINE T7 ULTRA kit (Ambion) following the manufacturer's instructions. The RNA products were gel-purified (Qiagen), and each embryo was co-injected with 180 pg of WT Atp1a3a mRNA or 45 pg of WT Atp1a3b mRNA together with 11 ng of α3a-SP-MO or 3 ng of α3b-SP-MO, respectively. Embryos were assessed at 60 hpf, grouped morphologically into three classes depending on the severity of the brain ventricle dilation (+, slight/no; ++, moderate; +++, severe) (see Fig. 2D, lower panel), and quantified as percentages of the total number of embryos. Experiments were repeated at least three times, and data are presented as mean ± S.D.
FIGURE 2.

Knockdown of Atp1a3a causes brain ventricle dilation. A, significant brain ventricle dilation occurred in embryos upon Atp1a3a KD mediated by α3a-MO or α3a-SP-MO as compared with std-MO-injected control embryo. This phenotype was rescued by co-injection of Atp1a3a mRNA. p53-MO co-injections with any of the MOs did not rescue the brain ventricle dilation phenotype. B, brain ventricles of Tg(gfap:GFP) line, non-MO-injected and α3a-MO-injected, were injected with rhodamine-conjugated dextran. Brain ventricles and the astrocytes are highlighted by red and green fluorescence, respectively. C, qRT-PCR tested efficiency of α3a-SP-MO-mediated KD in terms of changes in the relative expression level of Atp1a3a. Data are presented as mean ± S.E. of triplicate measurements. Concentrations of MOs are indicated. D, mean percentages ± S.D. of the α3a-SP-MO-injected embryos suffering from brain ventricle dilation of different severity, with (white columns, n = 117) and without (black columns, n = 96) Atp1a3a mRNA co-injection, are plotted. Embryos at the lower panel represent the extent of the brain ventricle dilation (VD) used as a criterion for grouping as severe (+++) (scale bar: ∼235 μm), moderate (++) (scale bar: ∼150 μm), and slight/no (+) (scale bar: ∼65 μm). *, p < 0.1; **, p < 0.01.
Atp1a3a RNA in Situ Hybridization and TH Immunostaining
WT TU zebrafish were raised in embryo medium with 1-phenyl-2-thiourea. RNA in situ hybridization was adapted from Schulte-Merker et al. (31), and immunostaining was subsequently performed as described previously (29) with minor modifications. In situ hybridization was applied on noninjected embryos using Atp1a3a antisense riboprobe, and the final labeling was maintained by Fast Red tablets (Sigma). Stained embryos were fixed in 4% paraformaldehyde, blocked in 10% heat inactivated goat serum/phosphate buffered saline (PBS)/0.1% Tween20 (HIGS/PBST), and incubated in anti-TH primary antibody (Millipore) at 1:200 final concentration overnight at 4 °C. Following the washes, embryos were incubated in goat anti-mouse secondary antibody conjugated to Alexa Fluor 488 at 1:1000 final concentration, for 4 h at room temperature. After serial washings, embryos are gradually transferred to 100% glycerol and imaged under Leica DM4000 and Zeiss LSM 710 T-PMT confocal microscope upon being mounted in 0.5% agarose at proper orientation.
Quantification of DA Neurons
Tyrosine hydroxylase (Th) antisense riboprobe was generated as Atp1a3b riboprobe, described above. In situ hybridization protocol to detect Th mRNA expression, applying labeling via Fast Red tablets, was adapted from Schulte-Merker et al. (31) and performed on WT, α3a-SP-MO-mediated Atp1a3a KD, and Atp1a3a mRNA-rescued Atp1a3a KD embryos. Embryos were observed under an inverted microscope, Olympus IX71. Bright field images of Th riboprobe hybridized WT (n = 10) and α3a-SP-MO-mediated Atp1a3a KD embryos (n = 10) were quantified in terms of dopaminergic (DA) neuron content. Due to the extent of the brain ventricle dilation, it was not possible to quantify the number of DA neurons by single-cell count, and thus, total staining of labeled DA neurons in these embryos was quantified using a semiquantitative open source image analysis software, ImageJ (National Institutes of Health, Bethesda, MD). Mean integrated density ± S.D. was plotted.
Whole-cell Patch Clamp Electrophysiology for Atp1a3a
Atp1a3a KD and control embryos at 48 hpf were sacrificed, skinned, and mounted dorsally to enable access to RB cells as described previously (32). Atp1a3a KD embryos were grouped into two groups according to brain ventricle dilation severity: ones with severe dilation (+++) and ones with slight/no (+) dilation. Electrodes were made to a tip resistance of 2.6–3.7 megaohms using a P-97 microelectrode puller (Sutter Instruments, Novato, CA) and filled with intracellular pipette solution (in mm: 135 KCl, 10 EGTA, and 10 HEPES; pH 7.4). RMP recordings were obtained from RB cells of these embryos together with WT and std-MO-injected control embryos using patch electrodes and an Axopatch-200B amplifier (Axon Instruments, Molecular Devices, Sunnyvale, CA). Recordings were performed at room temperature, using bath solution (in mm: 125 NaCl, 3 KCl, 10 CaCl2, and 5 HEPES; pH 7.4) under an inverted microscope, Zeiss Axioskop (Germany). Data are presented as mean ± S.E.
Touch Response
Mechanosensory stimulation was delivered to the embryo trunk with a needle. WT, std-MO-injected, α3a-MO-injected, and α3b-MO-injected embryos with brain ventricle dilation phenotype at 60 hpf were recorded under an Olympus IX71 microscope at 25 frames/s. Experiments were independently repeated at least three times.
Proteomics Analysis by Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)
30–50 of std-MO-, α3a-MO-, and α3b-MO-injected embryos, 60 hpf, were collected in Eppendorf tubes and lysed in lysis buffer (0.02 m Tris, 0.137 m NaCl, 1% Nonidet P-40, 10% glycerol, 1 mm PMSF, 1× Complete protease inhibitor mixture tablet (Roche Applied Science)) with the help of a homogenizer. Cell lysate was analyzed by iTRAQ LC-MS/MS as described previously (33). Briefly, the cell lysate was in-solution-digested using trypsin and labeled with the iTRAQ (reporter ions m/z 114 and 115) followed by fractionation of the peptide mixture using hydrophilic interaction chromatography and LC-MS/MS analysis of the peptide fractions. Raw data files were processed using the Proteome Discoverer software (version 1.3) integrated with the MASCOT database search program (version 2.2.3). Data were searched against the Non-redundant Database (nrDB) restricted to D. rerio.
RESULTS
Specific Expression of Atp1a3a and Atp1a3b Transcripts in the Developing Zebrafish Brain
As an initial step to understand the roles of the α3Na+/K+-ATPases, we assessed expression of Atp1a3a and Atp1a3b mRNA by qRT-PCR. We detected the Atp1a3a transcript at the 1000-cell (3 hpf) and 50% epiboly (5.3 hpf) stages. Thereafter it declined and was barely detectable by 75% epiboly (8 hpf) and at the 6-somite (12 hpf) stage, suggesting that these early detected transcripts were provided maternally. Atp1a3a reappeared at the prim-6 (25 hpf) and prim-22 (35 hpf) stages. The highest expression of Atp1a3a occurred at the pectoral fin stage (60 hpf), which was almost 6-fold higher than the relative expression of the Atp1a3a transcript in adult zebrafish (Fig. 1A). In contrast, the Atp1a3b mRNA was barely detectable from the 1000-cell to the 6-somite stage (Fig. 1A), suggesting lack of maternal Atp1a3b contribution. Similar to Atp1a3a, the Atp1a3b transcript appeared at prim-6 and onwards and also showed a marked increase at the pectoral fin stage (Fig. 1A). Further experiments were performed at 48–60 hpf due to the notable expression of both isoforms at these stages and the fact that the embryonic central nervous system (CNS) is well structured by these stages. At these time points, the MO is still effective.
FIGURE 1.

Expression of Atp1a3a and Atp1a3b mRNA in zebrafish embryos. A, Atp1a3a (black bars) and Atp1a3b (gray bars) mRNA expressions were quantified by qRT-PCR and normalized to Actb2 expression. Data are presented as mean ± S.E. of triplicate measurements. B, Atp1a3a mRNA expression analyzed by whole-mount in situ hybridization in 60 hpf zebrafish embryos; the inset shows sense probe hybridized control embryo. Atp1a3a is expressed in the brain and the spinal cord. The numbered vertical dashed lines, here and in C, show the positions of the transverse sections shown below in sections I–V. The abbreviations used are: C: cerebellum; Cg: cranial ganglia; D: diencephalon; E: epiphysis; H: hypothalamus; Hb: hindbrain; Mo: medulla oblongata; N: notochord; Oc: optic cup; T: tectum; Te: telencephalon; Tg: tegmentum; Sc: spinal cord. Scale bars represent 100 μm in whole-mount images and 50 μm in sections. C, Atp1a3b mRNA expression analyzed by whole-mount in situ hybridization in 60 hpf zebrafish embryos; the inset shows sense probe hybridized control embryo. Atp1a3b is expressed in specific brain regions.
To determine whether of Atp1a3a and Atp1a3b transcripts were present in the CNS, we then performed in situ hybridization. The lack of antibody that detects the zebrafish α3 isoforms did not allow us to directly test protein levels; however, we assume that the detected transcripts give rise to protein products and represent active Na+/K+-ATPase pumps in the plasma membrane of the cell.
We showed that Atp1a3a transcripts localize to several CNS structures, including the epiphysis, tegmentum, tectum, cerebellum, cranial ganglia, hindbrain, and spinal cord (Fig. 1B). A dorsal view of the head shows a dense accumulation of the Atp1a3a transcript throughout the brain. Transverse rostral sections illustrate in more detail Atp1a3a expression in several brain structures (Fig. 1B, section I). More caudal sections show Atp1a3a expression in different CNS regions including the cranial ganglia (Fig. 1B, section II) and spinal cord (Fig. 1B, sections III and IV).
In contrast to the widespread expression of Atp1a3a, the Atp1a3b transcript was detected in a more restricted but overlapping set of brain structures such as epiphysis, tegmentum, cranial ganglia, hindbrain and anterior spinal cord, evident in both lateral and dorsal views (Fig. 1C). Transverse sections show Atp1a3b expression in the hindbrain and cranial ganglia (Fig. 1C, sections I and II), in the rostral (Fig. 1C, section III) but not the caudal (Fig. 1C, section IV) spinal cord.
Knockdown of Atp1a3a Causes Brain Ventricle Dilation
To test for a role of the α3aNa+/K+-ATPase in zebrafish development, we used an antisense MO oligonucleotide-mediated KD approach. The translation-blocking MOs inhibit protein synthesis by binding to the translation initiation site, whereas the splice-blocking MOs bind to intron-exon boundaries and may inhibit proper splicing of the corresponding pre-mRNA, causing nonsense-mediated mRNA decay. In both MO approaches, we assume that the protein function is significantly reduced. The efficiency of Atp1a3a KD induced by α3a-SP-MO was determined by qRT-PCR, demonstrating up to 62% reduction in the Atp1a3a transcript with increasing MO concentration (Fig. 2C).
Marked brain ventricle dilation was observed upon KD of Atp1a3a using either translation-blocking (α3a-MO) or splice-blocking (α3a-SP-MO) MOs (Fig. 2A), and was further visualized by rhodamine-conjugated dextran injection into zebrafish brain ventricles (Fig. 2B). Use of the transgenic line, Tg(gfap:GFP), that expresses green fluorescent protein (GFP) in astrocytes provided an overall view of the CNS structure. A pressure to the brain structures from the ventricle dilation was evident upon Atp1a3a KD (Fig. 2B). Injection of α3a-SP-MO led to severe, moderate, or slight/no brain ventricle dilation in 54, 36, or 11% of embryos, respectively (Fig. 2D), whereas std-MO-injected embryos appeared morphologically normal (Fig. 2A).
To exclude that the observed brain ventricle dilation was caused by nonspecific MO-induced activation of p53-dependent apoptosis (34), control experiments were carried out. Co-injection of p53-MO with any of the MOs did not have an effect on the phenotype (Fig. 2A), suggesting that p53-dependent effects, e.g. apoptosis, did not cause the phenotypes of Atp1a3a KD. To further confirm the specificity of the Atp1a3a KD phenotypes, embryos were co-injected with in vitro-synthesized Atp1a3a mRNA. In the rescued embryos, the extent of the brain ventricle dilation was reduced as compared with Atp1a3a KD embryos, and severe, moderate, and slight/no ventricle dilation was present in 21, 29, and 50%, respectively (Fig. 2D). Taken together, these findings suggest that Atp1a3a KD had specific effects that led to the brain ventricle dilation in α3a- and α3a-SP MO-injected embryos.
Atp1a3b-deficient Embryos Phenocopy the Atp1a3a-deficient Embryos
We used a similar approach to assess the developmental role of the other ATP1A3 ortholog, Atp1a3b. qRT-PCR analysis showed that the α3b-SP-MO led to reductions in transcript levels of 22 and 66% when injected in amounts of 1.5 and 3 ng, respectively (Fig. 3C). Moreover, Atp1a3b KD embryos showed brain ventricle dilation phenotype similar to those produced by Atp1a3a KD embryos (Fig. 3, A and B). Atp1a3b KD caused severe brain ventricle dilation in 59% of the embryos, moderate dilation in 27% of the embryos, and no extraordinary ventricle dilation in 14% of the embryos, and std-MO-injected embryos appeared morphologically normal (Fig. 3, A and D). Co-injections of p53-MO ruled out the activation of p53-dependent apoptosis in Atp1a3b KD embryos (Fig. 3A). Upon co-injection with in vitro transcribed Atp1a3b mRNA, severe brain ventricle dilation was observed in 16% of the embryos, moderate dilation was observed in 24% of the embryos, and no ventricle dilation was observed in 61% of the embryos (Fig. 3D). Importantly, the brain ventricle dilation of Atp1a3a KD embryos was not rescued by co-injection of Atp1a3b mRNA, and conversely, Atp1a3b KD embryos were not rescued by co-injection of Atp1a3a mRNA (Fig. 3E).
FIGURE 3.

Knockdown of Atp1a3b phenocopies Atp1a3a knockdown. A, brain ventricle dilation was observed in embryos upon Atp1a3b KD mediated by α3b-MO- or α3b-SP-MO-injected embryo as compared with the std-MO-injected control embryo. This phenotype was rescued by co-injection of Atp1a3b mRNA. p53-MO co-injections with any of the MOs did not rescue the brain ventricle dilation phenotype. B, brain ventricles of Tg(gfap:GFP) line, non-MO-injected and α3b-MO-injected, were injected with rhodamine-conjugated dextran. Brain ventricles and the astrocytes are highlighted by red and green fluorescence, respectively. C, α3b-SP-MO-mediated KD efficiency was tested by qRT-PCR in terms of changes in the relative expression level of Atp1a3b. Data are presented as mean ± S.E. of triplicate measurements. Concentrations of MOs are indicated. D, mean percentages ± S.D. of the α3b-SP-MO-injected embryos suffering from brain ventricle dilation (VD) of different severity, with (white columns, n = 148) and without (black columns, n = 88) Atp1a3a mRNA co-injection, are plotted. The degree of brain ventricle dilation is as follows: +, slight/no; ++, moderate; +++, severe. **, p < 0.01; ***, p < 0.001. E, Atp1a3a mRNA did not rescue α3b-SP-MO-injected embryos; similarly, Atp1a3b mRNA did not rescue α3a-SP-MO-injected embryos from brain ventricle dilation.
The observation of similar phenotypes upon the use of two distinct MOs (Figs. 2A and 3A), and the fact that the nonsense-mediated mRNA decay of the targeted transcript by SP-MOs could be observed in a concentration-correlated manner (Figs. 2C and 3C), together verified the specificity of the MOs used in this study. In addition, our observations of consistent brain ventricle dilation phenotypes, even after p53-MO co-injection with each MO (Figs. 2A and 3A), and importantly, the significant rescue of the phenotypes by injection of the mRNA of the knocked down gene (Figs. 2D and 3D), further confirmed the specificity. It is important to note that full rescue of the brain ventricle dilation phenotype was not accomplished, most likely due to the limited stability of the injected exogenous mRNA in the cell.
Atp1a3a KD Leads to Depolarization of the Resting Membrane Potential of Rohon-Beard Neurons
RB cells are mechanosensory neurons localized in the dorsal spinal cord (35, 36) and are involved in response to touch, a behavior that involves both neuronal and muscular components. RB neurons are within the domain of Atp1a3a expression (Fig. 1B) and accessible due to their superficial localization. Combining the advantages of Atp1a3a being expressed in RB neurons and the accessibility of these cells, we measured electrophysiological changes at the cellular level in RB neurons of Atp1a3a KD embryos. The RMP of RB neurons was recorded using whole-cell patch clamp. The RMP of RB neurons in Atp1a3a KD embryos displaying severe brain ventricle dilation was significantly (p < 0.01) more depolarized (mean RMP: −46.6 ± 5.1 mV) as compared with the RMP from slight/no brain ventricle dilation-displaying Atp1a3a KD embryos (mean RMP: −70.4 ± 2.9 mV), which were comparable (p > 0.05) with the control groups, noninjected (mean RMP: −72.1 ± 1.7 mV), and std-MO-injected (mean RMP: −65.2 ± 3.1 mV) embryos (Fig. 4A). A schematic representation illustrates the consequence of the depolarized RMP in RB neurons in an Atp1a3a KD embryo (Fig. 4B).
FIGURE 4.

RB neurons are more depolarized in Atp1a3a KD zebrafish displaying severe brain ventricle dilation. A, RMP values of RB neurons from WT (n = 5), std-MO-injected (n = 6), and α3a-MO-injected embryos (n = 10) are plotted. The α3a-MO-injected embryos are divided into embryos displaying severe (+++) (n = 5) or slight/no (+) (n = 5) brain ventricle dilation (VD). The number of cells (n) recorded per group stems from at least three different animals. RMP data are presented as mean ± S.D. **, p < 0.01 between RMPs of α3a-MO-injected embryos with severe ventricle dilation and control groups, WT, and std-MO-injected embryos. B, schematic representation summarizing the depolarization of the RMP in RB neurons in an Atp1a3a KD embryo. The scheme covers the time frame of 48–60 hpf. The dashed cross marks a malfunctioning α3aNa+/K+-ATPase.
Atp1a3a and Atp1a3b KD Embryos Differ in Their Mechanosensory Responses
The onset of Atp1a3a and Atp1a3b expression coincides with the ability of the embryo to respond to tactile stimulation at around 27 hpf (prim-6 stage), where a marked increase in the expression of both isoforms was detected (Fig. 1A). Considering that RB cells mediate touch sensitivity for the embryo, and that KD of Atp1a3a depolarized the RB RMP, we tested for effects on tactile sensitivity. The RB-mediated touch response is assayed by applying tactile stimuli to the trunk of a motile embryo (37). Although both Atp1a3a KD and Atp1a3b KD embryos display abnormal spontaneous motility (data not shown), they are motile and suitable for assessment of tactile sensitivity.
At 60 hpf, the control embryos, noninjected (supplemental Movie 1) and std-MO-injected (supplemental Movie 2), responded by burst swimming as expected. Atp1a3a KD embryos (supplemental Movie 3) typically responded quickly to tactile stimulation but with a brief escape and circling movements that in one instance culminated in convulsion (supplemental Movie 3) (top). Atp1a3b KD also had a range of effects, some of which were similar to and others which were different from those produced by Atp1a3a KD (supplemental Movie 4). The most consistent difference was that Atp1a3b KD embryos responded with a delay in contrast to the quick response of Atp1a3a KD embryos. Similar to Atp1a3a KD embryos, Atp1a3b KD embryos also displayed brief distance recoils and, in one instance, convulsion (supplemental Movie 4) (right). Successive frame shots from touch response movies (supplemental Movies 1, 3, and 4) provide an overview of the embryonic movements and allow comparisons between the different experimental groups (Fig. 5). Taken together, the data indicate that Atp1a3a or Atp1a3b KD impair but do not prevent embryonic motility. In addition, the embryos retain the ability to respond to touch but, given the motility defects, it was difficult to assess whether there was any reduction in touch sensitivity, although this is indicated by RMP alterations in RB neurons in Atp1a3a KD and by delayed response in Atp1a3b KD embryos.
FIGURE 5.

Atp1a3a and Atp1a3b KD embryos respond to touch but have abnormal motility. Successive frame shots from touch response assay display three representative WT embryos with burst swimming response (top panel, supplemental Movie 1), a representative α3a-MO-injected embryo that kept swirling around itself (middle panels, supplemental Movie 3), and a α3b-MO-injected embryo that responded as a short distance recoil (bottom panel, supplemental Movie 4). Touch-stimulated embryos are marked with a color-coded asterisk, and the frame shot times are merged on images in seconds.
Atp1a3a KD Leads to Disorganization of the Brain but No Loss of Dopaminergic Neurons
A subset of dystonia subtypes responds to l-DOPA treatments that target DA neurons in the substantia nigra (38). However, RDP patients do not respond to l-DOPA treatment. To assess whether zebrafish α3 isoforms differ in their association to DA neurons as compared with their mammalian counterparts, we questioned the presence of zebrafish α3 isoforms in DA neurons. Atp1a3a co-localized with the DA neuron marker, TH (Fig. 6A), a marker previously used to identify DA neurons in zebrafish (39). Confocal images highlight the classical arrangement of TH-positive cells in the diencephalon, a region that shows widespread expression of Atp1a3a mRNA (Fig. 6A). The expression pattern of Atp1a3b (Fig. 1C) does not suggest co-expression of this isoform with DA neurons.
FIGURE 6.

Atp1a3a mRNA is present in DA neurons, but no loss of DA neuron was observed in Atp1a3a KD embryos. A, TH-positive DA neurons (green fluorescence) and Atp1a3a mRNA expression (red fluorescence) in the zebrafish brain are imaged along the dorsoventral axis (anterior to the left; posterior to the right). Scale bars represent 50 μm. Areas marked by squares are shown in the lower panel at higher magnification (scale bars represent 5 μm). B, Th mRNA expression analyzed by in situ hybridization in WT, α3a-SP-MO-injected, and Atp1a3a mRNA-rescued embryos. C, total staining of Th mRNA in WT (n = 10) and α3a-SP-MO-injected embryos (n = 10) was quantified using ImageJ, and mean values of integrated densities (IntDen) are plotted with standard deviations.
We then tested whether Atp1a3a KD affected DA neurons by comparing the distribution of Th transcripts in WT, α3a-SP-MO-mediated Atp1a3a KD, and mRNA-rescued Atp1a3a KD embryos (Fig. 6B). Although there were some slight differences in the pattern and intensity of the Th mRNA signal (Fig. 6B), quantitative analysis showed no significant difference (Fig. 6C). The variations in Th expression profile observed in Atp1a3a KD zebrafish correlate with the extent of brain ventricle dilation and the consequent spatial reorganization in the brain, but not with loss of DA neurons. These results support the view that Atp1a3a KD does not lead to DA neuron degeneration.
Several Proteins Are Up/Down-regulated When Either Atp1a3a or Atp1a3b Is Knocked Down
To investigate the cellular protein networks that potentially involve α3Na+/K+-ATPases, we carried out proteomics analyses. Candidate proteins affected in Atp1a3a or Atp1a3b KD embryos were identified using iTRAQ and listed together with their known tissue associations (supplemental Table S1). Interestingly, several candidates are of relevance to Na+/K+-ATPase and Atp1a3a or Atp1a3b KD phenotypes (Table 1), but only selected candidates are discussed below. It is interesting to note that cytoskeletal and muscle-associated proteins appear to be regulated by both α3 isoforms. In contrast, the level of ion-binding proteins and proteins involved in phosphate metabolism appear to depend on the α3b isoform.
We performed qRT-PCR to verify selected proteomic candidates as antibodies toward all of these zebrafish proteins were not available. We found that these transcripts were also down-regulated in Atp1a3a or Atp1a3b KD embryos, supporting the proteomics data at the transcript level (Fig. 7, A and B).
FIGURE 7.
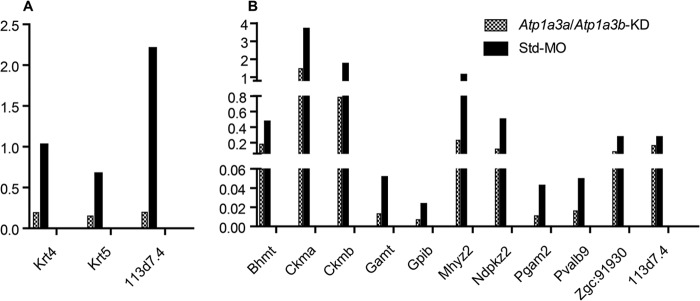
Relative mRNA expressions of some of the regulated proteins detected in proteomics assay. A and B, mRNA expressions of selected proteins regulated by α3a-MO-mediated Atp1a3a KD (A) or α3b-MO-mediated Atp1a3b KD (B) were quantified by qRT-PCR and normalized to Actb2 expression in embryos at 60 hpf. Data are presented as mean ± S.E. of triplicate measurements.
DISCUSSION
In this study, we analyzed zebrafish α3Na+/K+-ATPases to gain insight on specific functions of this particular ion pump and how we might use this model to elucidate the functions of the Na+/K+-ATPase in brain development. The Na+/K+-ATPase has a well known role as a modulator of membrane potential in neurons and is essential for generating an action potential. Consistent with the mammalian ATP1α3 expression, both ATP1A3 zebrafish orthologs are expressed primarily in the brain. Interestingly, the expression of the Atp1a3a mRNA is widely distributed in the brain, in contrast to a more restricted expression of the Atp1a3b mRNA, in line with the expression data available at The Zebrafish Model Organism Database. The Atp1a3a transcript was also abundant in the spinal cord and, to a lesser extent, in the heart. The latter observation is consistent with previous studies demonstrating ATP1α3 expression in the neonatal, but not adult, rat heart (40, 41).
Despite the distinct expression profiles of Atp1a3a and Atp1a3b transcripts, both Atp1a3a and Atp1a3b deficiencies cause brain ventricle dilation, rather than cellular swelling, indicating an altered fluid and electrolyte balance. Possibly, the subset of ventral CNS regions that express both Atp1a3a and Atp1a3b involves a pathological pathway that results in this phenotype A functional Na+/K+-ATPase, in a complex with aquaporins and glutamate transporters, and is important for maintaining water and ion balance in the brain (42). Interestingly, Na+/K+-ATPase, in particular Atp1a1, was previously demonstrated to be required for the zebrafish brain ventricle development (19, 43). However, our data suggest that Atp1a3a and Atp1a3b KD do not inhibit ventricle inflation, but the consequent phenotype is connected to dilated ventricles as a result of cerebrospinal fluid (CSF) accumulation. An increase or a decrease in the CSF volume can be pathological throughout life (44), and the Na+/K+-ATPase may serve to sense the ventricular volume in a homeostatic role. Consequently, even a slight disruption of Na+/K+-ATPase subunits could lead to significant changes in CSF volume. Overall, these data indicate a role of α3Na+/K+-ATPase in brain ventricle volume maintenance through its ion pump function. CSF composition is vital to brain health. The protein function of the α3Na+/K+-ATPase in neurons is likely connected to the extrusion of Na+ to the extracellular space coupled to the uptake of K+ into the intracellular cytoplasm. This exchange of Na+/K+ ions generates an electrochemical gradient, which, in turn, facilitates ion transport, for example of Cl−, HCO3−, and H2O, required to regulate CSF volume (45). Interestingly, a recent study showed that increases in aquaporin-1 (AQP1) and cation chloride transporters (Na+-K+-2Cl− cotransporter 1 (NKCC1)) expression under hyposmotic stress may be one of the molecular mechanisms underlying the pathophysiology of acute hyponatremia by increasing water transport across the blood-CSF barrier (46).
It is very interesting that the distinctively expressed α3 isoforms both result in brain ventricle dilation when knocked down, and moreover, that they are not able to cross rescue. This indicates that at some level, the ion pump function must be the major mechanism behind our observations despite the different proteomic changes when knocked down. Furthermore, we do not know at this stage to what extent other Na+/K+-ATPase isoforms are active in the cells expressing these α3 isoforms.
Modeling human diseases in other organisms requires complex analysis. Of particular relevance for this study is the fact that the water environment hosting zebrafish differs from other environments and thus requires other mechanisms for ion homeostasis to maintain osmolarity. However, we believe that it is not so extraordinary to observe a brain ventricle dilation phenotype in the Atp1a3 KD zebrafish, although this is not a manifestation observed in RDP patients harboring mutations in the ATP1A3 gene (47). Thus, we cannot rule out that the brain ventricle dilation is specific to zebrafish despite the conserved functions of the Na+/K+-ATPase. The CSF fills the ventricle of the brain, and the composition of the CSF influences neuronal activity and serves as a drainage pathway for the brain. The latter function might be significantly different in water-living animals. In summary, it appears that the α3Na+/K+-ATPase is a novel regulator of brain ventricle volume, at least in zebrafish.
Minor changes in ion composition in the extracellular and intracellular compartments of the brain can significantly affect neuronal function, which relies on precise ion gradients across their plasma membranes to trigger changes in membrane potentials underlying action potential generation and propagation. Therefore, ion and water transport in the brain are tightly regulated (48). The most studied role of the α3 isoform is its neuronal function, and failure of the Na+/K+-ATPase to maintain Na+ and K+ gradients leads to a decrease in both the RMP and the action potential and to altered neuronal excitability in several different neurons, such as rat hippocampus dentate interneurons, and pyramidal and Purkinje neurons (49). Consistent with this, we found that RB neurons had depolarized RMPs when Atp1a3a was knocked down. This result is important and additionally serves to strongly support the specificity of the Atp1a3 KD phenotypes. The change in the electrophysiological state of the RB neurons is thus predicted to alter the neuronal excitability, although future in vivo measurements of excitability are required to address this. Our data strongly support a neuronal function for α3 isoforms in zebrafish. We believe that it is important to keep in mind that the RMP recording is performed in RB neurons and the ventricle dilation is observed in the brain. We do not yet know the contributions from other Na+/K+-ATPase isoforms in these cell populations, and this could certainly explain why we could not demonstrate an altered RMP in the slight (if any) brain ventricle dilation-displaying embryos.
The production of any touch response in zebrafish can be divided into several steps from sensory perception to muscle activation. The mechanosensory neurons, in this case RB neurons, sense touch stimuli. Once triggered by sensory input, interneuronal networks located in the hindbrain and spinal cord produce the appropriate motor rhythm (50). Depolarization in RB neurons indicated abnormality in the sensory component of this type of behavior, although further experiments are needed to distinguish all the contributors of this phenotype. It is intriguing because the motor deficits observed in α3+/−KO mice were shown to have neuronal origin (16). Furthermore, inefficient central sensory-motor processing has been suggested to be a possible causative mechanism for dystonia, and numerous clinical phenomena suggest the primary involvement of the somatosensory system in this disorder (51, 52).
Intriguingly, a recent study reported Na+/K+-ATPase as an important player in locomotor behavior of frog tadpoles (53) and of Drosophila larvae (54). Also, in spinal network of neonatal rats, blocking of Na+/K+ pump activity disrupts rhythmic bursting of lumbar motor neurons (55, 56). Moreover, the pump function of Na+/K+-ATPase was recognized as a mechanism to gate sensory information entering the spinal cord, where it alters neuronal excitability (57). By a similar mechanism, the changes in neuronal excitability might account for the impaired spontaneous motility of the Atp1a3a and Atp1a3b KD embryos.
Treatment of RDP patients with l-DOPA has no effect, and the DA reuptake sites appear normal in such patients (58). Although studies in mouse brain did not detect the α3 isoform in DA neurons of substantia nigra (8), it is interesting that another subunit, the β subunit, ATP1β1, is down-regulated in DA neurons of patients with Parkinson disease (59). We detected Atp1a3a expression in zebrafish TH-positive cells. In line with this, a recent study identified Atp1a3a as a target of a cardiac glycoside (a Na+/K+-ATPase inhibitor; Neriifolin), which impairs DA neuronal survival (60). However, our results show that Atp1a3a KD does not result in loss of DA neurons in zebrafish, although we did observe reorganization in the DA neuron distribution profile. The latter is most likely caused by spatial restrictions due to the brain ventricle dilation, also noted in the Tg(gfap:GFP) Atp1a3a and Atp1a3b KD embryos.
We also identified novel proteomic changes associated with the α3 isoform deficiencies. This is in fact the first time a proteomic approach has been used to identify proteins up- or down-regulated in ATP1α3-deficient cells. Interestingly, a recent study explored proteomic changes in ATP1α2-deficient zebrafish (18), and some of the regulated proteins, e.g. parvalbumin and muscle creatine kinase, published in that study were also detected as regulated in Atp1a3b-deficient embryos, indicating that the link between these proteins and the Na+/K+-ATPase is most likely dependent on the Na+/K+-ATPase pump function, rather than an isoform-specific association. Hence, although preliminary, it provides an important initiative to be further assessed. To functionally address these candidate proteins in relation to the obtained Atp1a3a and Atp1a3b KD zebrafish and other animal models will be a future direction of this project.
Of particular interest is the down-regulation of parvalbumin detected in our proteomics assay of the Atp1a3a KD embryos. Parvalbumin is expressed preferentially in a subpopulation of GABAergic neurons in mice, overlapping with the α3Na+/K+-ATPase expression (8, 61, 62). Parvalbumin modulates short term synaptic plasticity (63) and thus compliments the signaling role of the α3Na+/K+-ATPase in synaptic plasticity and in dendritic growth in cortical neurons (64). Neuronal associations of α3Na+/K+-ATPase are further supported by the down-regulation of guanidinoacetate methyltransferase (GAMT). The guanidinoacetate is the principal metabolite accumulating in guanidinoacetate methyltransferase deficiency, which significantly inhibits Na+/K+-ATPase activity (65). It was proposed that such inhibition may be one of the mechanisms involved in the neuronal dysfunction observed in patients suffering from guanidinoacetate methyltransferase deficiency, which shows symptoms comparable with RDP: e.g. muscle weakness, epilepsy, and seizures (65, 66).
Although it is clear that zebrafish embryos will not develop a full range of complex, human-like disorders, they can be used to study certain biological markers (endophenotypes) of these disorders. Indeed, several features of zebrafish α3 isoforms are comparable with the mammalian counterparts. This study comprehensively examined the spatial distribution of the zebrafish ATP1A3 orthologs and is the first study to show that the two distinctively expressed α3 isoforms caused enlarged brain ventricle when gene functions were diminished. This was accompanied with abrupt embryonic motility, most likely linked to depolarized RMP, as shown for the spinal RB neurons, and this, combined with our proteomic data, highly promotes zebrafish as a relevant model to further assess α3Na+/K+-ATPase in brain development, neuronal excitability, and thus neuronal functions.
Acknowledgments
We thank C. Knoeckel and R. Moreno, Department of Physiology and Biophysics, University of Colorado, Denver, for the assistance in whole-cell patch clamp electrophysiology recordings and J. R. Nyengaard, Aarhus University, Department of Biomedicine for the help on sectioning.
This work was supported by grants from Aarhus University Forskningsfond (AUFF), and Danish National Research Foundation (DNRF) (Centre for Membrane Pumps in Cells and Disease (PUMPKIN)) (DNRF85).

This article contains supplemental Tables S1 and S2 and Movies 1–4.
- RDP
- rapid-onset dystonia-parkinsonism
- KD
- knockdown
- RB
- Rohon-Beard
- RMP
- resting membrane potential
- MO
- morpholino oligonucleotide
- std-MO
- standard control MO
- SP-MO
- splicing MO
- Tg
- transgenic
- qRT-PCR
- quantitative RT-PCR
- iTRAQ
- isobaric tags for relative and absolute quantitation
- DA
- dopaminergic
- TH
- tyrosine hydroxylase
- l-DOPA
- l-3,4-dihydroxyphenylalanine
- CSF
- cerebrospinal fluid
- hpf
- hours postfertilization
- Prim
- primordium of the lateral line.
REFERENCES
- 1. Blanco G. (2005) Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 25, 292–303 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan J. H. (2002) Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 [DOI] [PubMed] [Google Scholar]
- 3. Lingrel J., Moseley A., Dostanic I., Cougnon M., He S., James P., Woo A., O'Connor K., Neumann J. (2003) Functional roles of the α isoforms of the Na,K-ATPase. Ann. N.Y. Acad. Sci. 986, 354–359 [DOI] [PubMed] [Google Scholar]
- 4. Lingrel J. B., Kuntzweiler T. (1994) Na+,K+-ATPase. J. Biol. Chem. 269, 19659–19662 [PubMed] [Google Scholar]
- 5. McGrail K. M., Phillips J. M., Sweadner K. J. (1991) Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 11, 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shull G. E., Greeb J., Lingrel J. B. (1986) Molecular cloning of three distinct forms of the Na+,K+-ATPase α-subunit from rat brain. Biochemistry 25, 8125–8132 [DOI] [PubMed] [Google Scholar]
- 7. Schneider J. W., Mercer R. W., Caplan M., Emanuel J. R., Sweadner K. J., Benz E. J., Jr., Levenson R. (1985) Molecular cloning of rat brain Na,K-ATPase α-subunit cDNA. Proc. Natl. Acad. Sci. U.S.A. 82, 6357–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bøttger P., Tracz Z., Heuck A., Nissen P., Romero-Ramos M., Lykke-Hartmann K. (2011) Distribution of Na/K-ATPase α3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J Comp. Neurol. 519, 376–404 [DOI] [PubMed] [Google Scholar]
- 9. Brashear A., DeLeon D., Bressman S. B., Thyagarajan D., Farlow M. R., Dobyns W. B. (1997) Rapid-onset dystonia-parkinsonism in a second family. Neurology 48, 1066–1069 [DOI] [PubMed] [Google Scholar]
- 10. de Carvalho Aguiar P., Sweadner K. J., Penniston J. T., Zaremba J., Liu L., Caton M., Linazasoro G., Borg M., Tijssen M. A., Bressman S. B., Dobyns W. B., Brashear A., Ozelius L. J. (2004) Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 [DOI] [PubMed] [Google Scholar]
- 11. Brashear A., Dobyns W. B., de Carvalho Aguiar P., Borg M., Frijns C. J., Gollamudi S., Green A., Guimaraes J., Haake B. C., Klein C., Linazasoro G., Münchau A., Raymond D., Riley D., Saunders-Pullman R., Tijssen M. A., Webb D., Zaremba J., Bressman S. B., Ozelius L. J. (2007) The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 130, 828–835 [DOI] [PubMed] [Google Scholar]
- 12. Desfrere L., Karlsson M., Hiyoshi H., Malmersjö S., Nanou E., Estrada M., Miyakawa A., Lagercrantz H., El Manira A., Lal M., Uhlén P. (2009) Na,K-ATPase signal transduction triggers CREB activation and dendritic growth. Proc. Natl. Acad. Sci. U.S.A. 106, 2212–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobretsov M., Hastings S. L., Sims T. J., Stimers J. R., Romanovsky D. (2003) Stretch receptor-associated expression of α3 isoform of the Na+, K+-ATPase in rat peripheral nervous system. Neuroscience 116, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 14. Moseley A. E., Williams M. T., Schaefer T. L., Bohanan C. S., Neumann J. C., Behbehani M. M., Vorhees C. V., Lingrel J. B. (2007) Deficiency in Na,K-ATPase α isoform genes alters spatial learning, motor activity, and anxiety in mice. J. Neurosci. 27, 616–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clapcote S. J., Duffy S., Xie G., Kirshenbaum G., Bechard A. R., Rodacker Schack V., Petersen J., Sinai L., Saab B. J., Lerch J. P., Minassian B. A., Ackerley C. A., Sled J. G., Cortez M. A., Henderson J. T., Vilsen B., Roder J. C. (2009) Mutation I810N in the α3 isoform of Na+,K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc. Natl. Acad. Sci. U.S.A. 106, 14085–14090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeAndrade M. P., Yokoi F., van Groen T., Lingrel J. B., Li Y. (2011) Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism. Behav. Brain Res. 216, 659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kirshenbaum G. S., Clapcote S. J., Duffy S., Burgess C. R., Petersen J., Jarowek K. J., Yücel Y. H., Cortez M. A., Snead O. C., 3rd, Vilsen B., Peever J. H., Ralph M. R., Roder J. C. (2011) Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase α3 sodium pump. Proc. Natl. Acad. Sci. U.S.A. 108, 18144–18149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doganli C., Kjaer-Sorensen K., Knoeckel C., Beck H. C., Nyengaard J. R., Honoré B., Nissen P., Ribera A., Oxvig C., Lykke-Hartmann K. (2012) The α2Na+/K+-ATPase is critical for skeletal and heart muscle function in zebrafish. J. Cell Sci., in press [DOI] [PubMed] [Google Scholar]
- 19. Chang J. T., Lowery L. A., Sive H. (2012) Multiple roles for the Na,K-ATPase subunits, Atp1a1 and Fxyd1, during brain ventricle development. Dev. Biol. 368, 312–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barut B. A., Zon L. I. (2000) Realizing the potential of zebrafish as a model for human disease. Physiol Genomics. 2, 49–51 [DOI] [PubMed] [Google Scholar]
- 21. Lieschke G. J., Currie P. D. (2007) Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 8, 353–367 [DOI] [PubMed] [Google Scholar]
- 22. Friedrich R. W., Jacobson G. A., Zhu P. (2010) Circuit neuroscience in zebrafish. Curr. Biol. 20, R371–R381 [DOI] [PubMed] [Google Scholar]
- 23. Rajarao S. J., Canfield V. A., Mohideen M. A., Yan Y. L., Postlethwait J. H., Cheng K. C., Levenson R. (2001) The repertoire of Na,K-ATPase α and β subunit genes expressed in the zebrafish, Danio rerio. Genome Res. 11, 1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Westerfield D. (1995) The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio), pp. 787–801, University of Oregon Press, Eugene, OR [Google Scholar]
- 25. Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- 26. Doğanlı C., Kjærgaard T., Olsen A., Oxvig C., Füchtbauer E. M., Lykke-Hartmann K. (2010) Early developmental expression of Mus musculus zinc finger RNA-binding protein compared to orthologs in Caenorhabditis elegans and Danio rerio and subcellular localization of Mus musculus and Caenorhabditis elegans zinc finger RNA-binding protein in 2-cell Mus musculus embryos. DNA Cell Biol. 29, 713–727 [DOI] [PubMed] [Google Scholar]
- 27. Marshall O. J. (2004) PerlPrimer: cross-platform, graphical primer design for standard, bisulphite, and real-time PCR. Bioinformatics 20, 2471–2472 [DOI] [PubMed] [Google Scholar]
- 28. Chomczynski P., Mackey K. (1995) Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. BioTechniques 19, 942–945 [PubMed] [Google Scholar]
- 29. Thisse C., Thisse B. (2008) High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- 30. Gutzman J. H., Sive H. (2009) Zebrafish brain ventricle injection. J. Vis. Exp. 26, pii: 1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schulte-Merker S., Ho R. K., Herrmann B. G., Nüsslein-Volhard C. (1992) The protein product of the zebrafish homologue of the mouse T gene is expressed in nuclei of the germ ring and the notochord of the early embryo. Development 116, 1021–1032 [DOI] [PubMed] [Google Scholar]
- 32. Moreno R. L., Ribera A. B. (2009) Zebrafish motor neuron subtypes differ electrically prior to axonal outgrowth. J. Neurophysiol. 102, 2477–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beck H. C., Petersen J., Felthaus O., Schmalz G., Morsczeck C. (2011) Comparison of neurosphere-like cell clusters derived from dental follicle precursor cells and retinal Muller cells. Neurochem. Res. 36, 2002–2007 [DOI] [PubMed] [Google Scholar]
- 34. Robu M. E., Larson J. D., Nasevicius A., Beiraghi S., Brenner C., Farber S. A., Ekker S. C. (2007) p53 activation by knockdown technologies. PLoS Genet. 3, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clarke J. D., Hayes B. P., Hunt S. P., Roberts A. (1984) Sensory physiology, anatomy and immunohistochemistry of Rohon-Beard neurones in embryos of Xenopus laevis. J. Physiol. 348, 511–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ribera A. B., Nüsslein-Volhard C. (1998) Zebrafish touch-insensitive mutants reveal an essential role for the developmental regulation of sodium current. J. Neurosci. 18, 9181–9191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pineda R. H., Heiser R. A., Ribera A. B. (2005) Developmental, molecular, and genetic dissection of INa in vivo in embryonic zebrafish sensory neurons. J. Neurophysiol. 93, 3582–3593 [DOI] [PubMed] [Google Scholar]
- 38. Nemeth A. H. (1993) Dystonia Overview. in GeneReviews (Pagon R. A., Bird T. D., Dolan C. R., Stephens K., Adam M. P., eds), University of Washington, Seattle, WA: [PubMed] [Google Scholar]
- 39. Flinn L., Mortiboys H., Volkmann K., Köster R. W., Ingham P. W., Bandmann O. (2009) Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132, 1613–1623 [DOI] [PubMed] [Google Scholar]
- 40. Shyjan A. W., Levenson R. (1989) Antisera specific for the α1, α2, α3, and β subunits of the Na,K-ATPase: differential expression of α and β subunits in rat tissue membranes. Biochemistry 28, 4531–4535 [DOI] [PubMed] [Google Scholar]
- 41. Sweadner K. J., Farshi S. K. (1987) Rat cardiac ventricle has two Na+,K+-ATPases with different affinities for ouabain: developmental changes in immunologically different catalytic subunits. Proc. Natl. Acad. Sci. U.S.A. 84, 8404–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Illarionova N. B., Gunnarson E., Li Y., Brismar H., Bondar A., Zelenin S., Aperia A. (2010) Functional and molecular interactions between aquaporins and Na,K-ATPase. Neuroscience 168, 915–925 [DOI] [PubMed] [Google Scholar]
- 43. Lowery L. A., Sive H. (2005) Initial formation of zebrafish brain ventricles occurs independently of circulation and requires the nagie oko and snakehead/atp1a1a.1 gene products. Development 132, 2057–2067 [DOI] [PubMed] [Google Scholar]
- 44. Lowery L. A., Sive H. (2009) Totally tubular: the mystery behind function and origin of the brain ventricular system. BioEssays 31, 446–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johanson C. E., Duncan J. A., 3rd, Klinge P. M., Brinker T., Stopa E. G., Silverberg G. D. (2008) Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res. 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim J., Jung Y. (2012) Increased aquaporin-1 and Na+-K+-2Cl− cotransporter 1 expression in choroid plexus leads to blood-cerebrospinal fluid barrier disruption and necrosis of hippocampal CA1 cells in acute rat models of hyponatremia. J. Neurosci. Res. 90, 1437–1444 [DOI] [PubMed] [Google Scholar]
- 47. Bøttger P., Doğanlı C., Lykke-Hartmann K. (2012) Migraine- and dystonia-related disease-mutations of Na+/K+-ATPases: Relevance of behavioral studies in mice to disease symptoms and neurological manifestations in humans. Neurosci. Biobehav. Rev. 36, 855–871 [DOI] [PubMed] [Google Scholar]
- 48. Kahle K. T., Simard J. M., Staley K. J., Nahed B. V., Jones P. S., Sun D. (2009) Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology 24, 257–265 [DOI] [PubMed] [Google Scholar]
- 49. Dobretsov M., Stimers J. R. (2005) Neuronal function and α3 isoform of the Na/K-ATPase. Front. Biosci. 10, 2373–2396 [DOI] [PubMed] [Google Scholar]
- 50. Fetcho J. R. (1992) The spinal motor system in early vertebrates and some of its evolutionary changes. Brain Behav. Evol. 40, 82–97 [DOI] [PubMed] [Google Scholar]
- 51. Hallett M. (1995) Is dystonia a sensory disorder? Ann. Neurol. 38, 139–140 [DOI] [PubMed] [Google Scholar]
- 52. Fiorio M., Gambarin M., Valente E. M., Liberini P., Loi M., Cossu G., Moretto G., Bhatia K. P., Defazio G., Aglioti S. M., Fiaschi A., Tinazzi M. (2007) Defective temporal processing of sensory stimuli in DYT1 mutation carriers: a new endophenotype of dystonia? Brain 130, 134–142 [DOI] [PubMed] [Google Scholar]
- 53. Zhang H. Y., Sillar K. T. (2012) Short-term memory of motor network performance via activity-dependent potentiation of Na+/K+ pump function. Curr. Biol. 22, 526–531 [DOI] [PubMed] [Google Scholar]
- 54. Pulver S. R., Griffith L. C. (2010) Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nat. Neurosci. 13, 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rozzo A., Ballerini L., Abbate G., Nistri A. (2002) Experimental and modeling studies of novel bursts induced by blocking Na+ pump and synaptic inhibition in the rat spinal cord. J. Neurophysiol. 88, 676–691 [DOI] [PubMed] [Google Scholar]
- 56. Ballerini L., Bracci E., Nistri A. (1997) Pharmacological block of the electrogenic sodium pump disrupts rhythmic bursting induced by strychnine and bicuculline in the neonatal rat spinal cord. J. Neurophysiol. 77, 17–23 [DOI] [PubMed] [Google Scholar]
- 57. Parker D., Hill R., Grillner S. (1996) Electrogenic pump and a Ca2+-dependent K+ conductance contribute to a posttetanic hyperpolarization in lamprey sensory neurons. J. Neurophysiol. 76, 540–553 [DOI] [PubMed] [Google Scholar]
- 58. Brashear A., Mulholland G. K., Zheng Q. H., Farlow M. R., Siemers E. R., Hutchins G. D. (1999) PET imaging of the pre-synaptic dopamine uptake sites in rapid-onset dystonia-parkinsonism (RDP). Movement Disord. 14, 132–137 [DOI] [PubMed] [Google Scholar]
- 59. Simunovic F., Yi M., Wang Y., Macey L., Brown L. T., Krichevsky A. M., Andersen S. L., Stephens R. M., Benes F. M., Sonntag K. C. (2009) Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson's disease pathology. Brain 132, 1795–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sun Y., Dong Z., Khodabakhsh H., Chatterjee S., Guo S. (2012) Zebrafish chemical screening reveals the impairment of dopaminergic neuronal survival by cardiac glycosides. PloS One 7, e35645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baimbridge K. G., Celio M. R., Rogers J. H. (1992) Calcium-binding proteins in the nervous system. Trends Neurosci. 15, 303–308 [DOI] [PubMed] [Google Scholar]
- 62. Seto-Ohshima A. (1994) Review: calcium-binding proteins in the central nervous system. Acta Histochem. Cytochem. 27, 93–106 [Google Scholar]
- 63. Caillard O., Moreno H., Schwaller B., Llano I., Celio M. R., Marty A. (2000) Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 97, 13372–13377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Benarroch E. E. (2011) Na+, K+-ATPase Functions in the nervous system and involvement in neurologic disease. Neurology 76, 287–293 [DOI] [PubMed] [Google Scholar]
- 65. Zugno A. I., Stefanello F. M., Streck E. L., Calcagnotto T., Wannmacher C. M. D., Wajner M., Wyse A. T. S. (2003) Inhibition of Na+, K+-ATPase activity in rat striatum by guanidinoacetate. Int. J. Dev. Neurosci. 21, 183–189 [DOI] [PubMed] [Google Scholar]
- 66. Stöckler S., Isbrandt D., Hanefeld F., Schmidt B., von Figura K. (1996) Guanidinoacetate methyltransferase deficiency: The first inborn error of creatine metabolism in man. Am. J. Hum. Genet. 58, 914–922 [PMC free article] [PubMed] [Google Scholar]