

Background: CtBP is a transcriptional corepressor with a pronounced function in developmental processes as well as tumorigenesis.
Results: CCNH/CDK7 interacts with CtBP2 and promoted CtBP2-mediated migratory and invasive potential of cancer cells.
Conclusion: CCNH/CDK7 complex has been shown a novel pathway that regulates CtBP2 stability.
Significance: CCNH/CDK7-CtBP2 axis could potentially serve as a novel target for the development of anti-metastatic interventions.
Keywords: Cancer Biology, Cell Invasion, Cell Migration, Protein-Protein Interactions, Tumor Cell Biology, CCNH, CDK7, CtBP2
Abstract
CtBP2 has been demonstrated to possess tumor-promoting capacities by virtue of up-regulating epithelial-mesenchymal transition (EMT) and down-regulating apoptosis in cancer cells. As a result, cellular CtBP2 levels are considered a key factor determining the outcome of oncogenic transformation. How pro-tumorigenic and anti-tumorigenic factors compete for fine-tuning CtBP2 levels is incompletely understood. Here we report that the cyclin H/cyclin-dependent kinase 7 (CCNH/CDK7) complex interacted with CtBP2 in vivo and in vitro. Depletion of either CCNH or CDK7 decreased CtBP2 protein levels by accelerating proteasome-dependent CtBP2 clearance. Further analysis revealed that CCNH/CDK7 competed with the tumor repressor HIPK2 for CtBP2 binding and consequently inhibited phosphorylation and dimerization of CtBP2. Phosphorylation-defective CtBP2 interacted more strongly with CCNH/CDK7 and was more resistant to degradation. Finally, overexpression of CtBP2 increased whereas depletion of CtBP2 dampened the invasive and migratory potential of breast cancer cells. CtBP2 promoted the invasion and migration of breast cancer cells in a CCNH-dependent manner. Taken together, our data have delineated a novel pathway that regulates CtBP2 stability, suggesting that targeting the CCNH/CDK7-CtBP2 axis may yield a viable anti-tumor strategy.
Introduction
Carcinogenesis is a multifaceted pathological event wherein pro-oncogenic factors and tumor suppressors wrestle for the control of cancer cells. A more malignant phenotype of cancer cells is characterized by the ability to migrate to and colonize in distal sites in a process termed metastasis (1). Patients with metastasized tumors are expected to have a much shorter disease-free survival rate (2). Hence, the battle between oncoproteins and tumor suppressors may converge on metastasis in which oncoproteins promote, whereas tumor suppressors antagonize, migratory and invasive behavior of cancer cells.
Epithelial-mesenchymal transition (EMT)3 is a biologically conserved process defined by the replacement of an epithelial phenotype by a mesenchymal phenotype (3). Once a cancer undergoes EMT, it sheds connections with neighboring cells and the extracellular matrix and becomes more aggressive in migration and invasion, leading to enhanced metastasis. Accompanying EMT, there are changes in gene expression profile within cancer cells characterized by the loss of epithelial-specific markers and the activation of mesenchymal-specific genes (4). By differentially modulating gene expression, a host of transcriptional activators and repressors participate in EMT and by extension cancer metastasis (5).
C-terminal binding protein 2 (CtBP2) is a splice variant of the CtBP family that regulates a range of physiological and pathological processes (6). Among known CtBP2 functions is its ability to promote EMT, which facilitates cancer cell metastasis, by functioning as a transcriptional corepressor of epithelial-specific genes (7). Specifically, CtBP2 forms a complex with sequence-specific transcription factors and binds to proximal promoter region of E-cadherin, a prominent epithelial marker, to repress transcription (8). In accordance, CtBP-null mouse epithelial fibroblast cells exhibit marked up-regulation of epithelial markers, pointing to a role for CtBP in supporting cell migration and invasion (9). Indeed, higher CtBP2 expression has been noted in patients with malignant cancer compared with those with a more benign form and is correlated with a poorer prognosis (10–12). Thus, CtBP2 level and/or activity can be deemed as a predicting factor for cancer metastasis. However, the intricate mechanism underlying the regulation of CtBP2 levels during cancer metastasis is not well defined. Here we report that cyclin H/CDK7 complex increases cellular CtBP2 levels by altering its phosphorylation and proteasomal destruction. Therefore, the CCNH/CDK7-CtBP2 axis could potentially serve as a novel target for the development of anti-metastatic interventions.
EXPERIMENTAL PROCEDURES
Cell Lines, Cell Culture, and Transfections
HEK293T, human MDA-MB-231, and MCF-7 breast cancer cells were cultured in Dulbecco's modified Eagle's medium (DMEM; BioWhittaker) containing 10% fetal bovine serum (FBS; Cellgro, Mediatech), 2 mm l-glutamine, and 200 units each of streptomycin and penicillin (Cellgro)/ml. Cells were passaged with 0.1% trypsin and 0.04% EDTA in Hanks' balanced salt solution.
Cells were transfected at 70–90% confluence, utilizing 2 μl of Lipofectamine reagent (Invitrogen)/μg of DNA. Lipofectamine and DNA were incubated in serum-free DMEM for 5 min in separate tubes. After incubation, the contents of two tubes were combined, incubated for an additional 15 min, and applied onto cells.
Coimmunoprecipitaton and Immunoblotting
HEK293T cells were grown to confluence in 10-cm dishes and transiently transfected with 10 μg of the expression plasmids by using Lipofectamine reagent (Invitrogen) according to the manufacturer's instructions. At 48 h after transfection, the growth medium was aspirated, and cells were washed three times with cold phosphate-buffered saline (PBS). Cells were further lysed in plates by using 1 ml of radioimmunoprecipitation assay buffer (150 mm NaCl, 0.5% Nonidet P-40, 50 mm Tris (pH 8.0), 2 mm PMSF, Complete protease inhibitor mixture) and then collected in microcentrifuge tubes, where cell lysates were purged 10 times through the 27-gauge syringe, followed by a 30-min incubation with gentle rotation at 4 °C. After centrifugation at 4 °C for 30 min at 14,000 × g, the supernatant was used for coimmunoprecipitation with 1 μg of specific antibodies or control IgG (Santa Cruz Biotechnology), shaken for 2 h (4 °C), mixed with 30 μl of protein A/G (Santa Cruz Biotechnology), incubated for another 2 h (4 °C), and washed three times with washing buffer. Proteins bound to the beads were boiled in 60 μl of loading buffer followed by Western blotting.
The antibodies used for immunodetection were as follows: anti-CtBP2 (BD Biosciences; 1:1000), anti-β-actin (Sigma; 1:10,000). anti-FLAG (Sigma-Aldrich; 1:1000), anti-GFP (Santa Cruz Biotechnology; 1:800), anti-HIPK2 (Abcam; 1: 1000), EMT Antibody Sampler Kit (Cell Signaling).
Protein Purification and in Vitro Binding Assays
CCNH, CDK7, and CtBP2 sequences were inserted into a pGEX-6P-1 vector and transformed into Escherichia coli strain BL-21/DE3. Proteins were induced by adding 0.2 mm isopropyl-d-thiogalactopyranoside (Sigma) for 6 h (29 °C). GST pulldowns were performed as described previously. Cells were harvested by centrifugation and then resuspended in buffer (PBS, 1% Triton X-100, and 1 mm dithiothreitol (DTT)) for sonication. After centrifugation of the sonicated lysates, the supernatants were incubated with glutathione-Sepharose 4B beads (GE Healthcare) for purification. Forty-eight hours after transfection with the indicated plasmids, 293T cells were washed three times with ice-cold PBS and suspended in lysis buffer. The resulting supernatants were divided into three parts. One-tenth of the supernatant was used for input. Equal parts of the remaining supernatant were incubated with GST or GST-fused proteins for 2 h (4 °C). The beads were then washed with washing buffer (20 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm EDTA (pH 8.0), and 0.5% Nonidet P-40) three times and boiled in 40 μl of loading buffer for immunoblotting.
Cell Migration Assays
The wound closure assay was performed as described previously (7). A wound was induced on the confluent monolayer cells by scraping a gap with a micropipette tip, and the speed of wound closure was monitored by light microscopy. Cell migration was measured by the traveling distance from the original wound edge after 20 h of incubation.
In Vitro Invasion Assay
A 24-well Transwell plate (8-μm pore size, Corning) was used to measure the migratory and invasive ability of each cell line. For Transwell migration assays, 5 × 104 cells were plated in the top chamber, which was lined with a noncoated membrane. For invasion assays, chamber inserts were coated with 200 mg/ml Matrigel and dried overnight under sterile conditions. Then, 1 × 105 cells were plated in the top chamber. The mean values of triplicate assays for each experimental condition were used.
Statistical Analysis
Statistical analyses were performed using the Mann-Whitney U-test. All data are expressed as means ± S.E. Differences were considered significant if p < 0.05.
RESULTS
CtBP2 Interacts with CCNH/CDK7 in Vitro and in Vivo
To search for proteins that could interact with and hence regulate the activity of CtBP2, we employed a yeast two-hybrid screen with CtBP2 as bait. Using this strategy, we were able to identify CCNH/CDK7 as a potential binding partner for CtBP2 (data not shown). To verify this potential interaction, we transfected HEK293T cells with differentially tagged expression constructs for CCNH/CDK7 and CtBP2 followed by immunoprecipitation. As shown in Fig. 1, A and B, anti-GFP antibody brought down FLAG-tagged CCNH or CDK7 along with GFP-tagged CtBP2. To probe the possibility that CCNH/CDK7 could interact directly with CtBP2, we performed a GST pulldown assay using purified proteins. After incubation with FLAG-CCNH/CDK7-transfected 293T cell lysates, the precipitated proteins were separated by SDS-PAGE and detected by Western blotting using an anti-FLAG antibody. The results showed that both CCNH and CDK7 bound to GST-CtBP2 but not to GST alone (Fig. 1, C and D). A reciprocal experiment using GST-CCNH/CDK7 and FLAG-CtBP2 gave similar results (Fig. 1, E and F). We then sought to determine whether the CtBP2-CCNH/CDK7 interaction can be detected in vivo. Coimmunoprecipitation experiments were performed on nontransfected MDA-MB-231 cell lysates with anti-CCNH (Fig. 1G) or anti-CtBP2 antibody (Fig. 1H). CtBP2 coimmunoprecipitated specifically with the anti-CCNH antibody, but not in control precipitation, demonstrating a naturally occurring interaction between endogenous CtBP2 and CCNH/CDK7 in vivo. To further delineate the domains with which CtBP2 binds to CCNH/CDK7, we transfected HEK293T cells with either full-length (1–445) or truncated CtBP2 constructs followed by immunoprecipitation. As indicated in Fig. J, CtBP2 interacted with CCNH/CDK7 via its most N terminus between amino acids 1 and 82.
FIGURE 1.
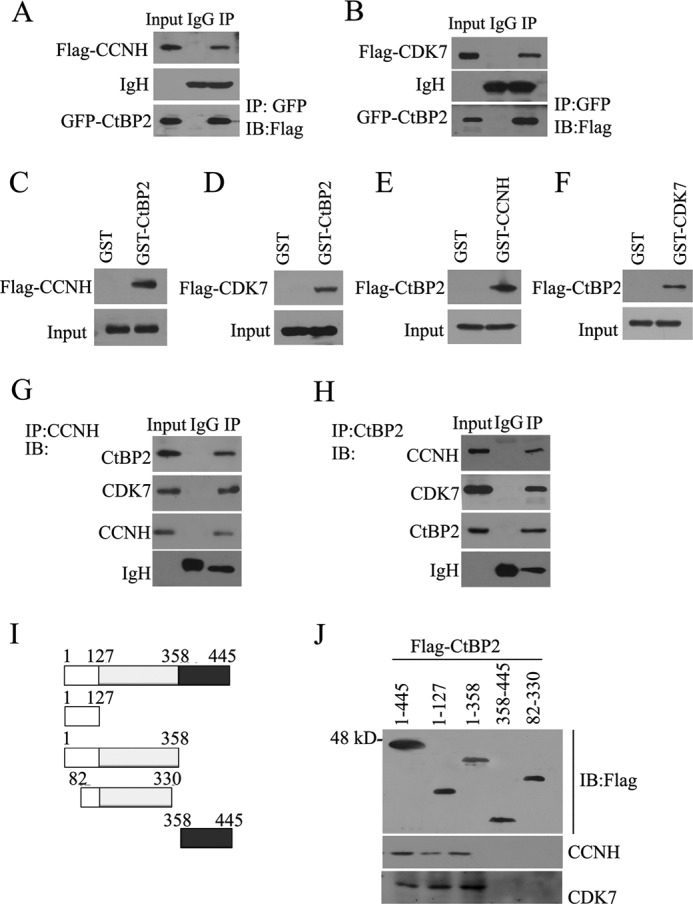
CtBP2 interacts with CCNH/CDK7 in vitro and in vivo. A and B, HEK293T cells were transfected with FLAG-CCNH, GFP-CtBP2, FLAG-CDK7, and GFP-CtBP2 as indicated. Proteins in the cellular lysates and immunoprecipitated (IP) proteins were subsequently analyzed by Western blotting (IB) the indicated antibodies. C–F, 293T cells were grown in 10-cm dishes and transfected with the pCDNA6/myc-His-3×FLAG-CCNH (C), pcDNA6/myc-His-3×FLAG-CDK (D), or pcDNA6/myc-His-3×FLAG-CtBP2 (E and F). Forty-eight hours later, the cells were harvested as described under “Experimental Procedures,” and the supernatant was divided into two equal parts and incubated with GST or GST-CtBP2 beads (C and D), GST or GST-CDK7 beads (E), GST or GST-CCNH (F) for 2 h. Protein-bound glutathione-Sepharose 4B beads were washed and boiled for 5 min and were detected by Western blotting using an anti-FLAG antibody. G and H, extracts from MDA-MB-231 cells were immunoprecipitated with antibody to CCNH (G) or to CtBP2 (H). Precipitates were then analyzed by immunoblotting using the indicated antibodies. J and I, endogenous CCNH or CDK7 was used to pull down the FLAG-CtBP2 deletion mutants indicated (I). Equal amounts of input proteins were confirmed by Western blotting with antibody for CtBP2. Coprecipitated CCNH or CDK7 was detected via immunoblotting with indicated antibodies.
CCNH/CDK7 Is Required to Maintain Cellular CtBP2 Levels by Blocking Proteasomal Degradation
Next, to determine how the interaction with CCNH/CDK7 might influence endogenous CtBP2 levels, we depleted cellular CCNH or CDK7 by shRNA mediated knockdown. Two independent shRNA clones targeting CCNH (ps4.1-CCNH-1 and -2) significantly down-regulated cyclin H expression in HEK293T cells (Fig. 2A). Of interest, this was a concomitant decrease in CtBP2 levels. Similarly, silencing CDK7 expression also resulted in a sharp loss of CtBP2 protein (Fig. 2B). We also examined the effect of CCNH/CDK7 elimination on CtBP2 protein levels in HepG2 hepatocellular carcinoma cells. Deletion of either cyclin H (Fig. 2C) or CDK7 (Fig. 2D) led to the down-regulation of CtBP2 protein expression.
FIGURE 2.
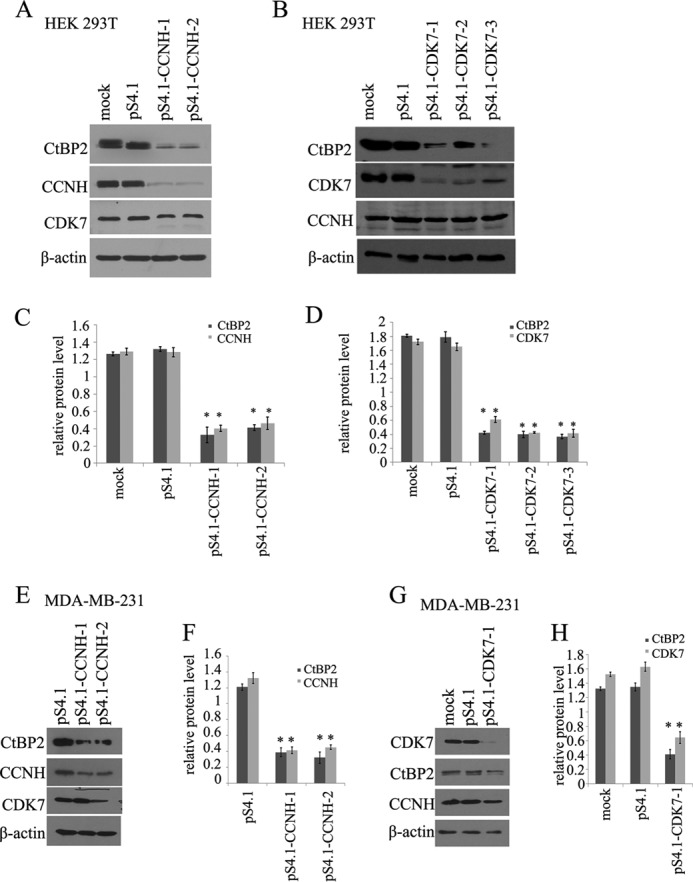
Knockdown of CCNH or CDK7 decreases levels of CtBP2. A and B, immunoblot of cell lysates from HEK293T cells expressing the empty vector or pS4.1-CCNH (A) or pS4.1-CDK7 (B). C and D, relative amount of protein for CtBP2, CCNH/CDK7. E and G, immunoblot of cell lysates from MDA-MB-231 cells expressing the empty vector or pS4.1-CCNH (E) or pS4.1-CDK7 (G). F and H, relative amount of protein for CtBP2 and CCNH/CDK7.
We then sought to determine whether reduction of CtBP2 protein expression by CCNH/CDK7 removal was a result of impaired protein stability. To this end, we used an inhibitor to eukaryotic translation, cycloheximide to stop translation and measured protein half-life by Western blotting. CtBP2 protein was degraded at a much faster rate when there was an insufficient amount of endogenous cyclin H or CDK7 rendered by shRNA, indicating that CCNH/CDK7 is probably indispensible for shielding CtBP2 from degradation (Fig. 3 A and B). To further test this hypothesis, we treated with cells with a 26S proteasome inhibitor, MG132. Indeed, the addition of MG132 restored CtBP2 protein levels even in the presence of CCNH/CDK7 shRNA (Fig. 3, C and D). Adding yet more support to this assertion is the observation that knockdown of either cyclin H or CDK7 significantly augmented ubiquitination of CtBP2, a prelude to protein degradation (Fig. 3E). Together, these data suggest that CCNH/CDK7 function to protect CtBP2 from proteasomal degradation.
FIGURE 3.
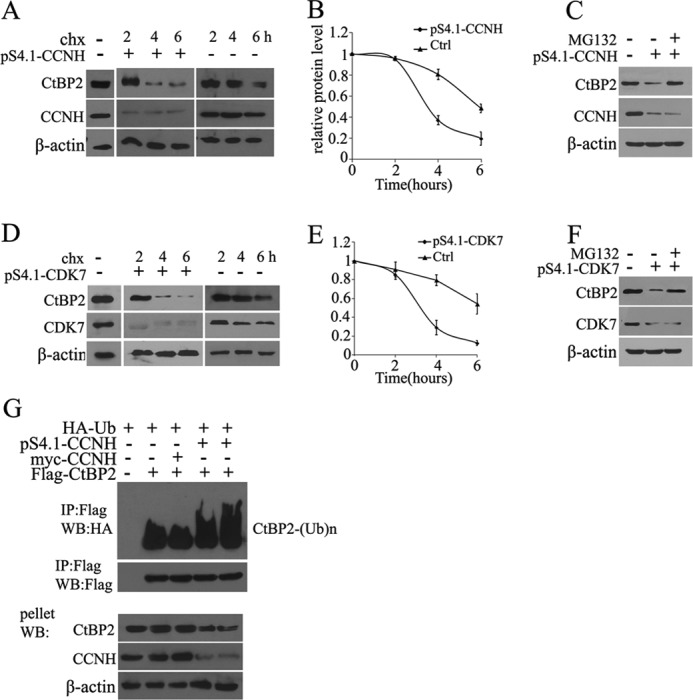
Knockdown of CCNH or CDK7 causes CtBP2 degradation. A and B, HEK293T cells were transfected with the empty vector or pS4.1-CCNH (A), and 40 h later incubated with cycloheximide (chx). Cells were lysed at the indicated times (in hours) after cycloheximide addition and Western blotted with CtBP2 antibody. The relative amount of protein for CtBP2 is plotted below. C and D, 293T cells were transfected with the empty vector or pS4.1-CDK7 and 40 h later incubated with cycloheximide. Cells were lysed at the indicated times (in hours) after cycloheximide addition and Western blotted with CtBP2 antibody. The relative amount of protein for CTBP2 is plotted below. E and F, cells were treated and analyzed as in A or C, except that 30 min prior to the addition of cycloheximide, MG-132 was added to inhibit the proteasome. G, HEK293T cells were transfected with FLAG-tagged CtBP2, myc-tagged CCNH or pS4.1-CCNH, together with a HA-tagged ubiquitin expression construct, as indicated. FLAG immunoprecipitates or the pellet fraction was analyzed by FLAG and HA Western blotting (WB) as indicated.
CCNH/CDK7 Disrupt the Interaction between CtBP2 and Homeodomain-interacting Protein Kinase 2 (HIPK2)
Having confirmed that CCNH/CDK7 affords CtBP2 an extended half-life, we next tackled the possible mechanisms. HIPK2 is a known modulator of CtBP2 degradation. It has been demonstrated that HIPK2 commits CtBP2 to proteasomal degradation by directly phosphorylating CtBP2 in cancer cells (13). We hypothesized that CCNH/CDK7 could somehow compete for the binding of CtBP2 with HIPK2, thus protecting CtBP2 from HIPK2-mediated degradation. To this end, we assessed the interaction between CtBP2 and HIPK2 in HEK293T cells either in the presence or absence of ectopic cyclin H. As depicted in Fig. 4, there was a significant reduction in the association between HIPK2 and CtBP2 when cyclin H (GFP-CCNH) was overexpressed. Therefore, CCNH/CDK7 stabilizes CtBP2 protein in part by interfering its interaction with HIPK2.
FIGURE 4.
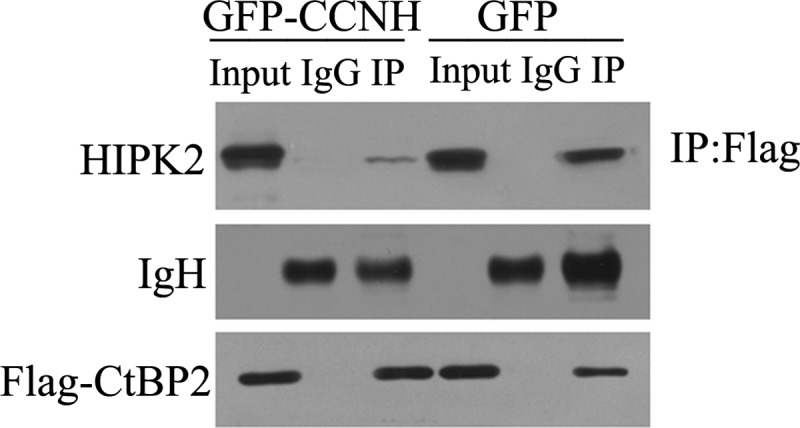
CCNH/CDK7 disrupt the interaction between CtBP2 and HIPK2. HEK293T cells were cotransfected with FLAG-tagged CtBP2, GFP, or GFP-tagged CCNH expression constructs, as indicated. Proteins were precipitated (IP) with anti-FLAG-agarose and analyzed by Western blotting using the indicated antibodies.
Interaction of CCNH/CDK7 with CtBP2 Changes Its Phosphorylation and Dimerization
We also evaluated other possibilities by which CCNH/CDK7 could impact CtBP2. CtBP2 is a target of post-translational modifications; among them, phosphorylation plays a key role in determining the ubiquitination and degradation of CtBP2 (14). To verify whether CCN/CDK7 could alter the phosphorylation levels of CtBP2, we purified CtBP2 from cells depleted with cyclin H or CDK7 and then probed CtBP2 phosphorylation by Western blotting with a pan-phosphoserine/phosphothreonine antibody. In the absence of CCNH or CDK7, CtBP2 phosphorylation was greatly elevated (Fig. 5, A and B).
FIGURE 5.
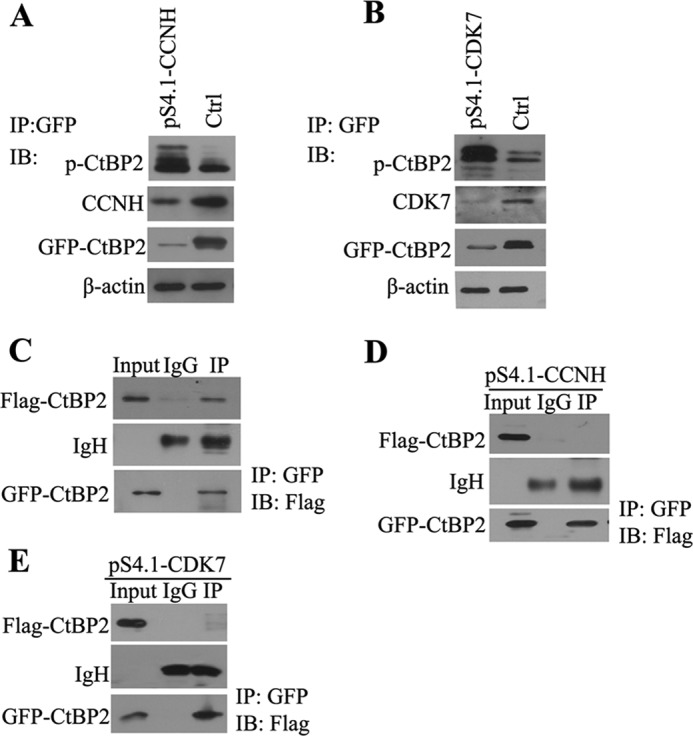
Interaction of CCNH/CDK7 with CtBP2 changes its phosphorylation and dimerization. A and B, HEK293T cells were cotransfected with GFP-tagged CtBP2 and pS4.1-CCNH (A) and pS4.1-CDK7 (B) expression constructs, as indicated. Proteins were precipitated (IP) with anti-FLAG-agarose and analyzed by Western blotting (WB) using the indicated antibodies. C, 293T cells were transfected with expression constructs as indicated. Proteins were isolated with anti-GFP-agarose and analyzed by Western blotting with a FLAG antibody. D and E, 293T cells were transfected with pS4.1-CCNH (D) or pS4.1-CDK7 (E) expression constructs. 48 hours later, proteins were isolated with anti-GFP-agarose and analyzed by Western blotting with a FLAG antibody.
CtBP2 activity is determined by its oligomerization status; dimerized CtBP2 is preferentially located in the nucleus and actively engaged in transcriptional repression (15, 16). We first verified that CtBP2 indeed formed a dimer in HEK293T cells by coimmunoprecipitation (Fig. 5C). shRNA-mediated silencing of cyclin H or CDK7, however, markedly disrupted the formation of a CtBP2 dimer, indicative of compromised repressor activity (Fig. 5, D and E). Collectively, these data demonstrate that CCNH/CDK7 antagonize CtBP2 phosphorylation and promote CtBP2 dimerization, both leading to enhanced CtBP2 activity.
Phosphorylation of Specific Residues within CtBP2 Alters Its Interaction with CCNH/CDK7 and Its Stability
Next, we tested the hypothesis that CtBP2 phosphorylation may influence its interaction with CCNH/CDK7. We mutated two key phosphorylation sites (Ser-164 by PKA and Ser-428 by HIPK2) in two forms: a defective form (Ser to Arg) and a constitutively active form (Ser to Asp). Of intrigue, whereas phosphorylation-defective CtBP2 (S164A and S428A) retained its ability to interact with CCNH/CDK7 (Fig. 6 A and B), constitutively phosphorylated CtBP2 (S164D and S428D) was unable to do so (Fig. 6, C and D).
FIGURE 6.
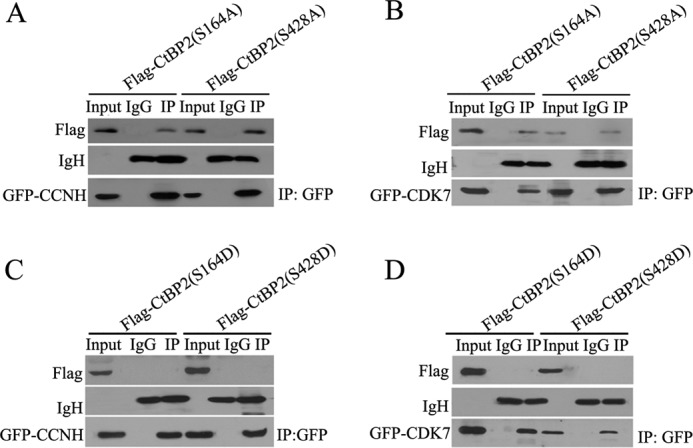
Nonphosphorylation mutants of CtBP2 interact with CCNH/CDK7. A and B, HEK 293T cells were cotransfected with FLAG-CtBP2(S164A) or FLAG-CtBP2(S428A), together with GFP-CCNH (A) or GFP-CDK7 (B) expression vectors, as indicated. Proteins were precipitated (IP) with anti-GFP-agarose and analyzed by Western blotting with FLAG antibody. C and D, mimic phosphorylation of CtBP2 disrupts its interaction with CCNH/CDK7. HEK293T cells were cotransfected with FLAG-CtBP2(S164D) or FLAG-CtBP2(S428D), together with GFP-CCNH (C) or GFP-CDK7 (D) expression vectors, as indicated. Proteins were precipitated with anti-GFP-agarose and analyzed by Western blotting with FLAG antibody.
In keeping with this observation, phosphorylation-defective CtBP2 exhibited a prolonged half-life compared with wild type CtBP2 (Fig. 7A). We were able to confirm that this extended protein stability of CtBP2 was indeed due to impaired proteasomal degradation (Fig. 7B). In agreement with this observation, SA mutants displayed significantly lower levels of ubiquitination compared with either wild type or SD mutants (Fig. 7C). Thus, CtBP2 phosphorylation may promote its own degradation by blocking the interaction with CCNH/CDK7.
FIGURE 7.

Phosphorylation of CtBP2. A and B, HEK293T cells were transfected with FLAG-CtBP2 or FLAG-CtBP2(S164A) or FLAG-CtBP2(S428A) and 40 h later incubated with cycloheximide (chx). Cells were lysed at the indicated times (in hours) after cycloheximide addition and Western blotted (WB) with the indicated antibodies. The relative amount of protein for CtBP2 is plotted below. C and D, cells were treated and analyzed as in A, MG-132 was added for 2 h to inhibit the proteasome. The relative amount of protein for CtBP2 is plotted below. E, 293T cells were transfected with FLAG-CtBP2 or FLAG-CtBP2(S164A) or FLAG-CtBP2(S428A) or FLAG-CtBP2(S164D) or FLAG-CtBP2(S428D), together with HA-ubiquitin (Ub). Proteins were precipitated with anti-FLAG-agarose and analyzed by Western blotting with HA antibody.
CtBP2 Promotes Breast Cancer Cell Migration and Invasion
CtBP-mediated repression of adhesion molecules such as E-cadherin suggests that CtBP is important in promoting EMT, a step that contributes to the malignant property of tumor cells due to the loss of intercellular adhesion in tumors, acquisition of motile and invasive phenotypes, and resistance to apoptosis (14). To determine whether CtBP2 can affect the expression of endogenous epithelial markers (E-cadherin), Western blot analysis was performed to detect alterations in the E-cadherin protein level after transfection with pS4.1-CtBP2. After CtBP2 knockdown in HEK293T and MDA-MB-231 cells, the expression of E-cadherin was significantly increased and the expression of mesenchymal markers (vimentin and N-cadherin) was markedly decreased (supplemental Fig. S1).
Finally, we made an attempt to examine the relevance of the newly identified CCNH/CDK7-CtBP2 axis in the regulation of cell migration and invasion. Overexpression of CtBP2 enhanced, whereas depletion of CtBP2 dampened, the invasion of breast cancer cells in Transwell assay (Fig. 8A). Similarly, cyclin H also conferred enhanced invasive capability to breast cancer cells (Fig. 8B). The ability of CtBP2 to promote cell invasion was impaired by the lack of cyclin H (Fig. 8C), suggesting that activation by the CCNH/CDK7 complex serves as a prerequisite for CtBP2 to exert its pro-metastatic function. We also employed wound healing assay to assess the ability of CtBP2 to influence cell migration and reached a similar conclusion that CtBP2 augmented breast cancer cell migration in a CCNH/CDK7-dependent manner (Fig. 8D).
FIGURE 8.

CtBP2 promotion of cell invasion is dependent on the CCNH expression. A–C, invasion activity of MDA-MB-231 cells transfected with the indicated expression vectors. Data are from a representative experiment performed in triplicate. Error bars represent S.E. The cell number is the number of migrated or invaded cells in five 50× microscopic fields per insert. D, migration activity of MDA-MB-231 cells transfected with indicated expression vectors. Cells with higher CtBP2 or CCNH expression showed more prominent migration activity, whereas lower CtBP2 or CCNH expression showed slower migration speed. MDA-MB-231 cells with higher CtBP2 expression but lower CCNH expression also showed slower migration speed.
CtBP2 repressors represent potentially attractive cancer drug targets, but are only useful as such if expressed in human tumors. Breast cancer and glioma tissues were thus analyzed for CtBP2 protein level by immunoblotting. Notably, the highly invasive samples contained comparatively high levels of CtBP2 and CCNH/CDK7, but unlike the low ones (supplemental Fig. S2).
DISCUSSION
Mounting evidence has implicated CtBP as a key promoter of carcinogenesis especially cancer metastasis (14). By interacting with a group of transcriptional repressors, which include Slug (17), ZEB1 (8), and Snail (18), CtBP participates in the down-regulation of E-cadherin gene, a hallmark of EMT that contributes to cancer metastasis. As such, expression of CtBP is critically related to the malignant transformation of a number of different cancers (19–23). The underlying mechanisms responsible for the modulation of CtBP levels during cancer metastasis, however, remain enigmatic. Our data as summarized here provide novel evidence that a CCNH/CDK7-CtBP2 axis controls cancer cell migration and invasion by fine-tuning CtBP2 protein levels in a phosphorylation-dependent manner.
Through molecular mapping, we demonstrate that CCNH/CDK7 interacts with CtBP2 via its most N-terminal domain (1–81). This apparently leads to the stabilization of CtBP2. One possible explanation is that interaction with CCNH/CDK7 shields CtBP2 from the binding and thus destruction by HIPK2. Indeed, the interaction between CtBP2 and HIPK2 is significantly weakened in the presence of exogenous cyclin H (Fig. 4). Bergman et al. have shown that unlike CtBP1, CtBP2 possesses a unique N-terminal domain that contains a strong nuclear localization signal mapped to amino acids 1–14 (24). Therefore, an equally plausible explanation is that by binding to the potential nuclear localization signal of CtBP2, CCNH/CDK7 may promote the nuclear localization of CtBP2 and protect it from proteasomal degradation in the cytoplasm.
CtBP2 undergoes extensive post-translational modifications that differentially regulate its subcellular localization, stability, and/or affinity for transcriptional factors (13, 25, 26). Several phosphorylation sites have been mapped for CtBP2 that are correlated with accelerated degradation. For instance, HIPK2 phosphorylates CtBP2 at serine 428 whereas PKA phosphorylates CtBP2 at 164 although it is likely that these two enzymes may act coordinately (27). Our data indicate that CCNH/CDK7 regulates CtBP2 stability by at least in part influencing its phosphorylation status (Fig.5), suggesting that cellular CtBP2 levels lie in the balance between CCNH/CDK7 and the kinases that phosphorylate and target CtBP2 for destruction. In support of this hypothesis, we have found that phosphorylation-defective, rather than default phosphorylated, CtBP2 interacts with CCN/CDK7. Therefore, elevated level of CCNH/CDK7 may decrease the amount of phosphorylatable CtBP2 and enhance CtBP-dependent transcriptional repression.
Both cyclin H and CDK7 have been implicated in the regulation of cancer metastasis. By screening a library of >10,000 small interfering RNAs, Collins et al. have reported that CDK7 may potentially promote the migration of ovarian cancer cells in vitro (28). In addition, high cyclin H expression is noted in cancers with lymphvascular space invasion (29). Our data tend to suggest that the ability of CCNH/CDK7 to promote cancer cell migration and invasion can be a function of CtBP2 stability because silencing of CCNH completely abrogates the up-regulation of migration and invasion by CtBP2 (Fig. 8). In an effort to validate the usefulness of CtBP2 as a therapeutic target in human cancer, we have shown that CtBP2 protein is overexpressed in highly invasive breast cancer and glioma tissues, likely due to concomitant CCNH/CDK7 overexpression and increased CtBP2 protein stability.
In summary, our data demonstrate for the first time that a cyclin H/CDK7 complex interacts with and regulates CtBP2 stability, thereby invoking enhanced migration and invasion of cancer cells. Given the potential significance of this interaction, the development of a small peptide that can block the interaction would likely be viable in the treatment of metastatic cancers in the future.
Acknowledgment
We thank Professor Yong Xu for helpful technical support in our work.
This work was supported by National Basic Research Program 973 of China Grants No. 2012CB822104 and No. 2011CB910604; the National Natural Science Foundation of China Grants 31070723, 81172879, 31270802, and 81201858; a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions; and by the Natural Science Foundation of Jiangsu province Grant BK2012231.

This article contains supplemental Figs. S1 and S2.
- EMT
- epithelial-mesenchymal transition
- CCNH/CDK7
- cyclin H/cyclin-dependent kinase 7
- CtBP2
- C-terminal binding protein 2
- HIPK2
- homeodomain-interacting protein kinase 2.
REFERENCES
- 1. Bogenrieder T., Herlyn M. (2003) Axis of evil: molecular mechanisms of cancer metastasis. Oncogene 22, 6524–6536 [DOI] [PubMed] [Google Scholar]
- 2. Eccles S. A., Welch D. R. (2007) Metastasis: recent discoveries and novel treatment strategies. Lancet 369, 1742–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scheel C., Weinberg R. A. (2011) Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 129, 2310–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zavadil J., Böttinger E. P. (2005) TGF-β and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774 [DOI] [PubMed] [Google Scholar]
- 5. Nieto M. A. (2011) The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 27, 347–376 [DOI] [PubMed] [Google Scholar]
- 6. Chinnadurai G. (2002) CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 9, 213–224 [DOI] [PubMed] [Google Scholar]
- 7. Zhang Q., Wang S. Y., Nottke A. C., Rocheleau J. V., Piston D. W., Goodman R. H. (2006) Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc. Natl. Acad. Sci. U.S.A. 103, 9029–9033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grooteclaes M. L., Frisch S. M. (2000) Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 19, 3823–3828 [DOI] [PubMed] [Google Scholar]
- 9. Grooteclaes M., Deveraux Q., Hildebrand J., Zhang Q., Goodman R. H., Frisch S. M. (2003) C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc. Natl. Acad. Sci. U.S.A. 100, 4568–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang L., Yang J., Ng S. K., Rodriguez N., Choi P. W., Vitonis A., Wang K., McLachlan G. J., Caiazzo R. J., Jr., Liu B. C., Welch W. R., Cramer D. W., Berkowitz R. S., Ng S. W. (2010) Autoantibody profiling to identify biomarkers of key pathogenic pathways in mucinous ovarian cancer. Eur. J. Cancer 46, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barroilhet L., Yang J., Hasselblatt K., Paranal R. M., Ng S. K., Rauh-Hain J. A., Welch W. R., Bradner J. E., Berkowitz R. S., Ng S. W. (September 3, 2012) C-terminal binding protein-2 regulates response of epithelial ovarian cancer cells to histone deacetylase inhibitors. Oncogene 10.1038/onc.2012.380 [DOI] [PubMed] [Google Scholar]
- 12. Birts C. N., Harding R., Soosaipillai G., Halder T., Azim-Araghi A., Darley M., Cutress R. I., Bateman A. C., Blaydes J. P. (2010) Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol. Cell 103, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Q., Nottke A., Goodman R. H. (2005) Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc. Natl. Acad. Sci. U.S.A. 102, 2802–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chinnadurai G. (2009) The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res. 69, 731–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verger A., Quinlan K. G., Crofts L. A., Spanò S., Corda D., Kable E. P., Braet F., Crossley M. (2006) Mechanisms directing the nuclear localization of the CtBP family proteins. Mol. Cell. Biol. 26, 4882–4894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhambhani C., Chang J. L., Akey D. L., Cadigan K. M. (2011) The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. EMBO J. 30, 2031–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tripathi M. K., Misra S., Khedkar S. V., Hamilton N., Irvin-Wilson C., Sharan C., Sealy L., Chaudhuri G. (2005) Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J. Biol. Chem. 280, 17163–17171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meloni A. R., Smith E. J., Nevins J. R. (1999) A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc. Natl. Acad. Sci. U.S.A. 96, 9574–9579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas G., Jacobs K. B., Yeager M., Kraft P., Wacholder S., Orr N., Yu K., Chatterjee N., Welch R., Hutchinson A., Crenshaw A., Cancel-Tassin G., Staats B. J., Wang Z., Gonzalez-Bosquet J., Fang J., Deng X., Berndt S. I., Calle E. E., Feigelson H. S., Thun M. J., Rodriguez C., Albanes D., Virtamo J., Weinstein S., Schumacher F. R., Giovannucci E., Willett W. C., Cussenot O., Valeri A., Andriole G. L., Crawford E. D., Tucker M., Gerhard D. S., Fraumeni J. F., Jr., Hoover R., Hayes R. B., Hunter D. J., Chanock S. J. (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 40, 310–315 [DOI] [PubMed] [Google Scholar]
- 20. Tsilidis K. K., Travis R. C., Appleby P. N., Allen N. E., Lindstrom S., Schumacher F. R., Cox D., Hsing A. W., Ma J., Severi G., Albanes D., Virtamo J., Boeing H., Bueno-de-Mesquita H. B., Johansson M., Quirós J. R., Riboli E., Siddiq A., Tjønneland A., Trichopoulos D., Tumino R., Gaziano J. M., Giovannucci E., Hunter D. J., Kraft P., Stampfer M. J., Giles G. G., Andriole G. L., Berndt S. I., Chanock S. J., Hayes R. B., Key T. J. (2012) Interactions between genome-wide significant genetic variants and circulating concentrations of insulin-like growth factor 1, sex hormones, and binding proteins in relation to prostate cancer risk in the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. Am. J. Epidemiol. 175, 926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peña C., García J. M., García V., Silva J., Domínguez G., Rodríguez R., Maximiano C., García de Herreros A., Muñoz A., Bonilla F. (2006) The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 119, 2098–2104 [DOI] [PubMed] [Google Scholar]
- 22. Straza M. W., Paliwal S., Kovi R. C., Rajeshkumar B., Trenh P., Parker D., Whalen G. F., Lyle S., Schiffer C. A., Grossman S. R. (2010) Therapeutic targeting of C-terminal binding protein in human cancer. Cell Cycle 9, 3740–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poser I., Bosserhoff A. K. (2004) Transcription factors involved in development and progression of malignant melanoma. Histol. Histopathol. 19, 173–188 [DOI] [PubMed] [Google Scholar]
- 24. Bergman L. M., Morris L., Darley M., Mirnezami A. H., Gunatilake S. C., Blaydes J. P. (2006) Role of the unique N-terminal domain of CtBP2 in determining the subcellular localisation of CtBP family proteins. BMC Cell Biol. 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao L. J., Subramanian T., Zhou Y., Chinnadurai G. (2006) Acetylation by p300 regulates nuclear localization and function of the transcriptional corepressor CtBP2. J. Biol. Chem. 281, 4183–4189 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Q., Yao H., Vo N., Goodman R. H. (2000) Acetylation of adenovirus E1A regulates binding of the transcriptional corepressor CtBP. Proc. Natl. Acad. Sci. U.S.A. 97, 14323–14328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang H., Fekete D. M., Andrisani O. M. (2011) CtBP2 down-regulation during neural crest specification induces expression of Mitf and REST, resulting in melanocyte differentiation and sympathoadrenal lineage suppression. Mol. Cell. Biol. 31, 955–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collins C. S., Hong J., Sapinoso L., Zhou Y., Liu Z., Micklash K., Schultz P. G., Hampton G. M. (2006) A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. Proc. Natl. Acad. Sci. U.S.A. 103, 3775–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kayaselcuk F., Erkanli S., Bolat F., Seydaoglu G., Kuscu E., Demirhan B. (2006) Expression of cyclin H in normal and cancerous endometrium, its correlation with other cyclins, and association with clinicopathologic parameters. Int. J. Gynecol. Cancer 16, 402–408 [DOI] [PubMed] [Google Scholar]