

Background: Lack of enzyme selectivity has been a major obstacle in developing MMP inhibitors into drugs.
Results: A fusion protein consisting of APP-IP and TIMP-2 has a highly selective inhibitory activity against MMP-2.
Conclusion: The exosite-assisted inhibitory mechanism makes APP-IP-TIMP-2 a highly selective inhibitor.
Significance: APP-IP-TIMP-2 may be developed as an effective drug for treatment of diseases in which MMP-2 activity is involved.
Keywords: Amyloid Precursor Protein, Fusion Protein, Matrix Metalloproteinase (MMP), Protease Inhibitor, Tissue Inhibitors Metalloprotease (TIMPS)
Abstract
Synthetic inhibitors of matrix metalloproteinases (MMPs), designed previously, as well as tissue inhibitors of metalloproteinases (TIMPs) lack enzyme selectivity, which has been a major obstacle for developing inhibitors into safe and effective MMP-targeted drugs. Here we designed a fusion protein named APP-IP-TIMP-2, in which the ten amino acid residue sequence of APP-derived MMP-2 selective inhibitory peptide (APP-IP) is added to the N terminus of TIMP-2. The APP-IP and TIMP-2 regions of the fusion protein are designed to interact with the active site and the hemopexin-like domain of MMP-2, respectively. The reactive site of the TIMP-2 region, which has broad specificity against MMPs, is blocked by the APP-IP adduct. The recombinant APP-IP-TIMP-2 showed strong inhibitory activity toward MMP-2 (Kiapp = 0.68 pm), whereas its inhibitory activity toward MMP-1, MMP-3, MMP-7, MMP-8, MMP-9, or MT1-MMP was six orders of magnitude or more weaker (IC50 > 1 μm). The fusion protein inhibited the activation of pro-MMP-2 in the concanavalin A-stimulated HT1080 cells, degradation of type IV collagen by the cells, and the migration of stimulated cells. Compared with the decapeptide APP-IP (t½ = 30 min), APP-IP-TIMP-2 (t½ ≫ 96 h) showed a much longer half-life in cultured tumor cells. Therefore, the fusion protein may be a useful tool to evaluate contributions of proteolytic activity of MMP-2 in various pathophysiological processes. It may also be developed as an effective anti-tumor drug with restricted side effects.
Introduction
The matrix metalloproteinases (MMPs)2 comprise a family of zinc-dependent endopeptidases capable of degrading protein components of extracellular matrix and play pivotal roles in tissue remodeling under physiological and pathological conditions such as morphogenesis, angiogenesis, tissue repair, and tumor invasion (1–4). The MMP family has been an attractive target for the development of anti-tumor therapies because the proteases are closely associated with tumor invasion and metastasis. However, a number of synthetic MMP inhibitors designed previously (5–8) have not been developed successfully as anti-tumor drugs, mainly because of unexpected side effects and lack of efficacy. The broad specificity of the MMP inhibitors is probably a major obstacle for developing safe and effective drugs. Recent studies (2, 9) have further suggested that some members of MMPs have anti-tumorigenic and -metastatic functions, and inhibition of their activities by broad-spectrum MMP inhibitors therefore offsets anti-tumor effect of the inhibitors, and even worse, stimulates tumor growth and metastasis. The activities of MMPs in vivo are regulated by a family of inhibitors known as tissue inhibitors of metalloproteinases (TIMPs). These physiological MMP inhibitors also have broad specificity against MMPs; the activities of almost all MMPs are susceptible to TIMP (TIMP-1 to TIMP-4) inhibition, and some members of a disintegrin and a metalloproteinase (ADAM) family are also inhibited by these inhibitors (10, 11). Lack of selectivity of TIMPs also makes it less feasible to use the physiological protein inhibitors for anti-tumor therapies. Therefore, design of selective inhibitors against target MMPs that have tumor-promoting effects is needed. Pioneer studies aiming to develop selective MMP inhibitors recently have been reported (12–18).
Among the MMP family, MMP-2 and MMP-9, also called type IV collagenases, are critical in the invasion of tumor cells across basement membranes, because type IV collagen is a major component of basement membranes (19–21). Although they have a common substrate, MMP-2 is categorized as a good target for anticancer drugs, whereas MMP-9 is not considered to be a safe target for these drugs (2).
Most MMPs including MMP-2 are secreted as inactive zymogens called pro-MMPs that are proteolytically activated. The activation of pro-MMP-2 is catalyzed almost specifically by membrane type MMPs localized on the cell surface. So far, six members of membrane type MMPs have been identified, and membrane type 1 MMP (MT1-MMP)-catalyzed pro-MMP-2 activation has been well characterized (22–25). Upon the activation of pro-MMP-2, the protease zymogen is first recruited on the cell surface, forming a ternary complex consisting of MT1-MMP, TIMP-2, and pro-MMP-2. In the complex, the N-terminal reactive site of TIMP-2 interacts with the active site of MT1-MMP to form the enzyme-inhibitor complex, whereas the C-terminal non-inhibitory region of TIMP-2 specifically binds to the hemopexin-like domain of pro-MMP-2. The protease zymogen in the ternary complex is then cleaved by the TIMP-2 free, non-inhibited form of MT1-MMP also located on the cell surface (22, 26, 27).
TIMPs use their N-terminal α-amino group and carbonyl oxygen of the main chain of the conserved Cys1 to coordinate the catalytic zinc ion of MMPs in their inhibitory actions (28–30). Our previous study (27) demonstrated that carbamylation of the α-amino group of Cys1 of TIMP-2 completely inactivates its inhibitory activity toward MMPs. The reactive site-modified TIMP-2 inhibits the MT1-MMP-mediated activation of pro-MMP-2, because the modified inhibitor does not interact with the active site of MT1-MMP but still has an affinity for the hemopexin-like domain of MMP-2, thereby interfering with the formation of the ternary complex on the cell surface. It has also been reported that addition of an extra alanine residue to the N terminus of TIMP-2 blocks its reactive site (31). On the other hand, we have identified a β-amyloid precursor protein-derived decapeptide having the ISYGNDALMP sequence (named APP-derived inhibitory peptide, APP-IP) as an MMP-2-selective inhibitor that interacts with the active site of the protease (32–34). These findings led us to speculate that a highly selective and strong inhibitor against MMP-2 can be designed by combining the MMP-2-selective peptide inhibitor, and the site of TIMP-2 specifically binds to the hemopexin-like domain of MMP-2.
In this study, we designed a fusion protein named APP-IP-TIMP-2, in which the sequence of APP-IP is added to the N terminus of TIMP-2 to replace the broad-specific reactive site of TIMP-2 with the MMP-2 selective inhibitory sequence. The fusion protein showed highly selective and strong inhibitory activity toward MMP-2. APP-IP-TIMP-2 may be developed as a safe and effective drug for treatment of diseases in which MMP-2 is involved.
EXPERIMENTAL PROCEDURES
Materials
The sources of materials used are as follows: pcDNA3.1/Zeo (−) and Lipofectamine LTX Reagent from Invitrogen (Carlsbad, CA); pGEM3z from Promega (Madison, WI); gelatin-Sepharose 4B, heparin-Sepharose CL-6B, and CNBr-activated Sepharose 4B from GE Healthcare UK Ltd. (Amersham Biosciences); the synthetic substrate for MMPs, 3163v (7-methoxycoumarin-4-yl)-acetyl-Pro-Leu-Gly-Leu-[Nβ-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl]-Ala-Arg amide), 3226v (7-methoxycoumarin-4-yl)-acetyl-Lys-Pro-Leu-Gly-Leu-[Nβ-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl]-Ala-Arg amide) and the synthetic MMPs inhibitor TAPI-1 (N-(R)-(2-(hydroxaminocarbonyl)methyl)-4-methylpentanoyl-l-naphthylalanyl-l-alanine-2-aminoethyl amide) from Peptide Institute, Inc. (Osaka, Japan); p-aminophenyl mercuric acetate (APMA) from Tokyo Kasei (Tokyo, Japan); purified human pro-MMP-1, human pro-MMP-8, and human ADAM17 from Calbiochem (La Jolla, CA); purified human pro-MMP-3, human pro-MMP-9, and the active catalytic domain of human MT1-MMP from EMD Millipore Co. (Billerica, MA); the plant lectin concanavalin A (Con A, type IV, substantially free of carbohydrates) was from Sigma; gelatin from Difco (Detroit, MI); bovine type IV collagen from Nitta gelatin (Osaka). The cDNA of human TIMP-2 cloned into pGM vector was constructed as described previously (35). TIMP-2 free form of pro-MMP-2 was purified from the conditioned medium (CM) of the human glioblastoma cell line T98G, as described previously (36). The recombinant active human MMP-7 was prepared as described previously (37). All custom oligo-DNA primers were provided by Rikaken Co., Ltd. (Tokyo). All other chemicals were of analytical grade or the highest quality commercially available.
Construction of Expression Vectors of Tissue Inhibitor of Metalloproteinases-2 (TIMP-2) N-terminally Fused with APP-IP-containing Sequence
To construct TIMP-2 of which the N-terminal side is fused with the amino acid sequences of APP-IP and spacers (see Fig. 1A), cDNA encoding TIMP-2 was first amplified by PCR using a sense primer with the EcoRI site: 5′-AAAGAATTCATGGGCGCCGCGGCC-3′ and antisense primer with the HindIII site: 5′-TTTAAGCTTAGGCCTGCTTATGGGTCC-3′, and pGM-TIMP-2 (35) as a template. The resultant PCR product was cleaved with EcoRI and HindIII and ligated into pGEM3z cleaved with the same pair of restriction enzymes. For introduction of the APP-IP-containing sequence between the signal sequence and the N terminus of TIMP-2 mature protein, the resultant pGEM-TIMP-2 was then cleaved with Eco52I and PstI, and ligated with two pairs of annealed oligonucleotides: 5′-GGCCGACGCCGCCGCAGGAGGAATTAGTT-3′ (sense) and 5′-CCATAACTAATTCCTCCTGCGGCGGCGTC-3′ (antisense), and 5′-ATGGTAATGATGCATTAATGCCAGGAGGAGGCTGCA-3′ (sense), and 5′-GCCTCCTCCTGGCATTAATGCATCATTA-3′ (antisense), respectively. The resultant pGEM-APP-IP-TIMP-2 was cleaved with EcoRI and HindIII, and the fragment encoding APP-IP-TIMP-2 fusion protein was ligated into the mammalian expression vector pcDNA3.1/Zeo (−) cleaved by the same pair of restriction enzymes.
FIGURE 1.

Schematic representation of interactions between APP-IP-TIMP-2 and MMP-2. A, the construction of APP-IP-TIMP-2 described under “Experimental Procedures” is schematically represented. The amino acid sequences of APP-IP (bold-face letters) and spacers (normal letters) inserted between the signal sequence and mature protein sequence of TIMP-2 (gray letters) are shown at the bottom of the scheme. The numbers shown at the top of the sequence represent the amino acid residue numbers of mature TIMP-2 protein. The SPase arrowhead represents the predicted site of cleavage by signal peptidase. SP, signal peptide region of TIMP-2 preprotein; i, the first methionine residue of TIMP-2 preprotein. B, the interaction between TIMP-2 and MMP-2 (top) and between APP-IP-TIMP-2 and MMP-2 (bottom) are schematically represented. CAT and HPX represent the catalytic and the hemopexin-like domains of MMP-2, respectively. N and C represent the N- and C-terminal regions of TIMP-2, respectively. C, APP-IP-TIMP-2 fusion protein was purified as described under “Experimental Procedures.” SDS-PAGE of the purified APP-IP-TIMP-2 (2 μg) was performed under reducing conditions followed by Coomassie Brilliant Blue R-250 staining. Lane 1, standard proteins (Bio-Rad low range); lane 2, the purified APP-IP-TIMP-2.
Expression of APP-IP-TIMP-2 in HT1080 Cells
The pcDNA-APP-IP-TIMP-2/Zeo vector constructed as described above was transfected into HT1080#10 cells, which had been cloned from HT1080 cells and found to express low levels of TIMP-2 protein (35), using Lipofectamine LTX Reagent according to the manufacturer's instructions. Stable transfectants (APP-IP-TIMP-2-HT1080#10) expressing the fusion protein were selected with zeocin. The selection was performed by culturing the cells for 4 weeks in a 1:1 mixture of Dulbecco's modified Eagles medium and Ham's F-12 medium (DME/F12) containing 300 μg/ml of zeocin.
Purification of APP-IP-TIMP-2 from CM of APP-IP-TIMP-2-HT1080#10 Cells
The APP-IP-TIMP-2-HT1080#10 cells were grown to confluency in 20 ml of the DME/F12 medium supplemented with 10% fetal bovine serum in 150-mm dishes. To prepare the CM, cells were rinsed three times with phosphate-buffered saline (PBS), and the culture was continued in 20 ml of serum-free DME/F12 medium. After 18 h, the medium was replaced with new serum-free DME/F12 medium (10 ml), and the culture was further continued for 48 h. After incubation, the CM was harvested, clarified by centrifugation, and stored at −40 °C until used for purification.
The frozen CM (1000 ml) was thawed and added with 561 g of ammonium sulfate to make an 80% saturated ammonium sulfate solution, and stirred at 4 °C for 15 h. The sample was then centrifuged at 9,000 rpm for 30 min at 4 °C. The resultant precipitates were dissolved in 50 ml of 20 mm Tris-HCl, pH 7.5, containing 150 mm NaCl and dialyzed extensively against the same buffer. After dialysis, the sample was clarified by centrifugation and then loaded on a heparin-Sepharose CL-6B column (bed volume, 5 ml) equilibrated previously with 20 mm Tris-HCl, pH 7.5, containing 150 mm NaCl. The column was washed with the equilibration buffer, and adsorbed proteins including APP-IP-TIMP-2 were eluted with 20 mm Tris-HCl, pH 7.5 containing 1 m NaCl.
To remove pro-MMP-2, the fraction eluted from the heparin-Sepharose column was loaded on a gelatin-Sepharose 4B column (bed volume, 5 ml) equilibrated previously with 20 mm Tris-HCl, pH 7.5, containing 1 m NaCl, and the flow-through fraction was collected. The gelatin-unbound fraction was next loaded on a Reactive Red-agarose column (bed volume, 5 ml) equilibrated previously with 20 mm Tris-HCl, pH 7.5, containing 1 m NaCl. The column was washed with the equilibration buffer, and adsorbed proteins were eluted with 20 mm Tris-HCl, pH 7.5, containing 2 m NaCl.
The proteins eluted from the Reactive Red-agarose column were further separated using reversed-phase high performance liquid chromatography. The sample was loaded on a Cosmosyl 5C4 column (4.6 × 100 mm) and eluted with a linear gradient of 0–80% acetonitrile containing 0.1% trifluoroacetic acid for 70 min at a flow rate of 0.5 ml/min. The column effluent was monitored at 280 nm. APP-IP-TIMP-2 eluted at about 52–54% acetonitrile was collected, freeze-dried, and dissolved in 50 mm HEPES, pH 7.5, containing 150 mm NaCl and 10 mm CaCl2.
As the preparation of the fusion protein still contained small amounts of TIMP-2, it was loaded on the MMP-7-Sepharose 4B column, in which 300 μg of active MMP-7 had been coupled with 1 ml of CNBr-activated Sepharose 4B, and the flow-through fraction was collected. The collected fraction was dialyzed against 50 mm HEPES, pH 7.5, containing 150 mm NaCl. As the MMP-7-unbound fraction contained a homogenous 27-kDa protein (Fig. 1C), it was used as a purified APP-IP-TIMP-2.
N-terminal Sequence Analysis
The N-terminal sequence of the purified APP-IP-TIMP-2 was analyzed by a commercial service (Nippi, Inc., Tokyo, Japan).
Preparation of the Active Form of MMP-2 and Its Hemopexin-like Domainless Form
600 μl of the TIMP-2 free form of pro-MMP-2 (0.93 μm) was incubated with 1 mm APMA at 37 °C for 2 h in 50 mm Tris-HCl, pH 7.5, containing 10 mm CaCl2 and 0.01% Brij 35. As described previously (32), this APMA treatment completely activated the pro-enzyme (66 kDa), and a part of the resultant active form of MMP-2 (57 kDa) was further converted into its hemopexin-like domainless form (41 kDa) by autocatalytic cleavage. The activation reaction was quenched by mixing with 5.4 ml of ice-cold buffer consisting of 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm CaCl2, and 0.01% Brij 35 (Tris-buffered saline containing calcium ion and Brij 35, TBS-Ca-Brij). To purify the active forms of MMP-2, the reaction mixture was applied to a gelatin-Sepharose 4B column (bed volume, 2 ml) previously equilibrated with TBS-Ca-Brij. After washing the column with the equilibration buffer, the adsorbed proteins were eluted with the same buffer containing 10% dimethyl sulfoxide. The eluted sample containing both the 57- and 41-kDa active forms of MMP-2 was dialyzed against TBS-Ca-Brij to remove dimethyl sulfoxide. To separate the 57- and 41-kDa forms, the dialyzed sample was applied to a heparin-Sepharose CL-6B (bed volume, 2 ml) equilibrated with TBS-Ca-Brij, and the flow-through fraction containing the hemopexin-like domainless form of MMP-2 was collected. After washing the column with the equilibration buffer, adsorbed sample (intact active form of MMP-2) was eluted with 50 mm Tris-HCl, pH 7.5, containing 500 mm NaCl, 10 mm CaCl2, and 0.01% Brij 35. The eluted sample was dialyzed against TBS-Ca-Brij.
Activation of Pro-MMP-1, Pro-MMP-3, Pro-MMP-8, or Pro-MMP-9
Pro-MMP-1 (0.47 μm), pro-MMP-3 (0.56 μm), or pro-MMP-8 (0.44 μm) was incubated at 37 °C for 2 h with 2 mm APMA in 50 mm Tris-HCl, pH 7.5, containing 10 mm CaCl2 and 0.01% Brij 35. Pro-MMP-9 (0.36 μm) was activated by incubation with 1 mm APMA at 37 °C for 1 h under the same buffer conditions.
Gelatin Zymography
Zymography was carried out on 10% polyacrylamide gels containing 1 mg/ml gelatin, as described previously (36).
Assay of Inhibitory Activity of APP-IP-TIMP-2
The active forms of MMP-1, MMP-2, the hemopexin-like domainless form of MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, the catalytic domain of MT1-MMP and ADAM17, of which concentrations are given in the legends of figures, were each incubated with various concentrations of APP-IP-TIMP-2 in 190 μl of TBS-Ca-Brij containing 0.01% bovine serum albumin at 37 °C for 15 min. Then 10 μl of 1.0 mm 3163v or 1.0 mm 3226v were added to the mixture, and the incubation was further continued for 30 min. The reaction was terminated by adding 20 μl of 0.5 m EDTA, pH 7.5. The amounts of the synthetic substrate hydrolyzed by the enzymes were measured fluorometrically with excitation at 326 nm and emission at 400 nm. The amount of the substrate hydrolyzed without enzyme was subtracted from the total amount of the hydrolyzed substrate.
Determination of the Apparent Inhibitor Constant for APP-IP-TIMP-2 Inhibition of Peptidolytic Activity of MMP-2
MMP-2 (6.3 pm) was incubated with various concentrations of APP-IP-TIMP-2 in 190 μl of TBS-Ca-Brij containing 0.01% bovine serum albumin at 25 °C for 1 h. Then 10 μl of 1.0 mm 3163v were added to the mixture, and the incubation was further continued at 37 °C for 4 h. The reaction was terminated by adding 20 μl of 0.5 m EDTA, pH 7.5. The amounts of the synthetic substrate hydrolyzed by MMP-2 were measured as described above. The data obtained were fitted to Equation 1, which is derived from the Morrison equation (38, 39),
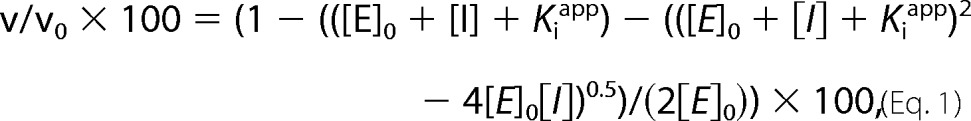 |
where v is the observed velocity of hydrolysis of 3163v by the enzyme, v0 is the velocity in the absence of inhibitor. Therefore, the left side of the equation represents the relative enzyme activity in percentage, [E]0 is the total concentration of enzyme, [I] is concentration of the added inhibitor, and Kiapp is the apparent equilibrium inhibition constant. The curve fitting was carried out without fixing the parameter of [E]0, using KaleidaGraph (Ver. 4.0) software. To determine the Kiapp value, the total concentration of MMP-2 (6.0 pm) was chosen so that the ratio of Kiapp/[E]0 is not less than 1/10 as recommended previously (40). Both the Kiapp and [E]0 values were obtained by computer-assisted non-linear curve fitting.
Analysis of Effect of APP-IP-TIMP-2 on Activation of Pro-MMP-2 in Con A-stimulated HT1080 Cells
HT1080 fibrosarcoma cell line was grown to semi-confluency in the DME/F-12 medium, supplemented with 10% fetal bovine serum. The cells were rinsed three times with serum-free DME/F-12 medium, and the culture was further continued in the presence of 70 nm APP-IP-TIMP-2 or 10 μm TAPI-1, or without inhibitor in serum-free DME/F-12 medium containing Con A (100 μg/ml). After 24 h, the resultant CM was collected, clarified by centrifugation, and dialyzed against distilled water at 4 °C. The sample was then lyophilized and dissolved in a small volume of an SDS-sampling buffer consisting of 50 mm Tris-HCl, pH 6.8, 2% SDS, and 10% glycerol. By these procedures, the initial CM was concentrated 10-fold. To examine the effects of inhibitors on the activation of proMMP-2, we analyzed the MMP-2 species in the conditioned media by gelatin-zymography (5 μl of the concentrated CM was loaded).
Migration Assays
HT1080 cells were cultured in 35-mm dishes in the DME/F-12 medium supplemented with 10% fetal bovine serum until they reached confluence. Prior to the migration assay, the cells were treated for 30 min with serum-free DME/F-12 medium containing mitomycin C (25 μg/ml) and Con A (100 μg/ml). Scratch wounds were then made in the confluent monolayer using a disposable plastic pipette tip. After gentle rinsing three times with PBS to remove detached cells, serum-free media containing 70 nm APP-IP-TIMP-2 or 10 μm TAPI-1, or that without inhibitor was added, and the cells were incubated at 37 °C for 24 h. After incubation, photographs were taken. The extent of cell migration in the presence or absence of inhibitors was quantified by measuring the areas of cells invaded into the scratch wounds with NIH Image J.
Preparation of Fluorescein Isothiocyanate (FITC)-conjugated Type IV Collagen
Type IV collagen (60 μg/ml) was incubated with 50 μm FITC in 50 mm carbonate buffer (pH 9.5) containing 150 mm NaCl at 25 °C for 1 h. After incubation, FlTC-conjugated type IV collagen was separated from free fluorescein by dialysis against PBS at 4 °C under dark conditions.
Analysis of Effect of APP-IP-TIMP-2 on Degradation of FITC-conjugated Type IV Collagen Coated on Glass Slide by Con A-stimulated HT1080#10 Cells
Each well of 8-well Lab-Tek chamber slides (Nunc, Naperville, IL) was incubated with 150 μl of FITC-conjugated type IV collagen (30 μg/ml) in PBS at 4 °C for 24 h to coat the protein. HT1080#10 cells were treated for 30 min with Con A (100 μg/ml) in DME/F-12 medium supplemented with 10% fetal bovine serum. The stimulated cells were detached from dishes using PBS containing 0.2% EDTA, and then resuspended in DME/F-12 medium supplemented with 10% fetal bovine serum at a density of 4 × 105 cells/ml. 250 μl of the cell suspension was inoculated per well of the protein-coated slide and incubated at 37 °C for 24 h in the presence of 70 nm APP-IP-TIMP-2 or 10 μm TAPI-1, or without inhibitor. After incubation, cells were removed by washing with PBS containing 0.2% EDTA. The fluorescence image of the FITC-conjugated type IV collagen that remained on the glass slide was analyzed using a fluorescence microscope (model BZ-8000; Keyence, Osaka, Japan).
Measurement of Stability of APP-IP or APP-IP-TIMP-2 in Culture of HT1080 Cells
HT1080 cells were cultured in 35-mm dishes in the DME/F-12 medium supplemented with 10% fetal bovine serum until they reached confluence. The cells were rinsed three times with serum-free DME/F-12 medium, and the culture was further continued for various lengths of time in the presence of 50 nm APP-IP-TIMP-2 or 5 μm APP-IP, or without inhibitor in 2 ml of serum-free DME/F-12 medium. After incubation, 20 μl of samples taken from each culture medium were used to measure their inhibitory activities toward the peptidolytic activity of MMP-2. The assay was performed as described in the legend of Fig. 7. The concentrations of APP-IP or APP-IP-TIMP-2 remaining in the culture media were estimated using the standard curve of the MMP-2 inhibitory activity of APP-IP or APP-IP-TIMP-2 plotted against known concentrations of each inhibitor.
FIGURE 7.

Stability of APP-IP or APP-IP-TIMP-2 in the culture of HT1080 cells. A, HT1080 cells were incubated with (●) or without (○) 5 μm APP-IP in serum-free DME/F-12 medium for indicated lengths of time. After incubation, 10 μl of samples taken from culture medium were mixed with 180 μl of MMP-2 solution consisting of 0.38 nm MMP-2, TBS-Ca-Brij, and 0.01% bovine serum albumin, and each mixture was incubated at 37 °C for 15 min. B, cells were incubated with (●) or without (○) 50 nm APP-IP-TIMP-2 for indicated lengths of time. After incubation, samples taken from culture medium were first diluted by 10-fold with TBS-Ca-Brij containing 0.01% bovine serum albumin, and 10 μl of the diluted samples were then mixed with 180 μl of the MMP-2 solution and incubated at 37 °C for 15 min. Then, 10 μl of 1.0 mm 3163v was added to the mixture, and the incubation was further continued for 30 min. The amount of 3163v hydrolyzed by MMP-2 in the absence of the cell culture-derived samples was taken as 100%. The enzyme activity is shown as the relative amount of 3163v hydrolyzed by the enzyme on the ordinate. The concentrations of APP-IP (C) or APP-IP-TIMP-2 (D) remaining in the culture media were estimated using the standard curve of the MMP-2 inhibitory activity of APP-IP or APP-IP-TIMP-2 plotted against known concentrations of each inhibitor. The concentration of each inhibitor before incubation was taken as 100%. The concentration of inhibitor in the cell culture is shown as the relative concentration on the ordinate.
RESULTS
Inhibitory Activities of APP-IP-TIMP-2 toward MMP-2 and Its Hemopexin-like Domainless Form
The N-terminal APP-IP and C-terminal TIMP-2 regions of APP-IP-TIMP-2 are designed to interact with the active site and the hemopexin-like domain of MMP-2, respectively, and three glycine residues are inserted between the two regions as a flexible connector (Fig. 1). The fusion protein was purified from the CM of the HT1080#10 cells stably transfected with an APP-IP-TIMP-2 expression vector as described under “Experimental Procedures.” When the N-terminal sequence of the purified fusion protein was analyzed, the AAGGIS sequence, corresponding to the 1st to 6th amino acid residue of the mature APP-IP-TIMP-2 protein (Fig. 1A), was determined.
To examine whether the interaction between the hemopexin-like domain of MMP-2 and TIMP-2 region of the fusion protein assists the MMP-2-directed inhibitory action of the APP-IP region, we prepared the active form of MMP-2 and its hemopexin-like domainless derivative by p-aminophenyl mercuric acetate treatment of pro-MMP-2 followed by separation on a heparin-Sepharose column chromatography. The catalytic activities of the proteases were measured using a synthetic peptidyl substrate, and effects of the fusion protein on the activities of the two forms of MMP-2 were compared. As shown in Fig. 2A, APP-IP-TIMP-2 sharply inhibited the activity of MMP-2, and the concentration of the fusion protein required for half-maximal inhibition (IC50 value) was 0.10 nm. Considering that the concentration of MMP-2 (0.20 nm) in the assay system was 2-fold higher than the IC50 value, the Kiapp value of the inhibition was thought to be much lower than the IC50 value. In contrast, the fusion protein at the concentration up to 0.5 nm showed no inhibitory effect toward the activity of the hemopexin-like domainless form of active MMP-2 (Fig. 2A), suggesting that the binding of the fusion protein with the hemopexin-like domain of MMP-2 effectively assists the inhibitory action of the APP-IP region.
FIGURE 2.

APP-IP-TIMP-2 inhibition of peptidolytic activity of MMP-2. A, the active form of MMP-2 (0.2 nm, ●) and the hemopexin-like domainless form of MMP-2 (0.2 nm, ○) were incubated with 50 μm 3163v at 37 °C for 30 min in the presence of indicated concentrations of APP-IP-TIMP-2. B, four different concentrations of MMP-2 (0.4 nm, ●; 0.2 nm, ○; 0.1 nm, ▴; 0.05 nm, ▵) were incubated with 50 μm 3163v at 37 °C for 15, 30, 60, and 120 min, respectively, in the presence of the indicated concentrations of APP-IP-TIMP-2. C, MMP-2 (6.0 pm) was incubated with 50 μm 3163v at 37 °C for 4 h in the presence of the indicated concentrations of APP-IP-TIMP-2 as described under “Experimental Procedures.” All reaction mixtures contained TBS-Ca-Brij and 0.01% bovine serum albumin. The amount of 3163v hydrolyzed by MMP-2 in the absence of APP-IP-TIMP-2 was taken as 100%. The enzyme activity is shown as the relative amount of 3163v hydrolyzed by the enzyme on the ordinate. The curve fitting of each data was carried out using Equation 1 as described under “Experimental Procedures.” The Kiapp and [E]0 values shown in C were obtained by computer-assisted non-linear curve fitting.
The results in Fig. 2A suggest that the APP-IP-TIMP-2 inhibition of the MMP-2 activity is a tight-binding inhibition in which the Kiapp value is lower than the concentration of enzyme. To verify this possibility, we examined the inhibitory effect of the fusion protein in the presence of various concentrations of MMP-2. We found that the IC50 value for the inhibition varied depending upon the concentration of MMP-2, and was decreased with reduced concentrations of the enzyme (Fig. 2B), being a typical pattern of the tight-binding inhibition (39). When data from the inhibition study were fitted to Equation 1, described under “Experimental Procedures,” the Kiapp value for the APP-IP-TIMP-2 inhibition of the activity of MMP-2 was derived to be 0.68 ± 0.08 pm (Fig. 2C). As the Kiapp value was about 1/8 of the concentration of total enzyme ([E]0 = 5.7 pm), whose parameter was also determined by curve fitting, the error in the Kiapp value was thought to be sufficiently small (40).
Specificity in Inhibitory Activity of APP-IP-TIMP-2
To examine enzyme specificity of the APP-IP-TIMP-2, we tested the inhibitory activities of the fusion protein toward various MMPs and ADAM17. As shown in Fig. 3, the peptidolytic activity of the catalytic domain of MT1-MMP was only slightly inhibited in the presence of 420 nm APP-IP-TIMP-2. The activities of MMP-1, MMP-3, MMP-7, MMP-8, MMP-9, and ADAM17 were not affected by the fusion protein at the concentration up to 420 nm (Fig. 3A), suggesting that the fusion protein is a highly selective inhibitor for MMP-2. These data also suggest that in the fusion protein, the reactive site of the TIMP-2 region, which has broad specificity against MMPs, is blocked completely by the N-terminally added sequence.
FIGURE 3.

Enzyme specificity in inhibitory activity of APP-IP-TIMP-2. A, MMP-1 (0.8 nm), MMP-2 (0.2 nm), MMP-3 (27 nm), MMP-7 (1.5 nm), MMP-8 (7.2 nm), MMP-9 (1.4 nm), and the catalytic domain of MT1-MMP (MT1cat, 1.0 nm) were incubated with 50 μm 3163v, and ADAM17 (2.4 nm) was incubated with 50 μm 3226v at 37 °C for 30 min in the absence or presence of APP-IP-TIMP-2 (420 nm). B, MMP-2 (0.2 nm, ●) and the catalytic domain of MT1-MMP (1.0 nm, ○) were incubated with 50 μm 3163v at 37 °C for 30 min in the presence of the indicated concentrations of APP-IP-TIMP-2. The amount of the peptide substrate (3163v or 3226v) hydrolyzed in the absence of APP-IP-TIMP-2 was taken as 100%. The enzyme activity is shown as the relative amount of the substrate hydrolyzed by the enzyme. The curve fitting of each data in B was carried out using Equation 1 as described under “Experimental Procedures.”
Effect of APP-IP-TIMP-2 on Activation of Pro-MMP-2 in Concanavalin A-stimulated HT1080 Cells
Our previous study (27) demonstrated that carbamylation of the α-amino group of the N-terminal Cys1 of TIMP-2 leads to loss of its inhibitory activity, and the resultant reactive site-modified TIMP-2 has an ability to inhibit the activation of intrinsic pro-MMP-2 in the Con A-stimulated HT1080 fibrosarcoma cells, in which activity of MT1-MMP is enhanced. It is assumed that the modified TIMP-2 interferes with the formation of the ternary complex consisting of MT1-MMP, TIMP-2, and pro-MMP-2 (26). As APP-IP-TIMP-2 is also a reactive site-modified TIMP-2, it is possible that the fusion protein has the ability to inhibit the activation of pro-MMP-2 in stimulated cells. To examine this possibility, HT1080 cells were stimulated by Con A in the absence or presence of APP-IP-TIMP-2 or TAPI-1, a broad-spectrum hydroxamate-based MMP inhibitor, and the pro- and active forms of MMP-2 in the resultant CM were analyzed by gelatin-zymography. As shown in Fig. 4A, gelatinolytic bands at 57 and 59 kDa, corresponding to the active and intermediate forms of MMP-2 but not that of pro-MMP-2 (66 kDa), were detected when the CM of the cells stimulated without inhibitor was analyzed. On the other hand, a faint band of the pro-form and a strong band of the intermediate form, but not that of the active one, were detected in analysis of the cells stimulated in the presence of APP-IP-TIMP-2. When the cells had been stimulated in the presence of TAPI-1, pro-MMP-2 was detected as a major MMP-2 form. These data are consistent with the view that the MT1-MMP-mediated activation of pro-MMP-2 was strongly inhibited by APP-IP-TIMP-2 in the step of the intermolecular auto-activation of MMP-2; the step is promoted strongly by the formation of the ternary complex (27). Meanwhile, TAPI-1 inhibited the catalytic activity of MT1-MMP, thereby inhibiting the MT1-MMP-catalyzed cleavage of pro-MMP-2, as schematically represented in Fig. 4B.
FIGURE 4.

Effect of APP-IP-TIMP-2 on processing of pro-MMP-2 in Con A-stimulated HT1080 cells. A, HT1080 cells were incubated for 24 h in serum-free medium containing Con A (100 μg/ml) with APP-IP-TIMP-2 (70 nm) or TAPI-1 (10 μm), or without inhibitor. The CMs were prepared from the incubated cells and subjected to gelatin zymography as described under “Experimental Procedures.” Arrowheads indicate the gelatinolytic bands of pro-MMP-2 at 66 kDa (upper), the intermediate form at 59 kDa (center), and the active form of MMP-2 at 57 kDa (lower). An arrow at 90 kDa indicates a gelatinolytic band of pro-MMP-9. Ordinate, molecular size in kDa. B, pro-MMP-2 (pro-form), the intermediate form, and the active form of MMP-2 are schematically represented. The numbers shown at the bottom of each scheme represent the amino acid residue numbers of pro-MMP-2. The MT1-MMP arrow and the MMP-2 arrow represent the sites of cleavage by MT1-MMP and MMP-2, respectively. SH represents the Cys residue interacting with catalytic zinc ion in pro-MMP-2.
Effects of APP-IP-TIMP-2 on Migration of Con A-stimulated HT1080 Cells and Degradation of Type IV Collagen by Stimulated Cells
Ikejiri et al. (41) found that a synthetic inhibitor having selectivity toward gelatinases (MMP-2 and MMP-9) inhibits migration of HT1080 cells in a scratch wound assay. To examine whether APP-IP-TIMP-2 inhibition of MMP-2 activity affects the migration of HT1080 cells, we monitored cell migration under the condition that activated intrinsic pro-MMP-2 and inhibited cell proliferation, which were achieved by pretreatment of cells with Con A and mitomycin C, respectively, and an effect of APP-IP-TIMP-2 or TAPI-1 on the cell migration was tested. As shown in Fig. 5A, APP-IP-TIMP-2 and TAPI-1 similarly inhibited the migration of the stimulated cells, and the rates of migration in the presence of the inhibitors were about 50% of that in the absence of the inhibitors. These results suggest that at least among MMPs, the proteolytic activity of MMP-2 mainly contributes to the migration of cells.
FIGURE 5.

Effects of APP-IP-TIMP-2 on migration of Con A-stimulated HT1080 cells. HT1080 cells were cultured in 35-mm dishes until they reached confluence. Cells were treated for 30 min with serum-free DME/F-12 medium containing mitomycin C (25 μg/ml) and Con A (100 μg/ml). Scratch wounds were then made in the confluent monolayer. The wounded cell cultures were then incubated at 37 °C for 24 h in serum-free medium containing 70 nm APP-IP-TIMP-2 or 10 μm TAPI-1, or without inhibitor (Control). Before (0 h) or after (24 h) incubation, photographs were taken (A). The extent of cell migration in the presence or absence of inhibitors was quantified by measuring the areas of cells invading the scratch wounds using NIH Image J (B). In B, the area of invaded cells (healed area) in the absence of inhibitor was taken as 100%. The extent of cell migration is shown as the relative healed area on the ordinate. Each bar represents the mean ± S.D. for six assays. Statistical significance was determined by an unpaired test. *, p < 0.01.
We further examined whether APP-IP-TIMP-2 inhibits the degradation of type IV collagen in the Con A-stimulated HT1080 cells. As shown in Fig. 6, when the stimulated cells were plated on the FITC-conjugated type IV collagen-coated slide and then incubated, several dark spots, whose sizes are similar to those of HT1080 cells, were observed in the fluorescence microscopic analysis. In contrast, the number of spots and their intensities were significantly reduced when the cells were plated and incubated in the presence of APP-IP-TIMP-2 or TAPI-1 (Fig. 6). These data are consistent with the view that pericellular MMP-2 is the major MMP capable of degrading type IV collagen in stimulated HT1080 cells.
FIGURE 6.

Effects of APP-IP-TIMP-2 on degradation of type IV collagen by Con A-stimulated HT1080#10 cells. FITC-conjugated type IV collagen was coated on 8-well glass slides as described under “Experimental Procedures.” HT1080#10 cells pretreated with Con A as described under “Experimental Procedures” were plated onto the slides coated with FITC-conjugated type IV collagen and incubated in DME/F-12 medium supplemented with bovine fetal serum in the absence (Control) or presence of APP-IP-TIMP-2 (70 nm) or TAPI-1 (10 μm) at 37 °C for 24 h. After incubation, the cells were removed, and the FITC-conjugated type IV collagen remaining on the slides was visualized as described under “Experimental Procedures.” Scale bar, 50 μm.
Stability of APP-IP-TIMP-2 in Culture of Tumor Cells
To examine the stability of APP-IP-TIMP-2 in the culture of tumor cells, the fusion protein or the decapeptide APP-IP was incubated with HT1080 cells, and time course of change in their inhibitory activities toward the peptidolytic activity of MMP-2 was investigated. As shown in Fig. 7, the inhibitory activity of the decapeptide was rapidly diminished during the incubation. The half-life of the inhibitory peptide was about 30 min. In contrast, the inhibitory activity of APP-IP-TIMP-2 was not changed even after 96-h incubation, suggesting that the TIMP-2 region of the fusion protein not only assists the inhibitory action of the APP-IP region but also endows the inhibitory sequence with high stability.
DISCUSSION
We designed a highly selective and strong inhibitor against MMP-2 by combining the MMP-2-selective peptide inhibitor APP-IP and physiological MMP inhibitor TIMP-2, which has a site capable of binding specifically to the hemopexin-like domain of MMP-2. Recent studies have suggested that the accessory domains of several metalloproteinases contribute to their recognition of substrates, thereby determining their substrate specificities. For instance, the interaction of the cysteine-rich/spacer domains of ADAM with thrombospondin motif 13 with its specific substrate von Willebrand factor is reported to be critical for the metalloproteinase-catalyzed cleavage of the protein substrate (42, 43). It is also suggested that Russell's viper venom factor X activator, a metalloproteinase belonging to the snake venom metalloproteinase family, uses its lectin-like domains to recognize and bind the γ-carboxyglutamic acid domain of coagulation factor X; the metalloproteinase activates the serine protease zymogen to its active form factor Xa by cleaving a specific peptide bond in the serine protease domain of the zymogen (44). Very recently, the crystal structure of an active site mutant of MMP-1 bound to a triple-helical collagen peptide has been determined (45). The studies revealed that an exosite in the hemopexin-like domain of MMP-1, which binds to residues of the substrate peptide 10 residues apart from the scissile bond, is critical for collagenolysis. It is likely that an appropriate distance between the exosite and the active site is important for proteases to recognize precisely the scissile peptide bond of their substrates. In the case of APP-IP-TIMP-2, the distance between the MMP-2 binding site in the TIMP-2 region and the reactive site APP-IP sequence is therefore assumed to be critical for the MMP-2 selective inhibitory action of the fusion protein. We have previously demonstrated that the APP-IP sequence added to the N terminus of the catalytic domain of MMP-2 works as an effective intramolecular inhibitor, whereas the C-terminally added one does not (33), also suggesting the importance of appropriate positioning and direction of the APP-IP sequence relative to the active site of MMP-2 for the inhibitory action. Considering that the interaction between the C-terminal region of TIMP-2 and the hemopexin-like domain of MMP-2 facilitates the enzyme inhibition (46), it is likely that the exosite interaction brings the α-amino group of N-terminal Cys1 of TIMP-2, the reactive site of the inhibitor, into close proximity of the catalytic zinc ion of MMP-2. We speculate that the N-terminal APP-IP region of the fusion protein is similarly presented to the catalytic site of MMP-2 with the assistance of the exosite interaction as schematically represented in Fig. 1B.
We found that APP-IP-TIMP-2 strongly inhibited the peptidolytic activity of MMP-2 with a Kiapp value of 0.68 pm. This value is about 40,000-fold lower than the IC50 value of APP-IP inhibition of the MMP-2 activity (30 nm) determined in the previous study (32). Therefore, it is likely that the exosite interaction effectively assists the binding of the APP-IP region of the fusion protein to the active site of MMP-2. In contrast, APP-IP (32) and APP-IP-TIMP-2 (Fig. 3B) similarly inhibited the activity of the catalytic domain of MT1-MMP with IC50 and Kiapp values of 2 μm and 1.7 μm, respectively, suggesting that the TIMP-2 region of the fusion protein neither support nor interfere with the interaction between the APP-IP region and the MMP. Besides the MMP-2 selective inhibitory activity of the APP-IP region, the MMP-2-specific exosite-assisted inhibitory mechanism makes the fusion protein highly selective inhibitor for MMP-2. The inhibitory activity of APP-IP-TIMP-2 toward MMP-1, MMP-3, MMP-7, MMP-8, MMP-9 (IC50 ≫ 0.42 μm), or MT1-MMP (Kiapp = 1.7 μm) was indeed six orders of magnitude or more weaker than that toward MMP-2 (Kiapp = 0.68 pm). These data also suggest that in the fusion protein, the N-terminal reactive site of TIMP-2, which has broad-spectrum inhibitory activity against MMPs, is completely blocked by the APP-IP adduct. It has been reported that addition of a single alanine residue to the N terminus of TIMP-3 is sufficient to inactivate its inhibitory activity toward MMPs, but insufficient to inactivate the inhibitory activity toward ADAM17 (47). Although TIMP-2 inhibition of ADAM17 activity has not been reported, it raises a possibility that TIMP-2 retains the inhibitory activity toward member(s) of the ADAM family even after the addition of the APP-IP sequence. More recently, however, it has also been reported that increasing number of alanine residues added to the N terminus of TIMP-3 causes stepwise loss of its ADAM17 inhibitory activity, and addition of eight alanine residues essentially inactivates the inhibitory activity (48). This report makes it unlikely that APP-IP-TIMP-2 retains the inhibitory activity of TIMP-2, because the fusion protein has a 17-residue extension including the APP-IP sequence on the N-terminal side of the TIMP-2 region (Fig. 1A).
In cell culture systems, and probably in vivo, APP-IP-TIMP-2 prevents exertion of proteolytic activity of MMP-2 by dual mechanisms; one is through the inhibition of the activation of pro-MMP-2 mediated by both MT1-MMP and TIMP-2, and the other is through the strong inhibition of the enzyme activity of the activated MMP-2. When one uses the fusion protein to probe the role of enzyme activity of MMP-2 in various pathophysiological processes, the latter mechanism may be critical, because it has been reported that the MT1-MMP-mediated activation of pro-MMP-2 is stimulated by claudins in a TIMP-2-independent manner (49). The MT2-MMP-catalyzed conversion of the pro-form of MMP-2 to its active form also does not require TIMP-2 (50). Considering that the inhibition of the MT1-MMP-mediated activation of pro-MMP-2 by the reactive site-modified TIMP-2 is based on a competitive binding of the modified or native TIMP-2 to the hemopexin-like domain of pro-MMP-2 (27), it is unlikely that the modified inhibitor prevents the TIMP-2-independent activations of pro-MMP-2. Therefore, the suppressive effect of APP-IP-TIMP-2 on the activation of pro-MMP-2 in vivo is assumed to be limited.
We demonstrated that APP-IP-TIMP-2 significantly inhibits the migration of Con A-stimulated HT1080 cells and degradation of type IV collagen by the stimulated cells. These effects of the fusion protein on the cells were quite similar to those of TAPI-1, a broad spectrum MMPs inhibitor. Therefore, it is likely that, at least among MMPs, the proteolytic activity of MMP-2 contributes mainly to the cell migration and the degradation of type IV collagen. The fusion protein may be a useful tool to evaluate specific contributions of the proteolytic activity of MMP-2 in various in vitro and in vivo systems. Although the gene knockout and knockdown approaches are also useful to probe roles of individual MMPs in pathophysiological processes, they do not distinguish between proteolytic and non-proteolytic functions of the proteases. Recent reports suggest that MT1-MMP (51) and MMP-9 (52) stimulate cell migration in a proteolysis-independent manner; the interactions between the dimerized forms of these MMPs and CD44 cause phosphorylation of EGF receptor, thus activating MAPK and PI3K signaling pathways. As both the intracellular signaling and the proteolysis support cell migration, the proteolytic and the non-proteolytic functions of MMPs need to be analyzed separately to evaluate their precise contributions.
As compared with the decapeptide APP-IP, the fusion protein APP-IP-TIMP-2 showed much higher stability in the cultured tumor cells. In general, peptides are susceptible to proteolytic cleavage by both exo- and endopeptidases. We have previously reported that APP-IP is not cleaved by MMP-2, but it is very susceptible to the endopeptidase Asp-N cleavage (32). Considering that the truncation of N- or C-terminal amino acid residues of APP-IP as well as the endopeptidase Asp-N cleavage of the peptide leads to drastic loss of its MMP-2-inhibitory activity (32), the observed rapid loss of the inhibitory activity of APP-IP in the tumor cell culture is possibly caused by the tumor cell-derived exo- and/or endoproteases other than MMP-2. The TIMP-2 region of APP-IP-TIMP-2 may protect the APP-IP region from the proteolytic cleavage by limiting accessibility of the proteases. Because of the instability of APP-IP in cell culture, usefulness of the peptide as an MMP-2-selective inhibitor has been very limited. Therefore, the highly enhanced stability of the APP-IP sequence endowed by its fusion with TIMP-2 also enhances the usefulness of the MMP-2-selective inhibitor.
Besides being a good target for anticancer drugs, MMP-2 is likely involved in some types of cardiovascular diseases. For instance, it has been reported that deletion of the MMP-2 gene prevents cardiac rupture after myocardial infarction in mice; proteolytic activity of MMP-2 is suggested to promote macrophage infiltration in the infarcted myocardial tissue (53). It has also been suggested that platelet-derived active MMP-2 amplifies platelet aggregation in response to weak stimuli, thus inducing arterial thrombosis (54). As APP-IP-TIMP-2 is a highly selective inhibitor for MMP-2, it may be developed as a safe and effective drug for treatment of diseases in which proteolytic activity of MMP-2 is involved.
This work was supported in part by Grant No. T2401 (to S. H.) from the 2012 Strategic Research Promotion of Yokohama City University, Japan, JST (Research for Promoting Technological Seeds, to S. H.), and an Extramural Collaborative Research Grant from the Cancer Research Institute, Kanazawa University, Japan (to S. H.).
- MMP(s)
- matrix metalloproteinase(s)
- TIMP(s)
- tissue inhibitor(s) of metalloproteinases
- APP
- β-amyloid precursor protein
- APP-IP
- APP-derived inhibitory peptide
- MT1-MMP
- membrane-type 1 MMP
- ADAM
- a disintegrin and a metalloproteinase
- APMA
- p-aminophenyl mercuric acetate
- Con A
- concanavalin A
- CM
- conditioned medium.
REFERENCES
- 1. Nagase H., Visse R., Murphy G. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573 [DOI] [PubMed] [Google Scholar]
- 2. Overall C. M., Kleifeld O. (2006) Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 3. Werb Z. (1997) ECM and cell surface proteolysis: regulating cellular ecology. Cell 91, 439–442 [DOI] [PubMed] [Google Scholar]
- 4. Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 5. Betz M., Huxley P., Davies S. J., Mushtaq Y., Pieper M., Tschesche H., Bode W., Gomis-Rüth F. X. (1997) 1.8-A crystal structure of the catalytic domain of human neutrophil collagenase (matrix metalloproteinase-8) complexed with a peptidomimetic hydroxamate primed-side inhibitor with a distinct selectivity profile. Eur. J. Biochem. 247, 356–363 [DOI] [PubMed] [Google Scholar]
- 6. Brandstetter H., Grams F., Glitz D., Lang A., Huber R., Bode W., Krell H. W., Engh R. A. (2001) The 1.8-A crystal structure of a matrix metalloproteinase 8-barbiturate inhibitor complex reveals a previously unobserved mechanism for collagenase substrate recognition. J. Biol. Chem. 276, 17405–17412 [DOI] [PubMed] [Google Scholar]
- 7. Jia M. C., Schwartz M. A., Sang Q. A. (2000) Suppression of human microvascular endothelial cell invasion and morphogenesis with synthetic matrixin inhibitors. Targeting angiogenesis with MMP inhibitors. Adv. Exp. Med. Biol. 476, 181–194 [DOI] [PubMed] [Google Scholar]
- 8. Finzel B. C., Baldwin E. T., Bryant G. L., Jr., Hess G. F., Wilks J. W., Trepod C. M., Mott J. E., Marshall V. P., Petzold G. L., Poorman R. A., O'Sullivan T. J., Schostarez H. J., Mitchell M. A. (1998) Structural characterizations of nonpeptidic thiadiazole inhibitors of matrix metalloproteinases reveal the basis for stromelysin selectivity. Protein Sci. 7, 2118–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Decock J., Thirkettle S., Wagstaff L., Edwards D. R. (2011) Matrix metalloproteinases: protective roles in cancer. J. Cell Mol. Med. 15, 1254–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amour A., Knight C. G., Webster A., Slocombe P. M., Stephens P. E., Knäuper V., Docherty A. J., Murphy G. (2000) The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 473, 275–279 [DOI] [PubMed] [Google Scholar]
- 11. Kashiwagi M., Tortorella M., Nagase H., Brew K. (2001) TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 276, 12501–12504 [DOI] [PubMed] [Google Scholar]
- 12. Rosenblum G., Meroueh S. O., Kleifeld O., Brown S., Singson S. P., Fridman R., Mobashery S., Sagi I. (2003) Structural basis for potent slow binding inhibition of human matrix metalloproteinase-2 (MMP-2). J. Biol. Chem. 278, 27009–27015 [DOI] [PubMed] [Google Scholar]
- 13. Gooyit M., Lee M., Schroeder V. A., Ikejiri M., Suckow M. A., Mobashery S., Chang M. (2011) Selective water-soluble gelatinase inhibitor prodrugs. J. Med. Chem. 54, 6676–6690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pochetti G., Montanari R., Gege C., Chevrier C., Taveras A. G., Mazza F. (2009) Extra binding region induced by non-zinc chelating inhibitors into the S1′ subsite of matrix metalloproteinase 8 (MMP-8). J. Med. Chem. 52, 1040–1049 [DOI] [PubMed] [Google Scholar]
- 15. Dublanchet A. C., Ducrot P., Andrianjara C., O'Gara M., Morales R., Compère D., Denis A., Blais S., Cluzeau P., Courté K., Hamon J., Moreau F., Prunet M. L., Tertre A. (2005) Structure-based design and synthesis of novel non-zinc chelating MMP-12 inhibitors. Bioorg. Med. Chem. Lett. 15, 3787–3790 [DOI] [PubMed] [Google Scholar]
- 16. Devel L., Rogakos V., David A., Makaritis A., Beau F., Cuniasse P., Yiotakis A., Dive V. (2006) Development of selective inhibitors and substrate of matrix metalloproteinase-12. J. Biol. Chem. 281, 11152–11160 [DOI] [PubMed] [Google Scholar]
- 17. Engel C. K., Pirard B., Schimanski S., Kirsch R., Habermann J., Klingler O., Schlotte V., Weithmann K. U., Wendt K. U. (2005) Structural basis for the highly selective inhibition of MMP-13. Chem. Biol. 12, 181–189 [DOI] [PubMed] [Google Scholar]
- 18. Devy L., Huang L., Naa L., Yanamandra N., Pieters H., Frans N., Chang E., Tao Q., Vanhove M., Lejeune A., van Gool R., Sexton D. J., Kuang G., Rank D., Hogan S., Pazmany C., Ma Y. L., Schoonbroodt S., Nixon A. E., Ladner R. C., Hoet R., Henderikx P., Tenhoor C., Rabbani S. A., Valentino M. L., Wood C. R., Dransfield D. T. (2009) Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 69, 1517–1526 [DOI] [PubMed] [Google Scholar]
- 19. Liotta L. A. (1986) Tumor invasion and metastases–role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer Res. 46, 1–7 [PubMed] [Google Scholar]
- 20. Collier I. E., Wilhelm S. M., Eisen A. Z., Marmer B. L., Grant G. A., Seltzer J. L., Kronberger A., He C., Bauer E. A., Goldberg G. I. (1988) H-ras oncogene-transformed human bronchial epithelial cells (TBE-1) secrete a single metalloprotease capable of degrading basement membrane collagen. J. Biol. Chem. 263, 6579–6587 [PubMed] [Google Scholar]
- 21. Wilhelm S. M., Collier I. E., Marmer B. L., Eisen A. Z., Grant G. A., Goldberg G. I. (1989) SV40-transformed human lung fibroblasts secrete a 92-kDa type IV collagenase which is identical to that secreted by normal human macrophages. J. Biol. Chem. 264, 17213–17221 [PubMed] [Google Scholar]
- 22. Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. (1994) A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370, 61–65 [DOI] [PubMed] [Google Scholar]
- 23. Pei D. (1999) Identification and characterization of the fifth membrane-type matrix metalloproteinase MT5-MMP. J. Biol. Chem. 274, 8925–8932 [DOI] [PubMed] [Google Scholar]
- 24. Pei D. (1999) Leukolysin/MMP25/MT6-MMP: a novel matrix metalloproteinase specifically expressed in the leukocyte lineage. Cell Res. 9, 291–303 [DOI] [PubMed] [Google Scholar]
- 25. Yana I., Seiki M. (2002) MT-MMPs play pivotal roles in cancer dissemination. Clin. Exp. Metastasis 19, 209–215 [DOI] [PubMed] [Google Scholar]
- 26. Strongin A. Y., Collier I., Bannikov G., Marmer B. L., Grant G. A., Goldberg G. I. (1995) Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 270, 5331–5338 [DOI] [PubMed] [Google Scholar]
- 27. Higashi S., Miyazaki K. (1999) Reactive site-modified tissue inhibitor of metalloproteinases-2 inhibits the cell-mediated activation of progelatinase A. J. Biol. Chem. 274, 10497–10504 [DOI] [PubMed] [Google Scholar]
- 28. Gomis-Rüth F. X., Maskos K., Betz M., Bergner A., Huber R., Suzuki K., Yoshida N., Nagase H., Brew K., Bourenkov G. P., Bartunik H., Bode W. (1997) Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389, 77–81 [DOI] [PubMed] [Google Scholar]
- 29. Fernandez-Catalan C., Bode W., Huber R., Turk D., Calvete J. J., Lichte A., Tschesche H., Maskos K. (1998) Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 17, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wisniewska M., Goettig P., Maskos K., Belouski E., Winters D., Hecht R., Black R., Bode W. (2008) Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 381, 1307–1319 [DOI] [PubMed] [Google Scholar]
- 31. Wingfield P. T., Sax J. K., Stahl S. J., Kaufman J., Palmer I., Chung V., Corcoran M. L., Kleiner D. E., Stetler-Stevenson W. G. (1999) Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J. Biol. Chem. 274, 21362–21368 [DOI] [PubMed] [Google Scholar]
- 32. Higashi S., Miyazaki K. (2003) Identification of a region of β-amyloid precursor protein essential for its gelatinase A inhibitory activity. J. Biol. Chem. 278, 14020–14028 [DOI] [PubMed] [Google Scholar]
- 33. Higashi S., Miyazaki K. (2008) Identification of amino acid residues of the matrix metalloproteinase-2 essential for its selective inhibition by β-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 283, 10068–10078 [DOI] [PubMed] [Google Scholar]
- 34. Hashimoto H., Takeuchi T., Komatsu K., Miyazaki K., Sato M., Higashi S. (2011) Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of β-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 286, 33236–33243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shofuda K., Moriyama K., Nishihashi A., Higashi S., Mizushima H., Yasumitsu H., Miki K., Sato H., Seiki M., Miyazaki K. (1998) Role of tissue inhibitor of metalloproteinases-2 (TIMP-2) in regulation of pro-gelatinase A activation catalyzed by membrane-type matrix metalloproteinase-1 (MT1-MMP) in human cancer cells. J. Biochem. 124, 462–470 [DOI] [PubMed] [Google Scholar]
- 36. Miyazaki K., Hattori Y., Umenishi F., Yasumitsu H., Umeda M. (1990) Purification and characterization of extracellular matrix-degrading metalloproteinase, matrin (pump-1), secreted from human rectal carcinoma cell line. Cancer Res. 50, 7758–7764 [PubMed] [Google Scholar]
- 37. Higashi S., Oeda M., Yamamoto K., Miyazaki K. (2008) Identification of amino acid residues of matrix metalloproteinase-7 essential for binding to cholesterol sulfate. J. Biol. Chem. 283, 35735–35744 [DOI] [PubMed] [Google Scholar]
- 38. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 39. Williams J. W., Morrison J. F. (1979) The kinetics of reversible tight-binding inhibition. Methods Enzymol. 63, 437–467 [DOI] [PubMed] [Google Scholar]
- 40. Murphy D. J. (2004) Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal. Biochem. 327, 61–67 [DOI] [PubMed] [Google Scholar]
- 41. Ikejiri M., Bernardo M. M., Bonfil R. D., Toth M., Chang M., Fridman R., Mobashery S. (2005) Potent mechanism-based inhibitors for matrix metalloproteinases. J. Biol. Chem. 280, 33992–34002 [DOI] [PubMed] [Google Scholar]
- 42. Soejima K., Matsumoto M., Kokame K., Yagi H., Ishizashi H., Maeda H., Nozaki C., Miyata T., Fujimura Y., Nakagaki T. (2003) ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood 102, 3232–3237 [DOI] [PubMed] [Google Scholar]
- 43. Akiyama M., Takeda S., Kokame K., Takagi J., Miyata T. (2009) Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc. Natl. Acad. Sci. U.S.A. 106, 19274–19279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takeda S., Takeya H., Iwanaga S. (2012) Snake venom metalloproteinases: structure, function and relevance to the mammalian ADAM/ADAMTS family proteins. Biochim. Biophys. Acta 1824, 164–176 [DOI] [PubMed] [Google Scholar]
- 45. Manka S. W., Carafoli F., Visse R., Bihan D., Raynal N., Farndale R. W., Murphy G., Enghild J. J., Hohenester E., Nagase H. (2012) Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. U.S.A. 109, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olson M. W., Gervasi D. C., Mobashery S., Fridman R. (1997) Kinetic analysis of the binding of human matrix metalloproteinase-2 and -9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem. 272, 29975–29983 [DOI] [PubMed] [Google Scholar]
- 47. Wei S., Kashiwagi M., Kota S., Xie Z., Nagase H., Brew K. (2005) Reactive site mutations in tissue inhibitor of metalloproteinase-3 disrupt inhibition of matrix metalloproteinases but not tumor necrosis factor-α-converting enzyme. J. Biol. Chem. 280, 32877–32882 [DOI] [PubMed] [Google Scholar]
- 48. Lim N. H., Kashiwagi M., Visse R., Jones J., Enghild J. J., Brew K., Nagase H. (2010) Reactive-site mutants of N-TIMP-3 that selectively inhibit ADAMTS-4 and ADAMTS-5: biological and structural implications. Biochem. J. 431, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyamori H., Takino T., Kobayashi Y., Tokai H., Itoh Y., Seiki M., Sato H. (2001) Claudin promotes activation of pro-matrix metalloproteinase-2 mediated by membrane-type matrix metalloproteinases. J. Biol. Chem. 276, 28204–28211 [DOI] [PubMed] [Google Scholar]
- 50. Morrison C. J., Overall C. M. (2006) TIMP independence of matrix metalloproteinase (MMP)-2 activation by membrane type 2 (MT2)-MMP is determined by contributions of both the MT2-MMP catalytic and hemopexin C domains. J. Biol. Chem. 281, 26528–26539 [DOI] [PubMed] [Google Scholar]
- 51. Zarrabi K., Dufour A., Li J., Kuscu C., Pulkoski-Gross A., Zhi J., Hu Y., Sampson N. S., Zucker S., Cao J. (2011) Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 286, 33167–33177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dufour A., Zucker S., Sampson N. S., Kuscu C., Cao J. (2010) Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J. Biol. Chem. 285, 35944–35956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsumura S., Iwanaga S., Mochizuki S., Okamoto H., Ogawa S., Okada Y. (2005) Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J. Clin. Invest. 115, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Momi S., Falcinelli E., Giannini S., Ruggeri L., Cecchetti L., Corazzi T., Libert C., Gresele P. (2009) Loss of matrix metalloproteinase 2 in platelets reduces arterial thrombosis in vivo. J. Exp. Med. 206, 2365–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]