

Background: Caspase-8 signaling can mediate apoptosis or survival depending on the cellular context.
Results: Inflamed microglia survive because of caspase-dependent suppression of RIPK1-mediated necroptosis for survival.
Conclusion: Caspase inhibition rescues neurons by triggering necroptosis of neurotoxic inflamed microglia.
Significance: We demonstrate a novel neuroprotective mechanism of caspase inhibitors, namely killing of neurotoxic microglia by necroptosis.
Keywords: Apoptosis, Caspase, Cell Death, Microglia, Necrosis (Necrotic Death), Necroptosis, Neurodegeneration, Neuroinflammation, Neuron, Phagoptosis
Abstract
Microglia are resident brain macrophages, which can cause neuronal loss when activated in infectious, ischemic, traumatic, and neurodegenerative diseases. Caspase-8 has both prodeath and prosurvival roles, mediating apoptosis and/or preventing RIPK1-mediated necroptosis depending on cell type and stimulus. We found that inflammatory stimuli (LPS, lipoteichoic acid, or TNF-α) caused an increase in caspase-8 IETDase activity in primary rat microglia without inducing apoptosis. Inhibition of caspase-8 with either Z-VAD-fmk or IETD-fmk resulted in necrosis of activated microglia. Inhibition of caspases with Z-VAD-fmk did not kill non-activated microglia, or astrocytes and neurons in any condition. Necrostatin-1, a specific inhibitor of RIPK1, prevented microglial caspase inhibition-induced death, indicating death was by necroptosis. In mixed cerebellar cultures of primary neurons, astrocytes, and microglia, LPS induced neuronal loss that was prevented by inhibition of caspase-8 (resulting in microglial necroptosis), and neuronal death was restored by rescue of microglia with necrostatin-1. We conclude that the activation of caspase-8 in inflamed microglia prevents their death by necroptosis, and thus, caspase-8 inhibitors may protect neurons in the inflamed brain by selectively killing activated microglia.
Introduction
Inflammation and activated microglia are implicated in the neuronal death and loss occurring in infectious, ischemic, traumatic, and neurodegenerative diseases (1). Microglia are resident brain macrophages, which become inflammatory activated in response to infection or tissue damage, producing proinflammatory cytokines (such as tumor necrosis factor α, TNF-α), becoming phagocytic, and in some circumstances inducing neuronal death. Numerous mechanisms of neuronal death have been described, including phagoptosis, necrosis, and apoptosis. Phagoptosis is a recently described mode of cell death caused by phagocytosis of otherwise viable cells and can occur progressively in vivo and in vitro when inflamed microglia phagocytose viable neurons that temporarily expose phosphatidylserine due to mild oxidative stress (2–5). Necrosis is a type of cell death associated with rupture of the plasma membrane, and can be either “unregulated” or “regulated” (6). Apoptosis is controlled by the intrinsic or extrinsic pathways. The intrinsic pathway is mediated by Bcl-2 family proteins causing release of cytochrome c from mitochondria leading to activation of caspase-9 via formation of the cytosolic apoptosome complex with Apaf-1. The extrinsic pathway is triggered by binding of death ligands (such as TNF-α) to death receptors of the tumor necrosis family (TNF), which results in assembly of a receptor-associated complex, allowing activation of the initiator caspase-8. Once activated by their respective upstream signals, caspase-8 and -9 may cleave and activate downstream executioner caspases -3 and -7, which, in turn, cleave a plethora of target proteins resulting in apoptotic death (7).
Novel non-apoptotic roles for caspase-8 have been described recently. For example, in response to TNF-α, caspase-8 can play a prosurvival role in complex with binding partner FLIPL (FLICE-like inhibitory protein long). This non-apoptotic caspase-8 activity is required to suppress a necrosis-like death mediated by receptor-interacting protein kinases (RIPK)2 1 and 3 (8–10). The specific RIPK1 inhibitor necrostatin-1 inhibits any necrotic death that is dependent on RIPK1 activity, and this regulated form of necrosis has been referred to as necroptosis or programmed necrosis (6, 9). Necrostatin-1 was identified as a compound that reduced infarct size in a rodent model of stroke, indicating that necroptosis may contribute to neuronal death following ischemic injury (11). In addition to suppression of necroptosis, caspase-8 has been demonstrated to be required for inflammatory activation of BV2, a transformed microglial cell line (12). We were therefore interested to test whether inflammatory stimuli would cause caspase-8 activation in primary microglia and whether such activity would play a role in activation or survival of these cells. Here, we demonstrate that caspase-8 activity is required for the survival of inflammatory activated microglia and that caspase inhibitors can exert a neuroprotective effect by killing neurotoxic microglia by necroptosis.
EXPERIMENTAL PROCEDURES
All experiments were performed in accordance with the UK Animals (Scientific Procedures) Act (1986) and approved by the Cambridge University local ethical committee.
Cell Culture and Treatments
Mixed neuronal/glial cultures were prepared from the cerebella of postnatal day 5–7 rats as described previously (13) and were allowed to mature in vitro for at least 6 days prior to treatment. Pure microglia were prepared from mixed cortical astroglial cultures as described previously (2). Reagents were procured as follows: lipopolysaccharide (LPS, Sigma), lipoteichoic acid (LTA, InvivoGen), tumor necrosis factor-α (TNF-α, Sigma), Z-Val-Ala-d,l-Asp(OMe)-fluoromethylketone (Z-VAD-fmk, Bachem). Z-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone (IETD), Ac-Ile-Glu-Thr-Asp-CHO (IETD-CHO), Ac-Ile-Glu-Thr-Asp-7-amino-4-methylcoumarin (IETD-AMC), Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone (DEVD), and GM6001 and necrostatin-1 were from Enzo Life Sciences. Neuronal and microglial cell survival was quantified as described previously (2). Briefly, live mixed neuronal glial cultures were incubated in culture medium with 1 μg/ml propidium iodide (PI) and 5 μg/ml Hoechst 33342. Cells with PI-positive nuclei were counted as necrotic, whereas cells with PI-negative but condensed and fragmented nuclei as shown by Hoechst staining were counted as apoptotic. Microglia were visualized with Alexa Fluor 488-labeled isolectin-B4 (1 μg/ml, Invitrogen). Anti-TNF-α (Abcam) blocking antibody was Fc-blocked with an F(ab′)2 fragment antibody (Jackson ImmunoResearch Laboratories). The soluble TNF-α level was assessed in culture medium using Quantikine Elisa Kits (R&D Systems). Caspase-8 activity was measured using the fluorogenic substrate Ac-IETD-AMC following a protocol from Ref. 14.
Statistical Analysis
Statistical analysis was performed using SPSS software. The normality of data was verified using the Shapiro-Wilk test. In vitro data were analyzed using one-way analysis of variance and post hoc Bonferroni test. Results were considered significant if p < 0.05.
RESULTS
Inhibition of Caspase-8 Activity Selectively Kills Inflammatory Activated Microglia in Primary Neuronal/Glial Cultures
We previously demonstrated that microglia can phagocytose live neurons upon induction of inflammatory signals and that caspase inhibitors can prevent this method of neuronal loss, although the exact mechanism of neuroprotection was unclear (2, 13). Recently, caspase-8 has been found to suppress RIPK1-mediated necroptosis (15), raising the possibility that caspase inhibitors may induce death of microglial cells following inflammatory activation, thereby sparing the neurons. To test this possibility, we prepared mixed cultures of neurons, astrocytes, and microglia from rat cerebella, and treated cultures with LPS, a Toll-like receptor 4 (TLR4) agonist, to induce inflammatory activation of glial cells. Cultures were stained with Hoechst 33342 and PI to examine viability and Alexa Fluor 488-labeled isolectin-B4 to visualize microglia (Fig. 1A). After 72 h of LPS treatment, we observed a significant increase in microglial numbers as well as altered morphology indicative of microglial activation (Fig. 1, A–D). A numbers of astrocytes were unaffected after 72 h of LPS treatment (Fig. 1E). In the absence of LPS, treatment of cultures with the broad-spectrum caspase inhibitor Z-VAD-fmk had no effect on number or viability of any cell type (Fig. 1, B and E). However, addition of Z-VAD-fmk to cultures pretreated with LPS resulted in a large increase in the number of PI-positive dead microglia (Fig. 1B). This finding suggested that caspases were preventing necrosis in the activated microglia. To test which caspases were required for microglial survival in LPS-stimulated cultures, we used two relatively selective caspase inhibitors; DEVD-fmk, which preferentially inhibits caspase-3 and -7, and the caspase-8 inhibitor IETD-fmk. Addition of DEVD-fmk had no effect on microglial survival in the presence or absence of LPS (Fig. 1, A and C). In contrast, addition of IETD to LPS-treated cultures resulted in almost complete death of microglia (Fig. 1, A and D). Of note, cell death induced by caspase inhibition in LPS-treated cultures was restricted to microglia as neither astrocytic nor neuronal viability or numbers were affected in these conditions (Fig. 1E and data not shown). In sum, the data indicated that in the presence of LPS, inhibition of a select set of caspases, most likely caspase-8, induced death of microglia specifically.
FIGURE 1.

Inhibition of caspase-8 selectively kills microglia following inflammatory activation. A, caspase inhibitors Z-VAD and IETD induce necrosis of microglia in mixed neuronal glial cultures. Microglia are labeled in green using Alexa Fluor 488-labeled isolectin-B4, nuclei are labeled in blue using Hoechst 33342, and necrotic cells have pink nuclei due to PI labeling. Necrotic microglia are indicated with white arrows. B–D, Z-VAD (50 μm) and IETD (50 μm) but not DEVD (50 μm) induce death of microglia in the presence of LPS. Microglia were treated for 48 h with 100 ng/ml LPS and then for a further 24 h with indicated caspase inhibitor before quantifying death. Data are means ± S.E. of % dead microglia (n = 3). E, astrocytes are not killed by caspase-8 inhibitors and inflammatory activation. Astrocytes were quantified by nuclear morphology following the same treatments as described in B–D. LTA was used at 50 μg/ml, TNF-α was added at 50 ng/ml. Data are means ± S.E. (n = 3). F, Z-VAD inhibits LPS- and TNF-α-induced IETDase cleavage activity in pure microglia. Pure microglia were treated with LPS or TNF-α for 24 h followed by 2 h in the presence of Z-VAD (50 μm). IETDase activity was then measured in cell extracts. Data represent mean ± S.E. (n = 4). G and H, TNF-α and LTA treatment render microglia susceptible to Z-VAD-induced necrotic death (n = 3). NS, not significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Burguillos and colleagues (12) recently demonstrated that LPS treatment could induce caspase-8 activity in the immortalized microglial cell line BV2. To test whether this occurred in primary cells, we prepared cultures of pure microglia from rat cortices and then made cell extracts following LPS treatment to test for caspase-8 activity using the specific fluorogenic substrate IETD-AMC (12). Indeed, LPS treatment induced a 2.6-fold increase in IETDase activity in extracts from pure microglia (Fig. 1F), similar to that previously reported using the BV2 cell line (12). The IETDase activity was inhibited by nanomolar concentrations of the inhibitor IETD-CHO, whereas increased DEVDase activity extracted from apoptotic UV-treated microglia was not inhibited by IETD-CHO (data not shown), indicating that increased IETDase activity indeed reflected increased caspase-8 activity and not activity of caspase 3/7. Importantly, a short 2-h incubation of LPS-treated microglia with Z-VAD-fmk prior to measurement was sufficient to inhibit the IETDase activity induced by 24-h stimulation with LPS (Fig. 1F). Thus, LPS activated caspase-8 in microglia, and this activity was inhibited by both IETD and Z-VAD.
The above data indicated a prosurvival role for caspase-8 activity following stimulation of microglia with the TLR4 ligand LPS. We next tested whether a similar response occurred in microglia activated by other proinflammatory stimuli. In addition to TLR4, the Toll-like receptor family member TLR2 has been implicated in the pathogenesis of inflammatory neurodegenerative processes (13). We tested whether TLR2 ligation would induce a similar response to LPS using the TLR2 ligand LTA. Following pretreatment of cultures with LTA for a 24-h addition of Z-VAD for a further 24 h resulted in the death of almost all microglia, whereas astrocytes were not affected (Fig. 1, H and E). We also tested TNF-α as a proinflammatory stimulus as its levels are elevated in many brain pathologies (16). Addition of TNF-α to mixed cultures had little effect on microglial numbers or viability alone; however, TNF-α pretreatment for 24 h followed by addition of Z-VAD for 24 h killed most microglia in culture (Fig. 1G). In addition, a significant 2-fold induction of IETDase activity was detected in cell extracts prepared from pure microglia following TNF-α, which was inhibited by further addition of Z-VAD (Fig. 1F). Thus, our data indicate that prosurvival caspase-8 activity was induced in microglia stimulated by a TLR4 ligand, a TLR2 ligand, and TNF-α.
TLR Ligation Induces Prosurvival Caspase Signaling Independently of TNF-α Signaling
Inflammatory activation of microglia with TLR ligands such as LPS and LTA results in the release of soluble TNF-α into the surrounding medium (2). As we also observed that treatment with TNF-α alone was capable of inducing the prosurvival caspase activity, we postulated that the LPS- and LTA-induced effects were secondary to TNF-α release and binding to microglia. To test whether TLR signaling could induce prosurvival caspase activity independently of TNF-α, we used GM6001, a matrix metalloprotease and TNF-α converting enzyme inhibitor shown previously to prevent TNF-α processing and release (17). Although pretreatment of cultures with GM6001 prevented TNF-α release following LPS treatment (Fig. 2B), it did not prevent microglial death induced by Z-VAD in LPS- and GM6001-treated cultures (Fig. 2A). Furthermore, although a TNF-α blocking antibody prevented Z-VAD-induced microglial death in cultures treated with recombinant TNF-α, it did not prevent the microglial death in cultures treated with LPS and Z-VAD, despite the fact that LPS induced release of TNF-α at lower concentrations than that of recombinant TNF-α added in our experiments (250–300 pg/ml TNF-α release following LPS versus 50 ng/ml recombinant TNF-α added) (Fig. 2, B and C). We conclude that although TNF-α signaling is sufficient to induce a prosurvival caspase response in microglia, TNF-α release is not required for LPS to trigger a similar response.
FIGURE 2.

A, matrix metalloprotease and TNF-α-converting enzyme (TACE) inhibitor GM6001 does not prevent LPS-induced sensitization of microglia to caspase inhibition. Cultures were preincubated for 1 h with 25 μm GM6001 or dimethyl sulfoxide for controls prior to addition of 100 ng/ml LPS for 24 h and subsequent addition of 50 μm Z-VAD for a further 24 h. Data represent mean values, and error bars represent S.E. of % dead microglia (n = 4). B, supernatants were collected prior to quantification of death in A, and TNF-α release was measured by ELISA. Data represent mean values, and error bars are S.E. (n = 4). C, cultures were treated with LPS or recombinant TNF-α (50 ng/ml) in the presence of either 2 μg/ml normal IgG or 2 μg/ml TNF-α blocking antibody for 24 h prior to addition of Z-VAD and quantification of cell death a further 24 h later. Data represent mean values, and error bars represent S.E. of % dead microglia (n = 3). * = p < 0.05, ** = p < 0.01, *** = p < 0.001. NS, not significant.
Caspase Inhibition Kills Inflammatory Activated Microglia by RIPK1-dependent Necroptosis
Recently, a number of studies have demonstrated a prosurvival role for caspase-8 activated by the TNF-α receptor or TLR3 (15). Caspase-8 in “survival mode” has been demonstrated to suppress a novel form of necrotic death, termed necroptosis, by suppressing the pronecrotic action of RIPK1. Necroptosis is specifically defined in molecular terms as a form of regulated necrosis, which is dependent on RIPK1 activity, and as such, is inhibitable by compounds that inhibit RIPK1 kinase activity such as necrostatin-1 (6). Necrostatin-1 was first discovered in a screen for regulators of necrotic death, and its death inhibiting activity is wholly dependent on inhibition of RIPK-1 activity (9, 11, 28). As shown in Fig. 1A, we found that microglia killed by caspase inhibition, and inflammatory stimuli were always PI-positive (Fig. 1A), indicating necrosis, particularly as Z-VAD inhibits apoptosis, and thus, PI-positive cells were unlikely to appear due to secondary necrosis following apoptosis. To test whether inflammatory activated microglia died by RIPK1-dependent necroptosis following caspase inhibition, we tested the effects of the inhibitor of RIPK1, necrostatin-1. Cultures were stimulated with LPS or TNF-α for 24 h prior to addition of necrostatin-1 (30 μm), and then 1 h later, caspase inhibitors were added. Remarkably, necrostatin-1 treatment almost completely prevented Z-VAD-induced microglial death in LPS-treated cultures (Fig. 3A). In addition, necrostatin-1 also prevented IETD-induced death of LPS-activated microglia (Fig. 3B). Interestingly, we found that IETD, in contrast to Z-VAD, induced a low but consistent amount of microglial death in unstimulated cultures, which was also completely rescued by necrostatin-1 treatment (Fig. 3, B and D). This low level of necroptosis induced by IETD may reflect a low level activation of microglia in unstimulated cultures. Necrostatin-1 treatment was also sufficient to inhibit caspase inhibitor-induced microglial death in TNF-α stimulated cultures (Fig. 3, C and D). Furthermore, we tested an inactive analog of necrostatin-1, necrostatin-1I, which does not inhibit RIPK1 or necroptosis in other settings (28). In contrast to necrostatin-1, necrostatin-1-I was unable to protect microglia against LPS- and Z-VAD-induced death, further supporting the finding that caspase inhibitor induced death of activated microglia was indeed executed by necroptosis (Fig. 3E). We conclude that both LPS and TNF-α induce prosurvival caspase-8 signaling in microglia that acts by suppressing RIPK1-dependent necroptosis.
FIGURE 3.

Caspase inhibitor-induced death of inflamed microglia is inhibited by RIPK1 inhibitor necrostatin-1. A–D, cultures were treated with 100 ng/ml LPS (A and B) or 50 ng/ml TNF-α (C and D) for 24 h prior to 1 h treatment with 30 μm necrostatin-1 followed by addition of Z-VAD (A and C) or IETD (B and D) for a further 24 h and quantification of cell death. Data represent mean values, and error bars are S.E. of % dead microglia (n = 3). E, cultures were treated with 100 ng/ml LPS for 24 h prior to 1 h treatment with 30 μm necrostatin-1 or necrostatin-1-I (an inactive analog of necrostatin-1) followed by addition of Z-VAD (50 μm) for a further 24 h and quantification of cell death (n = 3). NS, not significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
The Neuroprotective Effect of Caspase Inhibitors in a Model of Inflammatory Neurodegeneration Is Mediated by Inducing Necroptosis of Microglia rather than Direct Inhibition of Apoptosis in Neurons
We next tested whether caspase-dependent neuroprotection in a model of inflammatory neurodegeneration might be dependent on necroptotic killing of microglia rather than via direct protective effects on neurons. We have previously described microglial-dependent neuronal death in vitro and in vivo that is triggered by a number of proinflammatory stimuli, including LPS, LTA, and Aβ peptide, with neuronal death occurring between 48–72 h after addition of the inflammatory stimulus (2–5). Interestingly treatment with either Z-VAD or IETD prevented microglial-mediated death of neurons induced by LPS treatment (Fig. 4, A and B; for microglial data from these experiments, see Fig. 1, B and D). In contrast, the selective caspase-3 and -7 inhibitor DEVD did not prevent LPS-induced primary phagocytosis, correlating with the fact that it did not induce microglial death in LPS-activated cultures (Figs. 1C and 4C). Remarkably, necrostatin-1 rescue of microglia from Z-VAD-induced necroptosis was sufficient to restore neuronal loss following 72 h of LPS treatment (Fig. 4, D and E). These data demonstrate that caspase inhibitors can exert a neuroprotective effect by inducing necroptosis of microglia.
FIGURE 4.

Caspase inhibitors that kill microglia are able to prevent LPS-induced neuronal death in mixed neuronal/glial cultures over a 72-h time period. A–C, cultures were treated with 100 ng/ml for 48 h prior to addition of 50 μm Z-VAD (A), IETD (B), or DEVD (C) for a further 24 h prior to quantification of neuronal survival and death. Microglial survival from these experiments is presented in Fig. 1, B--D. Data represent mean values, and error bars are the S.E. of neuronal survival (n = 3). D and E, cultures were treated as described above, but where indicated, 30 μm necrostatin-1 was added 1 h prior to Z-VAD. Quantification of neuronal survival and death is shown in D (error bars represent S.E. of neuronal survival), whereas microglial survival and death is shown in E (error bars represent S.E. of microglial death) (n = 3). * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
DISCUSSION
We have found that inhibition of caspase-8 causes necroptosis in inflammatory activated microglia but not in neurons, astrocytes, or unactivated microglia. In a disease model where inflamed microglia exert a neurotoxic action, caspase-8 inhibition was sufficient to cause neuroprotection in a manner dependent on necroptosis of microglia. We considered the possibility that inhibition of caspase-8 might in principle protect neurons from inflammatory neuronal loss by either by blocking inflammation in the glia, blocking death in the neurons, or inducing death in the microglia. However, we show that inflammatory activation was not impeded by the caspase inhibitors that induced necroptosis, as the amount of LPS-induced inflammation measured by TNF-α in the culture medium (Fig. 2B) was not reduced. Moreover, we added the caspase inhibitors 24–48 h after adding the LPS specifically to avoid blocking prior to inflammatory activation. Most importantly, we found that necrostatin-1, which prevented necroptosis of the microglia, fully restored the neuronal loss prevented by caspase inhibition. We have previously shown that specifically killing microglia (with leucine methyl ester) prevents LPS and LTA induced neuronal loss in the model used here (2, 5). Thus, we conclude that by killing microglia, caspase inhibition is sufficient to prevent inflammatory neuronal loss in this model.
Administration of the relatively caspase-3/-7 specific inhibitor DEVD-fmk was not sufficient to kill inflammatory activated microglia. In addition, we reported previously that the caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone inhibited etoposide-induced apoptosis in neurons but did not affect microglial activation, proliferation, or survival following addition of LPS and thus did not prevent LPS-induced neuronal loss (4). In contrast Z-VAD-fmk and IETD-fmk, both of which prevented an increase in caspase-8 IETDase activity in activated microglia, induced necroptosis of activated microglia. Together, these data support the conclusion that inhibition of caspase-8 activity (rather than caspase-3/-7) is required to induce necroptosis of inflamed microglia and raise the intriguing possibility that further development of caspase family member-specific inhibitors may allow selective modulation of neuronal death or inflamed microglial survival in vivo.
It was recently demonstrated that LPS activates microglia via caspase-8, and it was concluded that inhibition of caspase-8 blocks inflammatory microglia-mediated neurotoxicity by preventing inflammatory activation of microglia (12). Interestingly, in contrast to our findings, Burguillos et al. (12) did not note any microglial death in response to caspase-8 inhibition. However, they were using a microglial cell line, BV2, rather than primary microglia, and we have repeated our experiments in BV2 cells and find that caspase-8 inhibition does not induce death in LPS-activated BV2 cells (data not shown). This difference might be the result of BV2 transformation by v-myc and v-raf or other genetic changes. In vivo, caspase-8 inhibition protected neurons in a MPTP model of Parkinson disease and reduced the density of activated microglia. This finding is compatible with caspase-8 inhibition causing necroptosis of microglia (12). Burguillos et al. (12) also reported that caspase-8 was activated in microglia from patients with Alzheimer and Parkinson disease, supporting the possibility that caspase-8 inhibition might be beneficial in these neurodegenerative diseases.
Caspase-8 inhibition by IETD has been shown to be protective in vivo against the delayed neuronal loss that occurs after focal cerebral ischemia in rats (a model of stroke) and also to reduce tissue damage following hypoxicischemic brain injury in newborn rats (18, 19). We have shown here that the caspase inhibitor Z-VAD can induce necroptosis of inflamed microglia by caspase-8 inhibition, and indeed, this inhibitor has been shown to reduce neuronal loss in a number of stroke models (11, 20–22). In addition the pan-caspase inhibitor Q-VD-OPh can also inhibit caspase-8 and exert neuroprotective effects in a number of neurodegenerative disease models (23). The mechanisms by which these caspase inhibitors exert neuroprotective effects merits further investigation given the recent findings of Burguillos et al. (12) and those presented in this paper.
We and others found that LPS, LTA, and TNF-α caused limited caspase-8 activation in microglia without inducing microglial apoptosis (12). In certain cell lines, LPS can activate caspase-8 via TLR4 recruitment of TRIF (TIR-domain containing adapter-inducing interferon-β), and TRIF recruitment of RIPK1 to form a complex with FADD, which recruits and activates procaspase-8 (24). Under these conditions, inhibition of caspase-8 results in the formation of an alternative complex (IIb) containing at least RIPK1 and RIPK3, which triggers necroptosis, and this can be prevented by necrostatin-1, which blocks the kinase activity of RIPK1. Active caspase-8 can cleave RIPK1 to block its activity (25) and cleave the deubiquitinase CYLD to block its activation of RIPK1 (26). TLR3 ligation in the presence of suppressors of cIAP activity results in recruitment and activation of a preformed cytosolic complex containing caspase-8 and RIP1K, termed the ripoptosome, which is capable of inducing both apoptotic and necroptotic death (27). Thus, it is possible that ligation of TLR2 and TLR4 in microglia may result in engagement of a ripoptosome-like complex, whereupon caspase-8 inhibition deinhibits RIPK1, allowing necroptosis to occur (Fig. 5).
FIGURE 5.
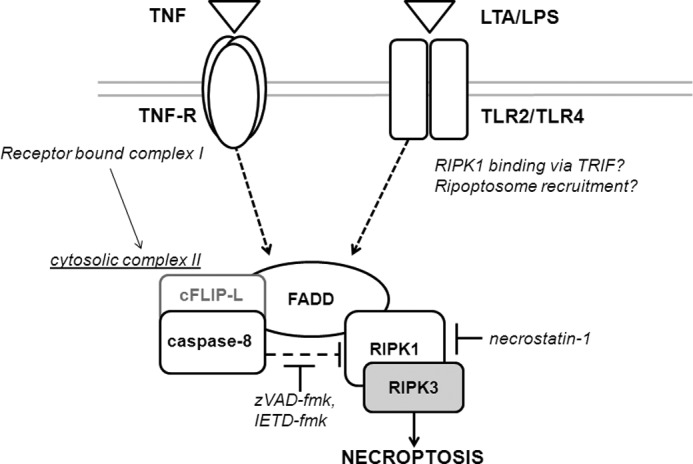
Inhibition of caspase-8 following TNF-receptor or TLR2/4 ligation results in execution of RIPK1-dependent necroptosis in microglia. Activation of TNF-receptor by TNF is known to induce formation of a large receptor-bound “complex I” that mediates activation of NF-κB. Complex I formation can be followed by formation in the cytosol of “complex II,” where caspase-8 in complex with cFLIP-L suppresses RIPK1 necroptotic activity. Inhibition of caspase-8 results in deinhibition of RIPK1 and formation of the necroptosis induction of necroptosis with RIPK3. TLR ligation also results in prosurvival caspase-8 activity, potentially by recruitment via the adaptor molecule TRIF of complex II or “ripoptosome”-like protein complexes. The RIPK-1 inhibitor necrostatin-1 prevents induction of necroptosis through inhibition of RIPK-1 kinase activity.
In conclusion, caspase-8 inhibition protects neurons in a model of inflammatory neuronal loss by specifically killing activated microglia by enabling necroptosis. It will be important to test whether caspase-8 inhibition is protective in vivo in models of neurological disease by similar mechanisms.
This work was supported by Wellcome Trust Grant RG50995.
- RIPK
- receptor-interacting protein kinase
- z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- IETD
- Z-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone
- AMC
- 7-amino-4-methylcoumarin
- PI
- propidium iodide
- TLR4
- Toll-like receptor 4
- DEVD
- Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone.
REFERENCES
- 1. Neher J. J., Brown G. C. (2007) Neurodegeneration in models of Gram-positive bacterial infections of the central nervous system. Biochem. Soc. Trans. 35, 1166–1167 [DOI] [PubMed] [Google Scholar]
- 2. Neher J. J., Neniskyte U., Zhao J. W., Bal-Price A., Tolkovsky A. M., Brown G. C. (2011) Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983 [DOI] [PubMed] [Google Scholar]
- 3. Neniskyte U., Neher J. J., Brown G. C. (2011) Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fricker M., Neher J. J., Zhao J. W., Théry C., Tolkovsky A. M., Brown G. C. (2012) MFG-E8 Mediates Primary Phagocytosis of Viable Neurons during Neuroinflammation. J. Neurosci. 32, 2657–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fricker M., Oliva-Martín M. J., Brown G. C. (2012) Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J. Neuroinflammation 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galluzzi L., Vitale I., Abrams J. M., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., Dawson T. M., Dawson V. L., El-Deiry W. S., Fulda S., Gottlieb E., Green D. R., Hengartner M. O., Kepp O., Knight R. A., Kumar S., Lipton S. A., Lu X., Madeo F., Malorni W., Mehlen P., Nuñez G., Peter M. E., Piacentini M., Rubinsztein D. C., Shi Y., Simon H. U., Vandenabeele P., White E., Yuan J., Zhivotovsky B., Melino G., Kroemer G. (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor R. C., Cullen S. P., Martin S. J. (2008) Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 [DOI] [PubMed] [Google Scholar]
- 8. Vanden Berghe T., van Loo G., Saelens X., Van Gurp M., Brouckaert G., Kalai M., Declercq W., Vandenabeele P. (2004) Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein FADD. J. Biol. Chem. 279, 7925–7933 [DOI] [PubMed] [Google Scholar]
- 9. Degterev A., Hitomi J., Germscheid M., Ch'en I. L., Korkina O., Teng X., Abbott D., Cuny G. D., Yuan C., Wagner G., Hedrick S. M., Gerber S. A., Lugovskoy A., Yuan J. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 [DOI] [PubMed] [Google Scholar]
- 12. Burguillos M. A., Deierborg T., Kavanagh E., Persson A., Hajji N., Garcia-Quintanilla A., Cano J., Brundin P., Englund E., Venero J. L., Joseph B. (2011) Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324 [DOI] [PubMed] [Google Scholar]
- 13. Kinsner A., Pilotto V., Deininger S., Brown G. C., Coecke S., Hartung T., Bal-Price A. (2005) Inflammatory neurodegeneration induced by lipoteichoic acid from Staphylococcus aureus is mediated by glia activation, nitrosative and oxidative stress, and caspase activation. J. Neurochem. 95, 1132–1143 [DOI] [PubMed] [Google Scholar]
- 14. Ciechomska I. A., Goemans C. G., Tolkovsky A. M. (2008) Molecular links between autophagy and apoptosis. Methods Mol. Biol. 445, 175–193 [DOI] [PubMed] [Google Scholar]
- 15. Green D. R., Oberst A., Dillon C. P., Weinlich R., Salvesen G. S. (2011) RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol. Cell 44, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venters H. D., Dantzer R., Kelley K. W. (2000) A new concept in neurodegeneration: TNFα is a silencer of survival signals. Trends Neurosci. 23, 175–180 [DOI] [PubMed] [Google Scholar]
- 17. Solorzano C. C., Ksontini R., Pruitt J. H., Auffenberg T., Tannahill C., Galardy R. E., Schultz G. P., MacKay S. L., Copeland E. M., 3rd, Moldawer L. L. (1997) A matrix metalloproteinase inhibitor prevents processing of tumor necrosis factor α (TNF α) and abrogates endotoxin-induced lethality. Shock 7, 427–431 [DOI] [PubMed] [Google Scholar]
- 18. Inoue S., Davis D. P., Drummond J. C., Cole D. J., Patel P. M. (2006) The combination of isoflurane and caspase 8 inhibition results in sustained neuroprotection in rats subject to focal cerebral ischemia. Anesth. Analg. 102, 1548–1555 [DOI] [PubMed] [Google Scholar]
- 19. Feng Y., Fratkin J. D., LeBlanc M. H. (2003) Inhibiting caspase-8 after injury reduces hypoxic-ischemic brain injury in the newborn rat. Eur. J. Pharmacol. 481, 169–173 [DOI] [PubMed] [Google Scholar]
- 20. Endres M., Namura S., Shimizu-Sasamata M., Waeber C., Zhang L., Gómez-Isla T., Hyman B. T., Moskowitz M. A. (1998) Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J. Cereb. Blood Flow Metab. 18, 238–247 [DOI] [PubMed] [Google Scholar]
- 21. Hara H., Friedlander R. M., Gagliardini V., Ayata C., Fink K., Huang Z., Shimizu-Sasamata M., Yuan J., Moskowitz M. A. (1997) Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. U.S.A. 94, 2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wiessner C., Sauer D., Alaimo D., Allegrini P. R. (2000) Protective effect of a caspase inhibitor in models for cerebral ischemia in vitro and in vivo. Cell Mol. Biol. 46, 53–62 [PubMed] [Google Scholar]
- 23. Venero J. L., Burguillos M. A., Brundin P., Joseph B. (2011) The executioners sing a new song: killer caspases activate microglia. Cell Death Differ. 18, 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaiser W. J., Offermann M. K. (2005) Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952 [DOI] [PubMed] [Google Scholar]
- 25. Rébé C., Cathelin S., Launay S., Filomenko R., Prévotat L., L'Ollivier C., Gyan E., Micheau O., Grant S., Dubart-Kupperschmitt A., Fontenay M., Solary E. (2007) Caspase-8 prevents sustained activation of NF-κB in monocytes undergoing macrophagic differentiation. Blood 109, 1442–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Donnell M. A., Perez-Jimenez E., Oberst A., Ng A., Massoumi R., Xavier R., Green D. R., Ting A. T. (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feoktistova M., Geserick P., Kellert B., Dimitrova D. P., Langlais C., Hupe M., Cain K., MacFarlane M., Häcker G., Leverkus M. (2011) cIAPs block ripoptosome formation, a RIP1/Caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Degterev A., Maki J. L., Yuan J. (2013) Activity and specificity of necrostatin-1, small molecule inhibitor of RIP1 kinase. Cell Death Differ. 20, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]