

Background: Diaminopimelate epimerase catalyzes a key step in the synthesis of meso-diaminopimelate and lysine.
Results: Solution and crystal studies show that diaminopimelate epimerase exists as an active dimer, whereas a monomeric mutant is catalytically inactive.
Conclusion: The diaminopimelate epimerase dimer is essential for function with evidence suggesting that dimerization attenuates subunit dynamics.
Significance: Structural insights into the design of antimicrobial agents to disrupt diaminopimelate epimerase dimerization are provided.
Keywords: Analytical Ultracentrifugation, Circular Dichroism (CD), Enzyme Mutation, Enzyme Structure, Molecular Dynamics, DAP Epimerase, Dimer, Sedimentation Equilibrium, Sedimentation Velocity
Abstract
Diaminopimelate (DAP) epimerase is involved in the biosynthesis of meso-DAP and lysine, which are important precursors for the synthesis of peptidoglycan, housekeeping proteins, and virulence factors in bacteria. Accordingly, DAP epimerase is a promising antimicrobial target. Previous studies report that DAP epimerase exists as a monomeric enzyme. However, we show using analytical ultracentrifugation, X-ray crystallography, and enzyme kinetic analyses that DAP epimerase from Escherichia coli exists as a functional dimer in solution and the crystal state. Furthermore, the 2.0-Å X-ray crystal structure of the E. coli DAP epimerase dimer shows for the first time that the enzyme exists in an open, active conformation. The importance of dimerization was subsequently probed by using site-directed mutagenesis to generate a monomeric mutant (Y268A). Our studies show that Y268A is catalytically inactive, thus demonstrating that dimerization of DAP epimerase is essential for catalysis. Molecular dynamics simulations indicate that the DAP epimerase monomer is inherently more flexible than the dimer, suggesting that dimerization optimizes protein dynamics to support function. Our findings offer insight into the development of novel antimicrobial agents targeting the dimeric antibiotic target DAP epimerase.
Introduction
Diaminopimelate (DAP)3 epimerase (EC 5.1.1.7) is a member of the pyridoxal 5′-phosphate-independent amino acid racemases. It catalyzes the stereoinversion of ll-DAP to meso-DAP in the lysine biosynthetic pathway in plants and bacteria (1–3). The products of this pathway, namely meso-DAP and lysine, are used in the cross-linking of the peptidoglycan cell wall of Gram-negative and Gram-positive bacteria, respectively. Lysine is also an important building block for the synthesis of housekeeping proteins and virulence factors (1–3). Consequently, DAP epimerase represents a promising target for the development of novel antimicrobial agents (4, 5).
DAP epimerase catalyzes the conversion of ll-DAP to meso-DAP using a two-base mechanism involving a pair of cysteine residues (Fig. 1) (6–8). The first cysteine is present in the thiolate form and acts as a base and abstracts a proton from ll-DAP, whereas the second cysteine acts as an acid and reprotonates the molecule, giving meso-DAP. The reaction goes through a planar carbanion-like transition state (Fig. 1).
FIGURE 1.
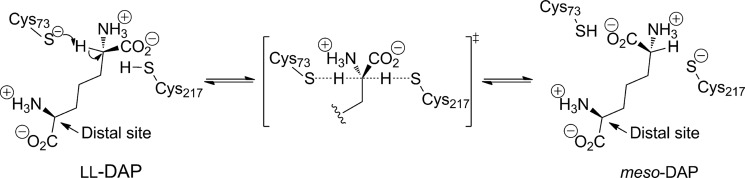
Reaction catalyzed by DAP epimerase and its mechanism.
Previous studies of DAP epimerase have reported a monomeric enzyme that adopts a symmetrical α/β domain structure with each domain contributing one of the two key active site cysteine residues located in the interdomain cleft (8–12). This unique fold was first observed for the Haemophilus influenzae enzyme and is now referred to as the DAP epimerase-like fold (9). Since the first structure of DAP epimerase was solved in 1998 (9), a number of structures have been determined that adopt the DAP epimerase-like fold. These include proline racemase (13), the isomerases PrpF (14, 15) and 3-methylitaconate Δ-isomerase (16), PhzF (17), and proteins of unknown or unconfirmed function (18, 19). Interestingly, the majority of these proteins have been found to adopt a dimeric quaternary structure with dimerization occurring through the N-terminal domain (13–15, 17–19).
Although previous studies of DAP epimerase suggest that the enzyme is monomeric (9, 11, 12), computational analyses of the crystal structures using Protein Interfaces, Surfaces and Assemblies (PISA) (20) suggest that the enzyme could adopt a dimeric architecture similar to the dimer observed in other DAP epimerase-like fold proteins. We therefore set out to examine the quaternary structure of bacterial DAP epimerase both in solution and the crystal state using the enzyme from Escherichia coli. In addition, we compared the function of the wild-type dimer and a monomeric mutant of E. coli DAP epimerase generated via site-directed mutagenesis.
Our studies reveal that DAP epimerase is a dimer in solution and the crystal state. The high resolution crystal structure of E. coli DAP epimerase shows that the dimeric enzyme adopts an open, active conformation not previously reported in the literature. Furthermore, mutagenesis studies targeting Tyr268 at the dimer interface yield a monomeric mutant (Y268A) that is catalytically inactive, demonstrating for the first time that dimerization of DAP epimerase is critical for catalysis.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
E. coli DAP epimerase was expressed and purified as described in Hor et al. (21), and the H. influenzae ortholog was expressed and purified as described in Cirilli et al. (9) with the exclusion of the ammonium sulfate precipitation step.
Generation of E. coli DAP Epimerase Mutant
The point mutation in the E. coli dapF gene contained in pET11a (21) was introduced using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The primer set 5′-GGCGGTACATGTCTACGCCGGATTTATTCATCTAC-3′ and 5′-GTAGATGAATAAATCCGGCGTAGACATGTACCGCC-3′ was designed to introduce the Y268A amino acid substitution. Mutagenesis was performed according to the manufacturer's instructions with successful mutation confirmed by dideoxynucleotide sequencing. The mutant was expressed at 16 °C in autoinduction medium (22) and purified using the same protocol as for the wild-type enzyme (21).
Mass Spectrometry
Mass spectrometric analyses were performed on an Agilent 6220 Accurate-Mass TOF LC/MS mass spectrometer coupled to an Agilent 1100 LC system (Agilent, Palo Alto, CA) as described previously (23, 24). Protein samples were left in their original buffer (typically 20 mm Tris, 150 mm NaCl, pH 8.0) and were loaded onto an Agilent Poroshell 300SB-C18 2.1 × 75-mm, 5-μm reverse phase column attached to the mass spectrometer.
Circular Dichroism Spectroscopy
CD spectroscopy was performed using an AVIV 410-SF CD spectrometer as reported previously (25–27). Wavelength spectra were collected between 190 and 250 nm in 20 mm Tris, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, pH 7.8 with 0.15 mg ml−1 enzyme using 1-mm quartz cuvettes with a step size of 0.5 nm and 2-s averaging time. Data were analyzed using the CONTINLL algorithm from the CDPro software package (28) using the SP43 database.
Analytical Ultracentrifugation
Absorbance-based sedimentation velocity and sedimentation equilibrium experiments were performed in a Beckman XL-I analytical ultracentrifuge with a four-hole An-60 Ti or eight-hole An-50 Ti rotor at 20 °C as described previously (26, 27, 29, 30). For sedimentation velocity experiments, double sector quartz cells were loaded with 380 μl of sample and 400 μl of reference (20 mm Tris, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, pH 7.8) with data collected at 40,000 or 50,000 rpm. For sedimentation equilibrium experiments, quartz cells were loaded with 100 μl of sample and 120 μl of reference (20 mm Tris, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, pH 7.8) with data collected at 10,000 and 16,000 rpm. Sedimentation velocity data were fitted with a continuous size distribution model using SEDFIT (31) or the enhanced van Holde-Weischet method (32) using UltraScan III (33, 34). Sedimentation equilibrium data were fitted to a monomer-dimer self-association model using SEDPHAT (35). Bead modeling was conducted using the SOMO method incorporated in the UltraScan III software suite (36, 37).
Enzyme Kinetic Assays
Enzyme activity of DAP epimerase was determined using the modified DAP epimerase-DAP dehydrogenase coupled spectrophotometric assay (38). Briefly, to an assay containing buffer, DAP and NADP+ were added to DAP dehydrogenase whereby an increase at 340 nm was observed until a plateau was attained that corresponded to the conversion of meso-DAP to tetrahydrodipicolinate until meso-DAP was depleted. DAP epimerase was then added to the resulting mixture, and the resulting rate was recorded. Assays were performed at 30 °C in a temperature-controlled Varian Cary 4000 UV spectrophotometer and incubated for 12 min before initiation of the reaction with enzyme. Assays were performed in duplicate or triplicate. A typical assay contained 100 mm Tris, pH 7.8, 0.1 mm ll-DAP, 0.44 mm NADP+, 1 mm DTT, 1.8 μm DAP dehydrogenase, and DAP epimerase.
X-ray Crystallography
The crystallization, data collection, and structure determination of wild-type E. coli DAP epimerase have been described previously (21). Y268A was crystallized using the hanging drop vapor diffusion method with drops containing 2 μl of protein solution (8.0 mg ml−1 in 20 mm Tris, 5 mm DTT, 5 mm tris(2-carboxyethyl)phosphine, pH 7.8) and 2 μl of precipitant solution (0.2 m sodium iodide, 18% (w/v) PEG 3350, 0.1 m Bis-tris propane, pH 6.5, 5 mm DAP) at 20 °C.
For x-ray data collection, the Y268A crystal was soaked in cryoprotectant solution containing reservoir solution made up in glycerol (20%, v/v) and directly flash cooled in liquid nitrogen. Intensity data were collected at −163 °C at the Australian Synchrotron (MX2 beamline). Data were collected in 0.5° oscillations for 250° using an ADSC Q315r image plate detector positioned 300 mm from the crystal with an exposure time of 0.5 s and 80% attenuation. The diffraction data were processed using the programs MOSFLM (39) and SCALA (40). Although 250° of data were initially collected, the resolution and the quality of the diffraction data decreased (as judged by an increased batch Rmerge); as such, only the first 80° data were analyzed. The Y268A structure was solved by molecular replacement using Phaser (41) using the wild-type E. coli DAP epimerase (Protein Data Bank code 4IJZ) as the search model.
Structural refinement of both wild-type and mutant structures was performed using REFMAC5 (40) and iterative model building with WINCOOT (42). Translation Libration Screw-motion refinement in REFMAC (43) was applied in the final rounds of refinement using three groups as determined using TLSMD (44, 45). Structure quality was assessed by the program MolProbity (46). X-ray data collection and refinement statistics for wild-type E. coli DAP epimerase and Y268A mutant structures are shown in Table 1. Interface analysis used PISA at the European Bioinformatics Institute authored by Krissinel and Henrick (20).
TABLE 1.
Refinement statistics for E. coli DAP epimerase wild-type structure (Protein Data Bank code 4IJZ) and Y268A mutant (Protein Data Bank code 4IK0)
r.m.s.d., root mean square deviation.
| Wild-type | Y268A | |
|---|---|---|
| Data collection | ||
| Space group | P41212 | P41212 |
| Cell parameters | ||
| a, b, c (Å) | 89.4, 89.4, 179.6 | 89.2, 89.2, 179.5 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 44.69–2.00 (2.11–2.00) | 51.59–2.05 (2.16–2.05) |
| Observed reflections | 215,760 (29,012) | 290,926 (42,700) |
| Unique reflections | 47,663 (6,683) | 46,324 (6,635) |
| Completeness (%) | 96.1 (93.7) | 100.0 (100.0) |
| Rmergea | 0.077 (0.339) | 0.126 (0.577) |
| Rpimb | 0.039 (0.137) | 0.055 (0.246) |
| Rrim | 0.087 (0.385) | 0.137 (0.628) |
| Mean I/σ(I) | 13.0 (4.2) | 10.4 (3.2) |
| Redundancy | 4.5 (4.3) | 6.3 (6.4) |
| Wilson B value (Å2) | 23.4 | 25.0 |
| Molecules in asymmetric unit | 2 | 2 |
| VM (Å3 Da−1) | 2.89 | 2.89 |
| Solvent content (%) | 57 | 57 |
| Refinement | ||
| Resolution | 44.37–2.00 (2.05–2.00) | 49.68–2.05 (2.10–2.05) |
| Rworkc | 0.161 (0.215) | 0.176 (0.210) |
| Rfreec,d | 0.200 (0.260) | 0.217 (0.239) |
| No. atoms | ||
| Protein | 4,289 | 4,240 |
| Ligands | 12 | 13 |
| Water | 516 | 395 |
| Mean B value (Å2) | ||
| Main chain | 22.4 | 23.6 |
| Side chain | 25.9 | 26.1 |
| Ligands | 46.0 | 52.7 |
| Water | 37.3 | 35.5 |
| r.m.s.d. from ideality | ||
| Bond length (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.64 | 1.65 |
| Ramachandran statistics (%) | ||
| Most favored regions | 97.3 | 98.2 |
| Additionally allowed regions | 2.7 | 1.6 |
| Disallowed regions | 0.0 | 0.2 |
a Rmerge = ΣhΣi|Ii(h) − I(h)|/ΣhΣiIi(h).
b Rpim = Σh[1/(N − 1)]½Σi|Ii(h) − I(h)|/ΣhΣiIi(h).
c R = Σ‖Fobs| − |Fcalc‖/Σ|Fobs| where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
d Rfree was calculated with 5% of the diffraction data and was selected randomly and omitted from the refinement. For the Y268A structure, the same 5% set of diffraction data was used as the wild-type E. coli DAP epimerase structure.
Molecular Dynamic Simulations
Simulations of the wild-type E. coli DAP epimerase crystal structure dimer and the monomer corresponding to chain A of the dimer were performed in the NPT ensemble at a temperature of 298 K and pressure of 1 atm using the molecular dynamics program NAMD (47). The CHARMM force field was used with CMAP corrections to backbone dihedral interactions (48). Cys-73 was deprotonated using the patch of Foloppe et al. (49). All simulations were solvated with TIP3P water, and ions were added at a concentration of 150 mm NaCl. A simulation time step of 2 fs was used with a total simulation length of 25 ns.
RESULTS
DAP Epimerase Is Dimeric in Solution
E. coli DAP epimerase was expressed and purified as described elsewhere (21). The enzyme was initially characterized by electrospray ionization-TOF MS to show that the mass of the recombinant protein (31.031.1 Da) was consistent with that of the amino acid sequence. CD spectroscopy showed that the enzyme was folded (Table 2), and subsequent analysis using the DAP epimerase-DAP dehydrogenase coupled assay (38) indicated that the enzyme was active (data not shown).
TABLE 2.
Secondary structure analysis of E. coli wild-type and mutant DAP epimerase
CD data were analyzed using CDPro (28) with the CONTINLL algorithm and the SP43 reference database. r.m.s.d., root mean square deviation.
| α-Helix | β-Strand | Turn | Random | r.m.s.d. | |
|---|---|---|---|---|---|
| % | % | % | % | ||
| WT | 18 | 31 | 21 | 30 | 0.059 |
| Y268A | 17 | 31 | 23 | 29 | 0.069 |
The quaternary structure of E. coli DAP epimerase was assessed by sedimentation velocity experiments performed in the analytical ultracentrifuge. The raw data (Fig. 2A) were fitted to an enhanced van Holde-Weischet model (32), indicating that E. coli DAP epimerase sediments with a standardized sedimentation coefficient (s20,w) of 4.3 S (Fig. 2B). This was complementary to the result obtained using the c(s) distribution (31, 50) (see Fig. 6A). Conversion of the c(s) profile to a c(M) distribution yields an apparent molecular mass of 61.3 kDa, which is consistent with E. coli DAP epimerase being a dimer in solution (Table 3).
FIGURE 2.

Sedimentation velocity analysis of E. coli and H. influenzae DAP epimerase. A, E. coli DAP epimerase sedimentation velocity raw data. Absorbance at 227 nm plotted as a function of radial position from the axis of rotation (cm) is shown. The raw data are presented as open circles. B, enhanced van Holde-Weischet integral distribution plot from extrapolation of E. coli DAP epimerase sedimentation velocity raw data shown in A. C, H. influenzae DAP epimerase sedimentation velocity raw data. Absorbance at 226 nm plotted as a function of radial position from the axis of rotation (cm) is shown. The raw data are presented as open circles. D, enhanced van Holde-Weischet integral distribution plot from extrapolation of H. influenzae DAP epimerase sedimentation velocity raw data shown in C.
FIGURE 6.

Sedimentation velocity and catalytic activity analyses of E. coli DAP epimerase wild type and Y268A mutant. A, continuous sedimentation coefficient (c(s)) distribution plotted as a function of sedimentation coefficient (s20,w) for wild-type enzyme (solid line) and Y268A (dashed line). Residuals are plotted for wild-type (black) and Y268A (gray). B, rate versus enzyme concentration plots of wild-type (dark circles) and Y268A (light circles) E. coli DAP epimerase. The initial rate (i.e. change in absorbance (Abs)) of the DAP epimerase reaction is plotted as a function of enzyme concentration. Kinetics were performed at a ll-DAP concentration of 0.140 and 0.0246 mm for Y268A and wild-type E. coli DAP epimerase, respectively. Error bars represent the S.D. of duplicate measurements.
TABLE 3.
Summary of hydrodynamic properties of E. coli DAP epimerase and Y268A mutant
| DAP epimerase | Theoretical molecular massa | s20,w | f/f0b | Experimental molecular massc | KD2→1 |
|---|---|---|---|---|---|
| kDa | S | kDa | |||
| E. coli | 62.0 | 4.3 | 1.3 | 61.3 | 22 nm |
| H. influenzae | 60.5 | 4.1 | 1.3 | 59.3 | NDd |
| Y268A | 61.9 | 2.9 | 1.2 | 29.9 | 54 μm |
a Molecular mass of a dimer based on amino acid composition.
b Frictional ratio derived using the ν bar method.
c Molecular mass derived experimentally using c(M) analysis.
d Not determined.
To determine the dimerization affinity of wild-type E. coli DAP epimerase, sedimentation equilibrium experiments were performed in the analytical ultracentrifuge at two rotor speeds (10,000 and 16,000 rpm) and various protein concentrations (0.32, 0.64, 1.9, and 5.8 μm). The resulting data were fitted by global nonlinear regression analyses to a monomer-dimer equilibrium, which yielded a dimer-monomer dissociation constant (KD2→1) of 22 nm (Table 3 and Fig. 3).
FIGURE 3.

Sedimentation equilibrium analysis of E. coli DAP epimerase. Sedimentation equilibrium data collected at 10,000 (triangles) and 16,000 rpm (circles) overlaid with the global nonlinear regression best fit to a monomer-dimer self-association model (global reduced χ2 = 0.104) are shown. Residuals are displayed above each panel for 10,000 rpm data (triangles) and 16,000 rpm data (circles). The experiment was conducted at initial protein concentrations of 0.32 (A) 0.64 (B), 1.9 (C), and 5.8 μm (D), yielding a KD2→1 of 22 nm.
In addition, sedimentation velocity experiments on the most well characterized DAP epimerase from H. influenzae (8–10, 51, 52) showed that this enzyme is also dimeric in solution with a standardized sedimentation coefficient (s20,w) of 4.1 S (Table 3 and Fig. 2, C and D). This result is consistent with the quaternary structure of E. coli DAP epimerase (and other proteins that adopt the DAP epimerase-like fold) and suggests that a dimer may be the true biologically relevant form.
E. coli DAP Epimerase Crystal Structure
We next set out to generate high resolution structural information for the E. coli DAP epimerase dimer. The recombinant enzyme was crystallized as reported recently (21), and the x-ray crystal structure was determined in the space group P41212 to a resolution of 2.0 Å by molecular replacement (Protein Data Bank code 4IJZ) (Fig. 4). The Ramachandran plot has 97.3% of residues in the most favored regions, 2.7% in the additionally allowed regions, and no residues in the disallowed regions. As expected, the structure adopts the DAP epimerase-like fold similarly to several other homologs (9–12).4 The monomer comprises two symmetrical domains with each domain consisting of a central α-helix surrounded by eight β-strands (Fig. 4B). These domains show pseudosymmetry with the N-terminal domain (residues 1–119 and 263–274) and C-terminal domain (residues 120–262) and are structurally similar with an root mean square deviation of 2.67 Å over 93 α-carbon pairs with ∼18% sequence identity between the two domains. Consistent with solution study results, E. coli DAP epimerase crystallized as a dimer in the asymmetric unit with the dimeric architecture structurally akin to DAP epimerase-like proteins with oligomerization taking place at the N-terminal domain (Fig. 4A).
FIGURE 4.

Crystal structure of wild-type E. coli DAP epimerase. A, dimer in the asymmetric unit. Monomers are in green and purple with the N-terminal domain in the dark color and the C-terminal domain in the light color. B, two symmetrical domains of E. coli DAP epimerase. The N-terminal domain is in dark green, the C-terminal domain is in light green, and active site Cys residues are in blue. C, overlay of the E. coli (Protein Data Bank code 4IJZ; green) and B. anthracis (Protein Data Bank code 2OTN; blue) DAP epimerase active sites. D, dimer interface in surface representation. E, dimer interface in schematic representation. Hydrogen bonds (red), salt bridges (blue), and other interface residues (yellow) are shown.
Significantly, the structure of E. coli DAP epimerase also crystallized in the open conformation with the two active site Cys residues existing in the non-disulfide-bonded reduced form with 9.5 and 13.3 Å between Cys residues in monomers A and B, respectively (Fig. 4, B and C). Accordingly, this crystal structure represents an active conformation of the enzyme (Fig. 4, B and C). This is in contrast to the inactive conformation observed in H. influenzae (Protein Data Bank codes 1BWZ and 1GQZ) and Mycobacterium tuberculosis DAP epimerase (Protein Data Bank code 3FVE) where a disulfide bridge between the active site Cys residues is observed (9–11). Only one other DAP epimerase structure, from Bacillus anthracis (Protein Data Bank code 2OTN), has been crystallized in the open conformation.4
Interestingly, SOMO bead model analysis (36, 37) of the dimer results in a computed sedimentation coefficient of 4.3 S and a frictional ratio of 1.3, which are identical to the experimentally determined values (Table 3). This suggests that the open dimer conformation observed in the crystal structure also forms in aqueous solution.
Active Site
The active site of E. coli DAP epimerase resides in a cleft between the two domains with each domain contributing one of the cysteine residues important for catalysis (Fig. 4, B and C). Superimposition of the active sites of E. coli and B. anthracis DAP epimerase structures indicates that the majority of active site residues are similarly oriented (Fig. 4C). However, minor changes are observed for Cys73, Gly74, and Asn75. The different arrangement of these residues can be explained by their location in the sequence. Cys73–Asn75 are located on a dynamic loop (51), which means there is increased flexibility in this region. This is supported by the higher temperature factor of these three residues of 35.2 Å2 compared with the average B factor for all residues of 21.8 Å2.
Dimer Interface
As for other proteins that adopt the DAP epimerase-like fold, the dimerization interface of E. coli DAP epimerase occurs between the N-terminal domains of the two monomers. The dimer interface of E. coli DAP epimerase buries 765 Å2 per monomer, which represents 6.1% of the total surface area of each monomer. This is similar to the H. influenzae and B. anthracis orthologs but is markedly less than other DAP epimerase-like fold proteins (Table 4). Analysis by PISA (20) reveals that the E. coli DAP epimerase dimer interface is composed of 13 hydrogen bonds and seven salt bridges. The locations of these interactions are depicted in Fig. 4, D and E.
TABLE 4.
Comparison of the dimer interface of proteins that adopt the DAP epimerase-like fold
Analysis was carried out using PISA (20).
| Protein | Protein Data Bank code | Buried surface area per monomer | Buried surface area |
|---|---|---|---|
| Å2 | % of total surface area | ||
| E. coli DAP epimerase | 4IJZ | 765 | 6.1 |
| H. influenzae DAP epimerase (inactive) | 1BWZ | 688 | 5.6 |
| H. influenzae DAP epimerase (open) | 2Q9J | 721 | 7.7 |
| M. tuberculosis DAP epimerase | 3FVW | 1086 | 8.9 |
| B. anthracis DAP epimerase | 2OTN | 857 | 5.9 |
| Shewanella oneidensis PrpF | 2PVZ | 2374 | 14.6 |
| Pseudomonas fluorescens PhzF | 1T6K | 1292 | 10.5 |
| Trypanosoma cruzi proline racemase | 1W62 | 1640 | 10.1 |
| E. coli YddE | 1QYA | 1045 | 9.7 |
The E. coli DAP epimerase dimer interface is contributed by three areas of the protein: residues 3–12 (N terminus), residues 36–43 (loop), and residues 266–273 (C terminus) (Fig. 4E). Residues of the C terminus, which form part of the N-terminal domain and represent the final β-strand of the monomer, interact in an antiparallel manner, creating a continuous β-sheet that weaves through the entire E. coli DAP epimerase dimer (Fig. 4A).
Engineering a Monomeric Variant
To probe the importance of dimerization, a point mutation targeting Tyr268 at the dimer interface was created. Care was taken in selecting Tyr268 because this residue is not close to the active site and would be unlikely to perturb the catalytic function of the enzyme. Interestingly, Tyr268 is involved in hydrogen bonding at the dimer interface with the same residue from the neighboring monomer. Therefore, Tyr268 was mutated to Ala, yielding the point mutant referred to herein as Y268A.
Y268A was found to be of the expected molecular mass (30,940.0 Da) as determined by mass spectrometry and shown by CD spectroscopy to have secondary structure proportions similar to those of the wild-type enzyme (Table 2). In addition, the crystal structure of Y268A (Protein Data Bank code 4IK0) was determined to 2.05-Å resolution, clearly showing that the Tyr to Ala mutation at position 268 was successful (Fig. 5A). In addition, x-ray crystallographic studies show that Y268A retains the secondary and tertiary structure of the wild-type enzyme with superimposition of the mutant and wild-type enzymes yielding a root mean square deviation of 0.220 Å over all 512 α-carbon atoms (Fig. 5B). In essence, the structure of the Y268A mutant in the crystal state is identical to that of the wild-type enzyme. Importantly, this demonstrates that the mutation does not alter the fold of the protein.
FIGURE 5.

X-ray crystal structure of Y268A. A, electron density map showing atoms around the mutated residue. The 2Fo − Fc electron density is displayed in gray and contoured to 1σ. B, overlay of Y268A (pink) and wild-type E. coli DAP epimerase (green).
The quaternary structure of Y268A was next investigated using sedimentation velocity analysis in solution. Analysis using the c(s) distribution (30, 31, 50) yielded a standardized sedimentation coefficient of 2.9 S, which is significantly lower than that of the wild-type enzyme at an equivalent initial protein concentration (Fig. 6A). Conversion of the c(s) distribution to a c(M) distribution shows that Y268A has an apparent molecular mass of 29.9 kDa, which correlates to a monomer (Table 3), suggesting that the propensity of the point mutant to dimerize has been significantly attenuated.
To quantitate the loss of dimerization affinity, sedimentation equilibrium experiments were performed at various initial protein concentrations of Y268A ranging from 1.6 to 15.9 μm. The resulting data generated at two rotor speeds (10,000 and 16,000 rpm) were globally fitted to a monomer-dimer equilibrium model. The resulting global nonlinear regression best fit yielded a dimer-monomer dissociation constant (KD2→1) of 54 μm (Table 3). This result indicates that the Ala substitution at position 268 has weakened the dimerization affinity by ∼2,400-fold. Accordingly, the Y268A point mutant enables the structure-function relationship of a monomer to be characterized in comparison with the wild-type dimer.
Y268A Shows Attenuated Activity
The enzymatic activity of Y268A was compared with that of wild-type E. coli DAP epimerase by screening at a fixed concentration of 2.5 μg ml−1 (∼80 nm). This enzyme concentration is significantly lower than the KD2→1 of the Y268A point mutant but higher than the wild-type dimerization dissociation constant. Interestingly, the catalytic activity of Y268A is markedly attenuated compared with the wild-type enzyme (∼1% at 80 nm enzyme concentration), which indicates that the E. coli DAP epimerase dimer is significantly more active than the monomeric form.
To support this inference, the relationship between catalytic activity and enzyme concentration for Y268A and wild-type enzyme was investigated. Not surprisingly, the plot of rate versus enzyme concentration for Y268A is nonlinear showing upward curvature, indicating that the mutant becomes more active as the enzyme concentration is increased (Fig. 6B). This phenomenon has been observed for other enzymes that show a dependence on quaternary structure for catalytic function (26, 27). By contrast, the plot of initial rate versus enzyme concentration for the wild-type enzyme is linear (Fig. 6B).
DISCUSSION
The DAP epimerase-fold was first identified in 1998 when the first structure of DAP epimerase from H. influenzae was determined (9). Since then a number of other proteins have been found to adopt this fold (13–19). Interestingly, the majority of DAP epimerase-like proteins have been reported to be dimeric, whereas DAP epimerase enzymes have been reported to be monomeric (9, 11, 12). Accordingly, the first aim of this study was to ascertain the biologically relevant quaternary structure of DAP epimerase in solution and the crystal state.
It was determined through sedimentation analyses that both E. coli and H. influenzae DAP epimerase are in fact dimers in solution at concentrations of 100–150 μg ml−1 (Fig. 2). Indeed, sedimentation equilibrium analyses show that the wild-type E. coli enzyme exists as a tight dimer with a dimer-monomer dissociation constant of 22 nm (Fig. 3 and Table 3).
In addition, the x-ray crystal structure of E. coli DAP epimerase determined to a resolution of 2.0 Å was crystallized in the active, open conformation, showing that the enzyme also exists as a dimer in the crystalline state (Fig. 4). The dimer adopts the same architecture observed in other proteins that adopt the DAP epimerase-like fold with dimerization facilitated by contacts in the N-terminal domain. In addition, although in previous studies H. influenzae DAP epimerase crystallized as a monomer in the asymmetric unit (8–10, 51), a dimer with the same architecture as that of E. coli DAP epimerase observed in this study (and other DAP epimerase-like fold proteins) can be generated using the appropriate symmetry operations. We believe that this dimer constitutes the catalytically active and biologically relevant form of the enzyme.
These results taken as a whole strongly suggest that DAP epimerase is in fact a dimer and that the oligomeric state of DAP epimerase has been misreported in the literature (9, 11, 12). Given that PISA analysis (20) indicates that DAP epimerase has an appreciably smaller dimer interface compared with other DAP epimerase-like proteins (Table 4), the significance of this dimerization interface and hence the oligomeric state of the protein may have been overlooked.
To probe the importance of dimerization, the crystal structure of E. coli DAP epimerase was used to design a monomeric mutant via site-directed mutagenesis targeting Tyr268, a key residue that forms significant stabilizing interactions at the dimer interface. Results show that the mutagenesis was successful, resulting in a mutant enzyme (Y268A) with a ∼2,400-fold loss in dimerization affinity compared with the wild-type enzyme (Table 3). Interestingly, enzyme kinetic analysis indicates that Y268A is significantly less active than the wild-type dimer (Fig. 6B). This cannot be due to loss in secondary or tertiary structure because CD spectroscopy (Table 2) and x-ray crystallography (Fig. 5) studies demonstrate that the secondary and tertiary structure of the mutant is preserved (Fig. 5). Accordingly, we propose that the loss of function found in Y268A is due to perturbation of the dimeric quaternary structure. However, because the active site in each monomer is located between the N- and C-terminal domains (Fig. 4B) and is distal from the dimerization interface (∼22 Å between Tyr268 and the closest active site Cys residue), loss of function in the monomeric state must be due to long distance effects.
We therefore hypothesize that dimerization plays an important role in optimizing conformational dynamics critical for function. In DAP epimerase, the binding of ligand triggers a large conformational change, bringing the N- and C-terminal domains together (8, 51). This event leads to the encapsulation of ligand and the reorganization of the active site with the formation of a network of hydrogen bonds at the catalytic center and the optimal positioning of the active site Cys residues (Fig. 1) (8, 51). The correct positioning of the two active site Cys residues (Cys73 and Cys217; E. coli numbering) is critical for function of the enzyme. If these two Cys residues are too far apart catalysis cannot take place; alternatively, if the two Cys residues are too close together disulfide formation will occur, rendering the enzyme inactive. Therefore, formation of the dimer is likely to aid in attenuating conformational flexibility at the interdomain junction where the active site is located to allow optimal functionality.
To test this hypothesis, molecular dynamic simulations were performed on both the monomeric and dimeric versions of DAP epimerase using the x-ray crystal structure of the E. coli enzyme (Protein Data Bank code 4IJZ). Preliminary results from the 25-ns simulations appear to indicate that the monomer is more flexible or “dynamic” than the dimer (Fig. 7). This is evident by the greater root mean squared fluctuations, particularly in the N-terminal domain (residues 1–119 and 263–274), when compared with the wild-type E. coli DAP epimerase dimer. The N-terminal domain includes a number of catalytic residues, including Cys73, Gly74, Asn75, Asn11, and Asn64, and is also the dimerization domain. Thus, the buttressing of two monomers to form a dimer may prevent increased movement of the active site residues, allowing for optimal catalytic function. However, future studies using experimental approaches to measure protein dynamics are required to confirm our preliminary molecular dynamics simulation results.
FIGURE 7.

Molecular dynamics simulations of E. coli DAP epimerase monomer and dimer. The root mean squared fluctuation (RMSF) is plotted as a function of residue number of monomer A for the wild-type dimer (gray line) and monomer (black line). Simulations were analyzed by aligning monomer A from all frames of the trajectories and computing the root mean squared fluctuation of monomer A.
Given that the findings in the study show that the monomer of DAP epimerase is significantly less active than the dimer, disruption of quaternary structure offers a new approach for the design of novel DAP epimerase inhibitors with potential antimicrobial activities. The use of small molecules or peptides to disrupt protein-protein interaction is an emerging area in drug design (54, 55). This approach has been successful in targeting homodimeric proteins, such as HIV protease and inducible nitric-oxide synthase, leading to the development of nanomolar inhibitors (53). Accordingly, the structure of E. coli DAP epimerase reported in this study permits structure-guided design of peptide or peptidomimetic molecules targeting the dimer interface, thus preventing dimerization of the enzyme to its active conformation.
In conclusion, this study has shown that DAP epimerase is dimeric in solution and that dimerization of the enzyme is required for optimal activity. Accordingly, our work offers insight into a new approach to aid in the development of inhibitors of DAP epimerase as novel antimicrobials.
Acknowledgments
We acknowledge the support and assistance of the friendly staff at the Bio21 Collaborative Crystallographic Centre at Commonwealth Scientific and Industrial Research Organisation Molecular and Health Technologies, Parkville, Melbourne, Australia. We thank John C. Vederas (University of Alberta) for generously providing us with the plasmid for H. influenzae DAP epimerase and members of the Perugini and Hutton laboratories for helpful discussions during the preparation of this manuscript. Part of this research was undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia.
This work was supported in part by Victorian Life Sciences Computation Initiative Grant VR0089 to its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, Australia.
The atomic coordinates and structure factors (codes 4IJZ and 4IK0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
M. H. Matho, K. Fukuda, E. Santelli, K. Jaroszewski, R. C. Liddington, and D. I. Roper, unpublished data.
- DAP
- diaminopimelate
- PISA
- Protein Interfaces, Surfaces and Assemblies
- Bis-tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
REFERENCES
- 1. Hutton C. A., Perugini M. A., Gerrard J. A. (2007) Inhibition of lysine biosynthesis: an evolving antibiotic strategy. Mol. Biosyst. 3, 458–465 [DOI] [PubMed] [Google Scholar]
- 2. Dogovski C., Atkinson S. C., Dommaraju S. R., Hor L., Hutton C. A., Gerrard J. A., Dobson R. C. J., Perugini M. A. (2009) in Biotechnology (Doelle H. W., Rokem S., eds) pp. 116–136, Eolss Publishers, Oxford, UK [Google Scholar]
- 3. Dogovski C., Atkinson S. C., Dommaraju S. R., Downton M., Hor L., Moore S., Paxman J. J., Peverelli M. G., Qiu T. W., Reumann M., Siddiqui T., Taylor N. L., Wagner J., Wubben J. M., Perugini M. A. (2012) in Biochemistry (Ekinci D., ed) pp. 225–262, InTech Open Access Publisher, Rijeka, Croatia [Google Scholar]
- 4. Hutton C. A., Southwood T. J., Turner J. J. (2003) Inhibitors of lysine biosynthesis as antibacterial agents. Mini Rev. Med. Chem. 3, 115–127 [DOI] [PubMed] [Google Scholar]
- 5. Cox R. J., Sutherland A., Vederas J. C. (2000) Bacterial diaminopimelate metabolism as a target for antibiotic design. Bioorg. Med. Chem. 8, 843–871 [DOI] [PubMed] [Google Scholar]
- 6. Antia M., Hoare D. S., Work E. (1957) The stereoisomers of diaminopimelic acid. Biochem. J. 65, 448–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiseman J. S., Nichols J. S. (1984) Purification and properties of diaminopimelic acid epimerase from Escherichia coli. J. Biol. Chem. 259, 8907–8914 [PubMed] [Google Scholar]
- 8. Pillai B., Cherney M. M., Diaper C. M., Sutherland A., Blanchard J. S., Vederas J. C., James M. N. (2006) Structural insights into stereochemical inversion by diaminopimelate epimerase: an antibacterial drug target. Proc. Natl. Acad. Sci. U.S.A. 103, 8668–8673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cirilli M., Zheng R., Scapin G., Blanchard J. S. (1998) Structural symmetry: the three-dimensional structure of Haemophilus influenzae diaminopimelate epimerase. Biochemistry 37, 16452–16458 [DOI] [PubMed] [Google Scholar]
- 10. Lloyd A. J., Huyton T., Turkenburg J., Roper D. I. (2004) Refinement of Haemophilus influenzae diaminopimelic acid epimerase (DapF) at 1.75 Å resolution suggests a mechanism for stereocontrol during catalysis. Acta Crystallogr. D Biol. Crystallogr. 60, 397–400 [DOI] [PubMed] [Google Scholar]
- 11. Usha V., Dover L. G., Roper D. I., Fütterer K., Besra G. S. (2009) Structure of the diaminopimelate epimerase DapF from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 65, 383–387 [DOI] [PubMed] [Google Scholar]
- 12. Pillai B., Moorthie V. A., van Belkum M. J., Marcus S. L., Cherney M. M., Diaper C. M., Vederas J. C., James M. N. (2009) Crystal structure of diaminopimelate epimerase from Arabidopsis thaliana, an amino acid racemase critical for l-lysine biosynthesis. J. Mol. Biol. 385, 580–594 [DOI] [PubMed] [Google Scholar]
- 13. Buschiazzo A., Goytia M., Schaeffer F., Degrave W., Shepard W., Grégoire C., Chamond N., Cosson A., Berneman A., Coatnoan N., Alzari P. M., Minoprio P. (2006) Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc. Natl. Acad. Sci. U.S.A. 103, 1705–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garvey G. S., Rocco C. J., Escalante-Semerena J. C., Rayment I. (2007) The three-dimensional crystal structure of the PrpF protein of Shewanella oneidensis complexed with trans-aconitate: insights into its biological function. Protein Sci. 16, 1274–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parsons J. F., Song F., Parsons L., Calabrese K., Eisenstein E., Ladner J. E. (2004) Structure and function of the phenazine biosynthesis protein PhzF from Pseudomonas fluorescens 2-79. Biochemistry 43, 12427–12435 [DOI] [PubMed] [Google Scholar]
- 16. Velarde M., Macieira S., Hilberg M., Bröker G., Tu S.-M., Golding B. T., Pierik A. J., Buckel W., Messerschmidt A. (2009) Crystal structure and putative mechanism of 3-methylitaconate-Δ-isomerase from Eubacterium barkeri. J. Mol. Biol. 391, 609–620 [DOI] [PubMed] [Google Scholar]
- 17. Blankenfeldt W., Kuzin A. P., Skarina T., Korniyenko Y., Tong L., Bayer P., Janning P., Thomashow L. S., Mavrodi D. V. (2004) Structure and function of the phenazine biosynthetic protein PhzF from Pseudomonas fluorescens. Proc. Natl. Acad. Sci. U.S.A. 101, 16431–16436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grassick A., Sulzenbacher G., Roig-Zamboni V., Campanacci V., Cambillau C., Bourne Y. (2004) Crystal structure of E. coli yddE protein reveals a striking homology with diaminopimelate epimerase. Proteins 55, 764–767 [DOI] [PubMed] [Google Scholar]
- 19. Liger D., Quevillon-Cheruel S., Sorel I., Bremang M., Blondeau K., Aboulfath I., Janin J., van Tilbeurgh H., Leulliot N. (2005) Crystal structure of YHI9, the yeast member of the phenazine biosynthesis PhzF enzyme superfamily. Proteins 60, 778–786 [DOI] [PubMed] [Google Scholar]
- 20. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 21. Hor L., Dobson R. C., Dogovski C., Hutton C. A., Perugini M. A. (2010) Crystallization and preliminary X-ray diffraction analysis of diaminopimelate epimerase from Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 23. Boughton B. A., Griffin M. D., O'Donnell P. A., Dobson R. C., Perugini M. A., Gerrard J. A., Hutton C. A. (2008) Irreversible inhibition of dihydrodipicolinate synthase by 4-oxo-heptenedioic acid analogues. Bioorg. Med. Chem. 16, 9975–9983 [DOI] [PubMed] [Google Scholar]
- 24. Boughton B. A., Hor L., Gerrard J. A., Hutton C. A. (2012) 1,3-Phenylene bis(ketoacid) derivatives as inhibitors of Escherichia coli dihydrodipicolinate synthase. Bioorg. Med. Chem. 20, 2419–2426 [DOI] [PubMed] [Google Scholar]
- 25. Davis A. J., Perugini M. A., Smith B. J., Stewart J. D., Ilg T., Hodder A. N., Handman E. (2004) Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana. J. Biol. Chem. 279, 12462–12468 [DOI] [PubMed] [Google Scholar]
- 26. Burgess B. R., Dobson R. C., Bailey M. F., Atkinson S. C., Griffin M. D., Jameson G. B., Parker M. W., Gerrard J. A., Perugini M. A. (2008) Structure and evolution of a novel dimeric enzyme from a clinically-important bacterial pathogen. J. Biol. Chem. 283, 27598–27603 [DOI] [PubMed] [Google Scholar]
- 27. Voss J. E., Scally S. W., Taylor N. L., Atkinson S. C., Griffin M. D., Hutton C. A., Parker M. W., Alderton M. R., Gerrard J. A., Dobson R. C., Dogovski C., Perugini M. A. (2010) Substrate-mediated stabilization of a tetrameric drug target reveals Achilles heel in anthrax. J. Biol. Chem. 285, 5188–5195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sreerama N., Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 29. Perugini M. A., Griffin M. D., Smith B. J., Webb L. E., Davis A. J., Handman E., Gerrard J. A. (2005) Insight into the self-association of key enzymes from pathogenic species. Eur. Biophys. J. 34, 469–476 [DOI] [PubMed] [Google Scholar]
- 30. Perugini M. A., Schuck P., Howlett G. J. (2000) Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J. Biol. Chem. 275, 36758–36765 [DOI] [PubMed] [Google Scholar]
- 31. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Demeler B., van Holde K. E. (2004) Sedimentation velocity analysis of highly heterogeneous systems. Anal. Biochem. 335, 279–288 [DOI] [PubMed] [Google Scholar]
- 33. Demeler B. (2005) in Analytical Ultracentrifugation: Techniques and Methods (Scott D. J., Harding S. E., Rowe A. J., eds) pp. 210–230, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 34. Demeler B. (2005) in Bioinformatics Basics: Applications in Biological Science and Medicine (Buehler L., Rashdid H., eds) 2nd Ed., pp. 226–255, CRC Press, Boca Raton, FL [Google Scholar]
- 35. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 [DOI] [PubMed] [Google Scholar]
- 36. Brookes E., Demeler B., Rocco M. (2010) Developments in the US-SOMO bead modeling suite: new features in the direct residue-to-bead method, improved grid routines, and influence of accessible surface area screening. Macromol. Biosci. 10, 746–753 [DOI] [PubMed] [Google Scholar]
- 37. Brookes E., Demeler B., Rosano C., Rocco M. (2010) The implementation of SOMO (SOlution MOdeller) in the UltraScan analytical ultracentrifugation data analysis suite: enhanced capabilities allow the reliable hydrodynamic modeling of virtually any kind of biomacromolecule. Eur. Biophys. J. 39, 423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cox R. J., Durston J., Roper D. I. (2002) Synthesis and in vitro enzyme activity of an oxa analogue of azi-DAP. J. Chem. Soc. Perkin 1. 1, 1029–1035 [Google Scholar]
- 39. Leslie A. G. W. (1991) in Crystallographic Computing 5: from Chemistry to Biology: Papers Presented at the International School in Crystallographic Computing Held at Bischenberg, France, July 29–August 5, 1990, pp. 50–61, Oxford University Press, Inc., Oxford, UK [Google Scholar]
- 40. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 41. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 44. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 45. Painter J., Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 46. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacKerell A. D., Bashford D., Bellott, Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 49. Foloppe N., Sagemark J., Nordstrand K., Berndt K. D., Nilsson L. (2001) Structure, dynamics and electrostatics of the active site of glutaredoxin 3 from Escherichia coli: comparison with functionally related proteins. J. Mol. Biol. 310, 449–470 [DOI] [PubMed] [Google Scholar]
- 50. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pillai B., Cherney M., Diaper C. M., Sutherland A., Blanchard J. S., Vederas J. C., James M. N. (2007) Dynamics of catalysis revealed from the crystal structures of mutants of diaminopimelate epimerase. Biochem. Biophys. Res. Commun. 363, 547–553 [DOI] [PubMed] [Google Scholar]
- 52. Koo C. W., Blanchard J. S. (1999) Chemical mechanism of Haemophilus influenzae diaminopimelate epimerase. Biochemistry 38, 4416–4422 [DOI] [PubMed] [Google Scholar]
- 53. Cardinale D., Salo-Ahen O. M., Ferrari S., Ponterini G., Cruciani G., Carosati E., Tochowicz A. M., Mangani S., Wade R. C., Costi M. P. (2010) Homodimeric enzymes as drug targets. Curr. Med. Chem. 17, 826–846 [DOI] [PubMed] [Google Scholar]
- 54. Wells J. A., McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 55. Gerrard J. A., Hutton C. A., Perugini M. A. (2007) Inhibiting protein-protein interactions as an emerging paradigm for drug discovery. Mini Rev. Med. Chem. 7, 151–157 [DOI] [PubMed] [Google Scholar]