

Background: MTDH is overexpressed in breast cancers and promotes resistance to multiple chemotherapeutic agents. We hypothesized that MTDH may contribute to TRAIL resistance in breast cancer.
Results: MTDH modulated TRAIL resistance in breast cancer cells through caspase-8 down-regulation and Bcl-2 up-regulation.
Conclusion: The findings uncovered novel mechanisms of TRAIL resistance mediated by MTDH.
Significance: MTDH can be considered as a novel target for overcoming TRAIL resistance in breast cancer.
Keywords: Apoptosis, Breast Cancer, Cancer Biology, Cancer Therapy, Metastasis, Oncogene, Metadherin
Abstract
Metadherin (MTDH), the newly discovered gene, is overexpressed in more than 40% of breast cancers. Recent studies have revealed that MTDH favors an oncogenic course and chemoresistance. With a number of breast cancer cell lines and breast tumor samples, we found that the relative expression of MTDH correlated with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) sensitivity in breast cancer. In this study, we found that knockdown of endogenous MTDH cells sensitized the MDA-MB-231 cells to TRAIL-induced apoptosis both in vitro and in vivo. Conversely, stable overexpression of MTDH in MCF-7 cells enhanced cell survival with TRAIL treatment. Mechanically, MTDH down-regulated caspase-8, decreased caspase-8 recruitment into the TRAIL death-inducing signaling complex, decreased caspase-3 and poly(ADP-ribose) polymerase-2 processing, increased Bcl-2 expression, and stimulated TRAIL-induced Akt phosphorylation, without altering death receptor status. In MDA-MB-231 breast cancer cells, sensitization to TRAIL upon MTDH down-regulation was inhibited by the caspase inhibitor Z-VAD-fmk (benzyloxycarbonyl-VAD-fluoromethyl ketone), suggesting that MTDH depletion stimulates activation of caspases. In MCF-7 breast cancer cells, resistance to TRAIL upon MTDH overexpression was abrogated by depletion of Bcl-2, suggesting that MTDH-induced Bcl-2 expression contributes to TRAIL resistance. We further confirmed that MTDH may control Bcl-2 expression partly by suppressing miR-16. Collectively, our results point to a protective function of MTDH against TRAIL-induced death, whereby it inhibits the intrinsic apoptosis pathway through miR-16-mediated Bcl-2 up-regulation and the extrinsic apoptosis pathway through caspase-8 down-regulation.
Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)3 is a member of the TNF super family that can initiate apoptosis via activation of the extrinsic apoptosis pathway (1, 2). It has emerged as a potential therapeutic agent due to its remarkable feature of selectively inducing apoptosis in cancer cells while sparing normal cells (3). TRAIL can effectively induce apoptosis in various types of cancer cells (4). Ongoing and completed phase I and phase II clinical trials with TRAIL are showing clinically promising outcomes with no apparent toxicity (5, 6). However, recent studies have indicated that a variety of cancer cells are resistant to the apoptotic effects of TRAIL (7–9), and it is notable that the majority of breast cancer cells are resistant to TRAIL-mediated apoptosis (10). The mechanisms underlying resistance to TRAIL are not fully understood, and the identification of TRAIL resistance factors could facilitate the development of more effective TRAIL-based cancer therapies.
A newly discovered gene, metadherin (MTDH, also known as AEG-1 or Lyric, a membrane protein with a large C-terminal cytoplasmic domain), is frequently overexpressed in primary tumor tissues, and its expression level is always associated with the progression and/or worse prognosis of various types of cancers, such as breast cancer (11, 12), prostate cancer (13), neuroblastoma (14), and hepatocellular carcinoma (15). MTDH is overexpressed in more than 40% of breast cancers and plays a role in breast cancer progression and metastasis (16–18). As a potent mediator in the development of malignancies, MTDH participates in multiple pathological steps, including malignant transformation, angiogenesis, and metastasis (19). In addition, a more general role for MTDH as a possible mediator of broad spectrum chemoresistance has been indicated recently (19, 20). NCI60 pharmacogenomic data revealed a significant correlation between MTDH overexpression and resistance of cancer cells to a broader spectrum of chemical compounds (19, 21). Subsequent in vitro and in vivo chemoresistance analyses showed that MTDH knockdown sensitizes several different breast cancer cell lines to paclitaxel, doxorubicin, and cisplatin (21, 22).
Considering the role of MTDH in drug resistance, we hypothesized that MTDH may be a potential target to overcome TRAIL resistance of breast cancer cells. Here, we demonstrate that MTDH contributes to TRAIL resistance in breast cancer cells both in vitro and in vivo through Bcl-2 up-regulation and/or caspase-8 down-regulation. Our findings suggest that MTDH inhibition may be used to restore TRAIL sensitivity in TRAIL-resistant breast cancers.
EXPERIMENTAL PROCEDURES
Production of Recombinant Human TRAIL (rhTRAIL)
rhTRAIL was produced essentially as described previously (23, 24). Briefly, M15 pRep4 bacteria with a modified inducible pQE9 expression plasmid (Qiagen) coding for the sequence MRGSHHHHHHGSEQKLISEEDLNLQ (His6-Myc) followed by amino acids 95–281 of human TRAIL were used (25). Bacteria were grown to an optical density of 0.7 at 600 nm, induced with 50 μg/ml of isopropyl-1-thio-β-d-galactopyranoside for 18 h at 37 °C, harvested, and suspended in B-PER lysis buffer (Thermo Fisher Scientific) according to the manufacturer's instructions. After centrifugation, the cleared supernatant was filtered at 0.22 μm and loaded onto a HiTrap chelating column (GE Healthcare). Nonspecific proteins were removed by washing with 2 column volumes of TBS (10 mm Tris-HCl, pH 7.4, 140 mm NaCl) containing 10 mm imidazole followed by 2 column volumes of TBS, 50 mm imidazole. rhTRAIL was then eluted with TBS, 0.5 m imidazole and dialyzed against TBS in a dialysis bag with a 15-kDa molecular mass cutoff.
Patients and Samples
A consecutive series of surgically resected primary invasive breast carcinomas was retrieved from the Qilu Hospital of Shandong University, Jinan, China. For all participants in this study, written informed consent was obtained as delineated by the protocol that was approved by the Ethical Committee of Shandong University. None of the patients received chemotherapy or irradiation therapy prior to the surgery. Surgically resected breast cancer tissues were quickly divided into two samples; one was for histoculture drug response assay (HDRA) of TRAIL, and the other was stored at −80 °C for the following Western blotting analysis.
HDRA
HDRA was conducted according to the previous study (26). Briefly, different concentrations of rhTRAIL were dissolved in DMEM/high glucose medium (Life Technologies) containing 20% FBS (Tian Jin Hao Yang Biological Manufacture Co., Ltd., Tianjin, China) and penicillin-streptomycin-amphotericin B. Then 1 ml of solution/well was added into a 24-well plate. The cut-off concentration used to distinguish in vitro sensitivity and resistance was500 ng/ml for rhTRAIL. The pieces of tumor tissues were placed on the gelatin foam of each well and were incubated for 7 days. Then 100 μl of 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) solutions, containing 50 mm sodium succinate, was added to each well. After the plates were incubated for a further 16 h, the medium was removed from each well, and 500 μl of dimethyl sulfoxide (DMSO) per well was added to extract MTT formazan. After 2 h, 100 μl of solution was extracted from each well and transferred to 96-well multiplate. The relative survival was calculated as T/C × 100, where T is mean absorbency of the treated wells per gram tumor, and C is mean absorbency of the control wells per gram of tumor.
Cells and Culture Conditions
The breast cancer cell lines MCF-7 and MDA-MB-231 were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and the cells were routinely cultured in DMEM/high glucose medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Jurkat cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS and antibiotics. All the cells were cultured at 37 °C, 5% CO2.
Plasmid, siRNA, and Transfection
The overexpression and knockdown plasmids were constructed as described previously (22). Briefly, the cDNA of MTDH was cloned into the multiple cloning site of the pcDNA3.1 vector (Invitrogen), and the plasmid was controlled by sequencing. The 19-nucleotide sequence 5′-ATGAACCAGAATCAGTCAGC-3′ was used to generate MTDH shRNA. The 60-nucleotide oligonucleotides were annealed and inserted into the pSUPER.retro.puro vector (OligoEngine, Seattle, WA). Small interfering RNAs (siRNAs) utilized for Bcl-2 and caspase-8 knockdown were purchased from Cell Signaling Technology (Danvers, MA). Mimics and inhibitor of mir-16 and the corresponding negative control (Shanghai GenePharma, Shanghai, China) were used to achieve overexpression or knockdown of miR-16 (sequence can be found in supplemental Table S1). Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. When necessary, cells were selected with 0.3 mg/ml G418 for pcDNA 3.1 based-vectors or 0.5 μg/ml puromycin for pSUPER.retro.puro-based vectors. Transiently transfected cells were harvested at 48 h for mRNA and at 72 h for protein analysis.
Real-time PCR and Quantitative PCR Analysis
Total RNA was extracted from cultured cells with TRIzol reagents (Invitrogen) according to the manufacturer's protocol. The concentration of total RNA was quantified by measuring the absorbance at 260 nm. Total RNA was reverse transcribed to cDNA by using the PrimeScript RT reagent kit (Takara, Dalian, China). The first-strand cDNA of miRNA was synthesized in two steps using the miRcute miRNA first-strand cDNA synthesis kit (Tiangen Biotech, Beijing, China). Real-time PCR was performed with the SYBR Green Premix Ex Taq II (Takara) with the Applied Biosystems StepOne Plus real-time PCR system (Applied Biosystems, Carlsbad, CA). The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control for detection of mRNA expression level, whereas U6 was used as endogenous control for microRNA expression analysis. Relative quantification analysis was performed using the comparative CT (2−ΔΔCT) method. Primer information used in the study can be found in supplemental Table S2.
Flow Cytometry Analysis
The phycoerythrin-conjugated anti-DR4, anti-DR5, and control mouse IgG are from eBioscience (San Diego, CA). The membrane protein levels of DR4 and DR5 were analyzed by flow cytometry as described previously (27). Vehicle- or TRAIL-treated cells were incubated with the phycoerythrin-conjugated anti-DR4 or anti-DR5 monoclonal antibody (1:100 dilution) for 1 h at 4 °C. After three washes, cells were analyzed immediately on a FACScan (BD Biosciences).
Western Blot Analysis
Cells were collected and lysed with radio immunoprecipitation assay buffer (PBS, 1% Nonidet P-40, 0.1% SDS, 5 mm EDTA, 0.5% sodium deoxycholate, 1 mm sodium orthovanadate) with protease inhibitors. Equal amounts of proteins were resolved on SDS-PAGE gels and then transferred to a PVDF membrane (Millipore, Bedford, MA). After blocking with 5% nonfat milk, the membrane was incubated overnight at 4 °C with the primary antibody (ImmunoWay Biotechnology Co., Newark, DE) and then with horseradish peroxidase-coupled secondary antibody. Signal was detected with enhanced chemiluminescence (ECL).
Cell Viability Assay
Cell viability was assessed by the MTT assay. MCF-7 cells and MDA-MB-231 cells were plated at a density of 3 × 103 and 1 × 103 cells/well in 96-well plates. After incubation at 37 °C, 20 μl of MTT (5 mg/ml in PBS) was added to each well, and cells were incubated for another 4 h at 37 °C. The supernatants were carefully removed, and 100 μl of DMSO was added to each well. The plates were gently shaken for 10 min, and the absorbance values were measured at 490 nm with a microplate reader (Bio-Rad).
In Vivo Experiments
MDA-MB-231 vector and MDA-MB-231 shMTDH cells (5 × 106 cells in 100 μl of PBS:Matrigel (1:1, v/v)) were injected subcutaneously into each flank of 4–5-week-old BALB/c nu/nu mice as described (22). Three weeks later, mice were treated intraperitoneally four times per week with rhTRAIL at 7.5 mg/kg or with the corresponding drug vehicle (TBS) (28, 29). Tumor growth was monitored weekly by size measurement. Both maximum (L) and minimum (W) length of the tumor were measured using a slide caliper, and the tumor volume was calculated as ½ LW2. When mice were sacrificed, tumors were dissected and embedded in paraffin, and 4-μm slides were subjected to terminal nucleotidyl transferase-mediated nick end labeling assay (Beyotime Institute of Biotechnology, Beijing, China). These experiments were approved by the Animal Care and Use Committee of Shandong University.
Immunoprecipitation of the Death-inducing Signaling Complex (DISC)
Biochemical analysis of the DISC was performed as described previously (30, 31). The stimulation mix containing 1 μg/ml His6-TRAIL and 2 μg/ml anti-His6 mouse monoclonal antibody (Cell Signaling Technology) was incubated for 30 min at 37 °C before use. Then 1 × 107 MDA-MB-231 cells or MCF-7 cells were washed with prewarmed DMEM and treated with the stimulation mix for 30 min at 37 °C. The stimulation was terminated by adding 25 ml of ice-cold PBS. Cells were harvested at 4 °C and lysed with 1 ml of lysis buffer. For the unstimulated control, the cells were lysed before the addition of the stimulation mix. His6-TRAIL-induced DISC was immunoprecipitated with protein A/G Plus-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and subjected to Western blot analysis.
MicroRNA Array Analysis
Total RNA isolation was performed with TRIzol reagents (Invitrogen) according to manufacturer's protocol. The concentration of total RNA was quantified by measuring the absorbance at 260 nm.
A microarray containing 873 miRNA probes was made (designed according to miRBase release 12.0). miRNA probe design, RNA labeling, and microarray hybridization were performed as described elsewhere (32, 33). Briefly, total RNA (2.5 μg) was labeled with pCp-DY647 (Dharmacon, Lafayette, CO). After hybridization, we scanned arrays with the LuxScan 10K microarray scanner (CapitalBio, Beijing, China), and analyzed images with GenePix Pro 6.0 software (Axon Instruments, Foster City, CA). For data processing, we first performed background subtraction and quantile normalization and then excluded miRNAs from the analysis when they were not detectable in both samples. We used Biometric Research Branch-ArrayTools version 3.8.0 (linus.nci.nih.gov/BRB-ArrayTools.html) for microarray data analysis.
Statistical Analysis
The software SPSS version 18.0 was used for statistical analysis. Student's t test and one-way analysis of variance analysis were used to determine significance. IC50 was calculated with probit regression analysis. All error bars represent the S.E. of three experiments. Differences with p < 0.05 were considered significant.
RESULTS
Generation of Recombinant Human TRAIL
A soluble form of His-tagged rhTRAIL was isolated by chelation affinity purification, and protein purity was confirmed by SDS-PAGE and Coomassie Blue staining (supplemental Fig. S1). The protein killed TRAIL-sensitive Jurkat cells with potency comparable with that of a commercial product (data not shown).
High Expression of MTDH Was Correlated with TRAIL Resistance in Breast Cancer Cells and Breast Tumor Samples
To explore whether relative expression of MTDH correlates with TRAIL resistance in breast cancer, we detected TRAIL sensitivity and MTDH expression in a variety of breast cancer cell lines and the nontransforming MCF-10A breast cells. A previous study indicated that TRAIL selectively induced apoptosis in cancer cells while sparing normal cells (3), which may explain why MCF-10A is most resistant to TRAIL although it has the lowest level of MTDH. HS578T and MCF-7 breast cancer cells showed relatively high expression levels of MTDH and were more resistant to TRAIL treatment. Nevertheless, the other cell lines expressing relatively low levels of MTDH were more sensitive to TRAIL (Fig. 1, A and B). We chose the most resistant MCF-7 cell line and the most sensitive MDA-MB-231 cell line in the following study.
FIGURE 1.

Expression of MTDH correlates with TRAIL resistance in breast cancer cell lines and breast tumor samples. A, relative survival of each breast cell line was tested with MTT assay after a 48-h treatment with rhTRAIL. Points are the average of three independent experiments; error bars represent S.E. B, expression level of MTDH was compared with Western blotting among these cell lines. β-Actin was used as the endogenous control. C, the sensitivity to rhTRAIL studied in the MTDH-negative (−) group was significantly higher than that in the positive (+) group (p = 0.0225). D, the expression of MTDH was determined with Western blotting, and β-actin was used as the loading control. N, paired normal tissue; T, tumor tissues.
To further evaluate the contribution of MTDH to TRAIL resistance, we conducted HDRA. We collected 16 breast tumor tissues and the paired normal tissues for HDRA from patients diagnosed with invasive ductal carcinoma. As is shown in Fig. 1, C and D, the susceptibility of MTDH-negative tumors to TRAIL was significantly higher than that of MTDH-positive tumors (p = 0.0225). The HDRA results suggested a correlation between MTDH expression and TRAIL resistance in breast carcinomas.
MTDH Modulated TRAIL Resistance Both in Vitro and in Vivo
To determine whether MTDH modulates TRAIL resistance in breast cancer cells, we generated stable MCF-7 cells overexpressing MTDH and stable MDA-MB-231 cells with shRNA-mediated knockdown of MTDH. Efficient overexpression of MTDH in MCF-7 cells and knockdown of MTDH in MDA-MB-231 was validated by real-time PCR and Western blotting (Fig. 2, A and B).
FIGURE 2.

Stable overexpression and knockdown of MTDH in breast cancer cell lines and MTDH contributes to TRAIL resistance in vitro. A, stable MDA-MB-231 cells expressing an MTDH-targeting shRNA (shMTDH) or the control pSUPER.retro.puro empty vector (Vector) (left) and stable MCF-7 cells transfected with an MTDH expression plasmid (MTDH) or the control pcDNA3.1 empty vector (Vector) (right) were analyzed for MTDH mRNA levels by real-time PCR after 2 weeks of selection. Columns represent average of three independent experiments; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus cells transfected with control vector. B, the transfection efficiency was confirmed by Western blot analysis. Cell extracts were prepared for Western blotting of MTDH, and β-actin was used as the endogenous control. The figure shown is representative of three independent experiments. C, MCF-7 vector cells and MCF-7 MTDH cells were treated with TRAIL for the indicated time points. The effect of MTDH on TRAIL-induced apoptosis was measured at different doses of TRAIL by the MTT assay and was plotted as a percentage of change relative to the control. D, sensitization of the MDA-MB-231 cell line by MTDH knockdown (shMTDH) was measured at different doses of TRAIL by the MTT assay. Points are the average of three independent experiments; error bars represent S.E.; *, p < 0.05; **, p < 0.01.
To investigate the role of MTDH in TRAIL-induced apoptosis, we tested whether exogenous expression or knockdown of MTDH could regulate TRAIL resistance in breast cancer cell lines. MCF-7 cells overexpressing MTDH showed enhanced resistance to TRAIL at various concentrations of rhTRAIL (Fig. 2C). Consistently, we found that resistance to rhTRAIL was significantly attenuated by knockdown of MTDH in MDA-MB-231 cells (Fig. 2D), indicating that MTDH expression may promote resistance to rhTRAIL. We also calculated the potencies to directly compare the sensitivity to TRAIL of each cell lines (Table 1).
TABLE 1.
Effect of rhTRAIL on cell viability assessed by the MTT method (ng/ml)
The effect of rhTRAIL on cell viability by the MTT assay is represented. The IC50 ± S.D. values were calculated based on the results of three independent experiments.
| 24 h | 48 h | 72 h | |
|---|---|---|---|
| MCF-7 vector | 8.586 ± 0.934 | 1.041 ± 0.017 | 0.602 ± 0.221 |
| MCF-7 MTDH | 43.045 ± 1.634 | 3.524 ± 0.547 | 1.992 ± 0.299 |
| MDA-MB-231 vector | 17.374 ± 1.240 | 11.126 ± 1.046 | 22.292 ± 1.384 |
| MDA-MB-231 shMTDH | 9.036 ± 0.956 | 4.712 ± 0.673 | 5.605 ± 0.749 |
Next, we studied the effect of MTDH on TRAIL sensitivity in vivo using a xenograft model. MDA-MB-231 cells knocked down or not for MTDH were injected subcutaneously into each flank of nude mice. Administration of TRAIL (7.5 mg/kg, four times per week) or the drug vehicle was initiated 3 weeks later. As shown in Fig. 3, A and B, MDA-MB-231 cells knocked down for MTDH grew slower than wild type controls in the absence of treatment. Administration of TRAIL further significantly suppressed tumor growth in MDA-MB-231 shMTDH but had no visible effect on control cells. Apoptosis induced by TRAIL in tumor tissues was measured by a terminal nucleotidyl transferase-mediated nick end labeling (TUNEL) assay. TUNEL-positive cells were detected at higher frequency in tumors derived from MDA-MB-231 shMTDH cells than in those derived from control cells (Fig. 3, C and D), in good agreement with the observation that TRAIL selectively impaired growth of tumors knocked down for MTDH. These data indicated that MTDH suppression may function as an effective sensitizer to TRAIL-based therapy in breast cancer.
FIGURE 3.

MTDH contributes to TRAIL resistance in vivo. A, MDA-MB-231 vector and MDA-MB-231 shMTDH cells were injected subcutaneously into each flank of BALB/c nu/nu mice. Three weeks later, mice were treated intraperitoneally with the drug vehicle or with rhTRAIL at 7.5 mg/kg, four times per week. Tumor volume was measured weekly by direct caliper. Error bars represent S.E.; *, p < 0.05 versus control vehicle-treated mice. B, representative tumors isolated from mice 5 weeks after implantation with control (Vector) or MTDH-depleted (shMTDH) MDA-MB-231 cells and treated with rhTRAIL or vehicle as described in panel A. C, representative TUNEL staining (red fluorescence) of MDA-MB-231 breast cancer xenografts shown in panel B. D, quantification of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells. Columns represent average of three independent experiments; error bars represent S.E.; *, p < 0.05; **, p < 0.01, significantly different from respective control.
MTDH Did Not Regulate Death Receptor (DR) or c-FLIP Protein Levels but Increased Bcl-2 and Decreased Caspase-8 Expression Levels
As both in vitro and in vivo analyses pointed to a role of MTDH in the resistance of breast cancer cell lines to TRAIL, we examined expression levels of proteins known to either favor or counteract TRAIL-induced cell death (5). TRAIL binds to two death domain-containing receptors (TRAILR1/DR4 and TRAILR2/DR5) that can trigger cell death and two decoy receptors (TRAILR3/DcR1 and TRAILR4/DcR2) proposed to be protective. The expression of DcR1 and DcR2 at the mRNA level, detected by real-time PCR, showed no correlation with MTDH expression (data not shown). Surface DR4 and DR5 expression was assessed by flow cytometry, and the results showed that DR4 and DR5 remained unchanged (Fig. 4). By Western blot, DR4 and DR5 levels were not affected by MTDH knockdown in MDA-MB-231 cells or by MTDH overexpression in MCF-7 cells (Fig. 5A), suggesting that MTDH has no major impact on TRAIL receptor expression. FLIPS, a cellular inhibitor of caspase-8 activation, was detected in MDA-MB-231 cells but was not regulated by MTDH levels. In contrast, expression levels of caspase-8, the initiator caspase immediately downstream of death receptors, negatively correlated with those of MTDH. Conversely, levels of the antiapoptotic protein Bcl-2, but not those of the proapoptotic protein Bax, correlated with those of MTDH. In addition, and in line with the observation that MTDH expression reduces cell sensitivity to TRAIL, we detected enhanced processing of several downstream executors of TRAIL-induced death in MTDH low cells. MTDH depletion sensitized MDA-MB-231 cells to TRAIL-induced processing of caspase-3 and poly(ADP-ribose) polymerase-2 (Fig. 5B). Similarly, MTDH overexpression in MCF-7 cells attenuated caspase-8 and poly(ADP-ribose) polymerase-2 processing and activated more phospho-Akt in response to TRAIL treatment (Fig. 5C).
FIGURE 4.

Flow cytometry analysis of surface protein levels of DR4 and DR5 after MTDH overexpression and knockdown. MTDH overexpression or knockdown did not alter surface DR4 (top panel) and DR5 (bottom panel) protein levels in the presence or absence of TRAIL or vehicle, as determined by flow cytometry. IgG was used as the negative control.
FIGURE 5.
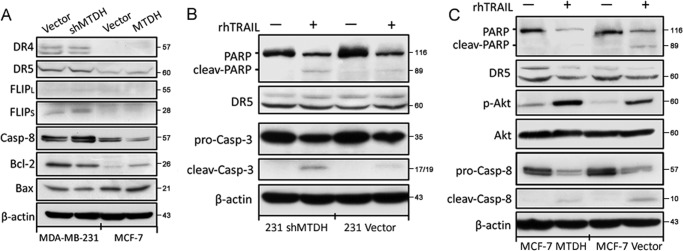
MTDH does not regulate DR or c-FLIP protein levels but increases Bcl-2 and decreases caspase-8 expression levels. A, expression of the indicated proteins was examined in MTDH knockdown (shMTDH) or overexpressing (MTDH) cells, and in their respective controls (Vector), by Western blot analysis. The blot for β-actin was performed to confirm equal loading of samples. casp-8, caspase-8. B, MDA-MB-231 vector and shMTDH cells were treated for 12 h with (+) or without (−) TRAIL at 40 ng/ml and then analyzed by Western blotting for the indicated proteins. C, MCF-7 cells stably expressing MTDH (MTDH) and their control (Vector) were treated for 24 h with (+) or without (−) TRAIL at 100 ng/ml and analyzed by Western blotting for the indicated proteins. p-Akt: phospho-Akt.
Inhibition of TRAIL-induced Apoptosis Depended on the Down-regulation of Caspase-8 and Decreased Recruitment of Caspase-8 into the DISC
To study the implication of caspases in TRAIL-induced death, cells with varying levels of MTDH were treated with TRAIL in the presence of the pan-caspase inhibitor Z-VAD-fmk. Z-VAD-fmk partially prevented death induced by TRAIL in MCF-7 and in MDA-MB-231 cells (Fig. 6, A and B). Notably, the decreased viability due to MTDH depletion in MDA-MB-231 cells was entirely prevented by adding Z-VAD-fmk before TRAIL treatment, suggesting that MTDH prevents a caspase-dependent process in these cells (Fig. 6A). However, the protective effect of MTDH overexpression in MCF-7 cells was additive with that of Z-VAD-fmk, indicating that factors other than caspases might be inhibited by MTDH in MCF-7 cells (Fig. 6B).
FIGURE 6.

Inhibition of TRAIL-induced apoptosis depends on the down-regulation of caspase-8 and on decreased recruitment of caspase-8 into the DISC. A, MDA-MB-231 vector cells and MDA-MB-231 shMTDH cells were treated with 100 μm Z-VAD-fmk (ZVAD). One hour later, cells were treated with (+) or without (−) TRAIL at 20 ng/ml for an additional 24 h. Cell viability was quantified with the MTT assay. B, MCF-7 vector and MCF-7 MTDH cells were pretreated for 1 h with vehicle or 100 μm Z-VAD-fmk μm and then exposed to vehicle or TRAIL at 80 ng/ml for an additional 24 h. Cell viability was monitored with the MTT assay. C, MDA-MB-231 vector cells and MDA-MB-231 shMTDH cells were transfected with control (MOCK) or caspase-8 siRNA (si-Casp-8). After 2 days, cells were treated for 24 h with vehicle or TRAIL at 20 ng/ml. The efficiency of caspase-8 (Casp-8) knockdown was assessed by Western blotting using β-actin as a loading control. Cell viability was quantified with the MTT assay. D, MDA-MB-231 vector or shMTDH cells (left panel) and MCF-7 vector or MTDH cells (right panel) were stimulated with His6-tagged TRAIL for 30 min. Cells were lysed, His6-TRAIL was immunoprecipitated (IP) with anti-His mouse mAb, and the co-immunoprecipitated DR4, DR5, or caspase-8 was detected by Western blotting. The negative control (−) was processed identically, except that TRAIL was added to cells after lysis. β-Actin was probed in cell lysates to ensure that similar amounts of lysate were used in the immunoprecipitation. A–C, columns represent average of three independent experiments; error bars represent S.E.; *, p < 0.05,**, p < 0.01 when compared with controls.
We observed that caspase-8 was negatively regulated by MTDH in breast cancer cells. We therefore knocked down caspase-8 in both MBA-MB-231 vector and MTDH-targeting shRNA (shMTDH) cells. As observed for Z-VAD-fmk, an efficient knockdown of caspase-8 protected TRAIL-exposed MDA-MB-231 cells against the increase in apoptosis observed after MTDH depletion (Fig. 6C).
Finally, we tested the recruitment of caspase-8 in TRAIL-induced DISC. His6-TRAIL-induced DISC was immunoprecipitated from MTDH-proficient or -deficient MCF-7 and MDA-MB-231 cells. There was more caspase-8 in the DISC of cells expressing little MTDH than in their respective controls expressing more MTDH (Fig. 6D). This recruitment was specific as no caspase-8 was recruited when MDA-MB-231 or MCF-7 cells were lysed prior to the addition of His6-TRAIL, although His6-TRAIL was still able to co-immunoprecipitate DR4 and DR5 under these conditions. We also observed increased recruitment of DR5 into the DISC of MDA-MB-231 shMTDH cells when compared with vector control cells, but no noticeable changes for other receptors in MDA-MD-231 and MCF-7 cells (Fig. 6D). Taken together, these results indicate that the depletion of MTDH leads to higher caspase-8 expression and recruitment into the DISC, which could explain the observed sensitization of the cells to TRAIL.
MTDH Up-regulated Bcl-2 Protein Levels through Suppression of MicroRNA-16
MTDH and Z-VAD-fmk protected MCF-7 cells from TRAIL by mechanisms that were at least partially different (Fig. 6B), suggesting that factors distinct from caspases regulate TRAIL resistance in these cells. The antiapoptotic protein Bcl-2 regulates both caspase-dependent and caspase-independent events leading to cell death (34). As Bcl-2 expression levels positively correlated with those of MTDH, we addressed the role of Bcl-2 in the regulation of TRAIL-induced cell death in MCF-7 cells.
The benefit of MTDH expression in MCF-7 cells was lost after knockdown of Bcl-2 in these cells, in accord with the hypothesis that the Z-VAD-fmk-independent protective effect of MTDH in MCF-7 cells is mediated by Bcl-2 (Fig. 7A). We next wondered whether MTDH may promote Bcl-2 expression by regulating levels of miRs. For this purpose, we applied miR arrays to detect altered miRs due to MTDH overexpression or knockdown (supplemental Fig. S2). The candidate microRNAs targeting Bcl-2 were identified based on three microRNA target prediction tools: TargetScanHuman 5.1, PicTar, and miRBase. In our study, microRNAs that were predicted by at least two of the methods were defined as promising candidates that target Bcl-2. The reverse target prediction analysis indicated that there are potentially six microRNAs that target Bcl-2. The expression levels of six candidate miRs were determined by microRNA array (supplemental Table S3), and the quantitative PCR method was adopted to confirm the changes in cells overexpressing MTDH or knocked down for MTDH (Fig. 7B). The consistency of the two methods was best observed for miR-16, whose levels inversely correlated with those of MTDH. In accordance with miR array data, miR-16 showed a decreased expression to 0.65-fold in MCF-7 cells overexpressing MTDH (Fig. 7B). In contrast, MTDH knockdown in MDA-MB-231 cells increased miR-16 expression to 1.51-fold. Based on these results and miR-16 target prediction (Fig. 7, C–E), we hypothesized that MTDH could down-regulate the expression of miR-16, thus increasing Bcl-2 expression in breast cancer cells.
FIGURE 7.

MTDH up-regulates Bcl-2 by reducing expression of microRNA-16. A, MCF-7 vector or MTDH cells were transfected with either control (MOCK) or Bcl-2 siRNAs (si-Bcl-2). After 2 days, cells were treated with vehicle or TRAIL at 20 ng/ml, as indicated. The transient knockdown of caspase-8 was validated by Western blotting, using β-actin as loading control. B, the abundance of miR-148a, miR-15a, miR-16, miR-21, miR-365b, and miR-7 was monitored by real-time PCR in MCF-7 vector and MTDH cells and MDA-MB-231 vector and shMTDH cells. Fold change represents the relative quantification of each microRNA versus control cells. C, the alignment of the miR-16 targeting sequences located in the 3′-untranslated region of the Bcl-2 genes from six organisms (Hsa, human; Ptr, chimpanzee; Mml, mouse; Rno, rat; Cfa, dog; Gga, chicken). The evolutionarily conserved nucleotides are highlighted in light gray. D, the predicted miR-16 targeting sequence located in the 3′-untranslated region (3′-UTR) of Bcl-2 mRNA. The seed region is highlighted in light gray. E, structure of predicted duplex. Minimum free energy (mfe) is −24.1 kcal/mol. F, MCF-7 vector and MTDH cells were transfected with 50 nm miR-16 mimics (+) or with a control oligonucleotide (−). MDA-MB-231 vector and shMTDH cells were transiently transfected with 50 nm miR-16 inhibitor (+) or with a control oligonucleotide (−). miR-16 expression levels monitored by real-time PCR were normalized either to expression levels in MCF-7 vector cells or to those in MDA-MB-231 vector cells. G, the MCF-7 and MDA-MB-231 cell lines were treated as described in panel F. Cell lysates were analyzed by Western blotting to detect the effect of miR-16 on the expression of Bcl-2. NC: negative control. H, MCF-7 vector or MTDH cells were transfected with either miR-16 mimic control or miR-16 mimics. After 2 days, cells were treated with vehicle or TRAIL at 40 ng/ml. I, a schematic model of the mechanisms through which MTDH modulates TRAIL-induced apoptosis in breast cancer cells. FADD, Fas-Associated protein with death domain; Casp-8, caspase-8; PARP, poly(ADP-ribose) polymerase-2. In A, B, F, and H, columns represent average of three independent experiments; error bars represent S.E.; *, p < 0.05.
To determine whether miR-16 could regulate Bcl-2 expression, we used miR-16 mimics or inhibitors to overexpress or knock down miR-16, respectively (Fig. 7F). Overexpression of miR-16 impaired expression of Bcl-2 in MCF-7 MTDH cells at the protein level (Fig. 7G, left). In contrast, suppression of miR-16 in MDA-MB-231 cells increased Bcl-2 expression in both control and shMTDH cells (Fig. 7G, right). To draw a closer association with miR-16 and consequent Bcl-2 expression, we tested the TRAIL resistance of MCF-7 cells after miR-16 manipulation. After transfection with miR-16 mimics, TRAIL resistance of MCF-7 MTDH cells was not higher than that of MCF-7 vector cells, indicating that regulation of Bcl-2 via miR-16 contributes to the TRAIL-resistant role of MTDH (Fig. 7H). Taken together, our data suggest that MTDH may facilitate TRAIL resistance by suppressing miR-16 and therefore up-regulating Bcl-2 protein expression (Fig. 7I).
DISCUSSION
Recently, we identified MTDH as a novel oncogene through a bioinformatic strategy (22). Further functional analyses revealed that MTDH increased chemoresistance in cancer cells. According to our previous studies, MTDH does not alter drug accumulation in cells but instead helps cells to survive antineoplastic stresses, for example by suppressing the proapoptotic protein BNIP3 (22). In addition, MTDH exerts resistance to doxorubicin through up-regulation of multidrug resistance gene 1 (MDR1) protein at both translational and post-translational levels. In the present study, we explored whether MTDH could modulate TRAIL resistance and therefore act as a crucial mediator in the extrinsic apoptosis pathway in breast cancer cells. This work provided evidence that MTDH contributes to TRAIL resistance in breast cancer cells.
It has been widely recognized that resistance can occur at different levels in the TRAIL death signaling pathway, from receptors to downstream caspases and Bcl-2 family members (35). Various molecular mechanisms can account for TRAIL resistance, including differential expression of death receptors, enhanced DISC recruitment, overexpression of c-FLIP, high levels of Akt and NF-κB activity, and defects in the release of mitochondrial proteins (36, 37). Most human breast cancer cell lines are highly resistant to TRAIL-induced apoptosis despite the expression of the death receptors DR4 and/or DR5 (38), indicating that TRAIL resistance in breast cancer cells is likely mediated by defects downstream of death receptor activation in the TRAIL signaling pathway (29). In agreement with this, our result showed that MTDH hardly altered the expression of DR4 (when it was expressed at all) and DR5 in MDA-MB-231 and MCF-7 cells. MTDH apparently had little impact on c-FLIP expression. c-FLIPL and c-FLIPS, which are potent inhibitors of DR4- and DR5-mediated apoptosis, were mostly undetectable in the cell lines analyzed. Only FLIPS was detected in MDA-MB-231 cells, but its levels were not regulated by MTDH.
Our observation that TRAIL-induced Akt phosphorylation in MCF-7 cells was increased in cells overexpressing MTDH is consistent with a previous study indicating that MTDH activates cell survival through a PI3K-Akt signaling pathway (39). Activation of the PI3K-Akt axis may thus be one of the underlying mechanisms for MTDH-induced TRAIL resistance (40). Importantly, our data also revealed that MTDH enhanced TRAIL resistance of breast cancer cells through both down-regulation of caspase-8 and reduced recruitment of caspase-8 in the DISC. How MTDH decreases caspase-8 expression is presently unclear and requires further investigation. Whether reduced recruitment of caspase-8 in the DISC simply reflects its lower expression levels in MTDH-expressing cells or whether it is the result of a more complex mechanism also remains to be studied. Along these lines, we observed in MDA-MB-231 cells that although DR5 expression in total extracts was independent on MTDH expression levels, DR5 was enriched in the DISC of TRAIL in MTDH knockdown cells. It would be of interest to investigate whether MTDH favors intracellular retention of DR5 at the expense of the surface-expressed receptor. In any case, enhanced caspase-8 recruitment to the DISC correlated with increased sensitivity of the cells to TRAIL.
Furthermore, the antiapoptotic protein Bcl-2 has been shown previously to protect breast carcinoma cell lines from TRAIL-induced cytotoxicity (41). Interestingly, we found that Bcl-2 was up-regulated by overexpression of MTDH and down-regulated when MTDH was silenced. Moreover, endogenous Bcl-2 in MCF-7 cells accounted for most of the resistance to TRAIL provided by MTDH overexpression. MicroRNAs have emerged as endogenous regulatory RNAs that pair to the mRNAs of target genes to direct their post-transcriptional repression (42). We identified miR-16 as a potential link between the expression levels of MTDH and Bcl-2. Further investigation and validation will be required to determine the role played by other microRNAs identified in our study in their ability to regulate Bcl-2 expression and resistance to cell death. Interestingly, our results indicate that the protection provided by MTDH and Bcl-2-in MCF-7 cells was not mimicked by a pan-caspase inhibitor, suggesting that MTDH acts on a caspase-independent pathway in these cells. This is somewhat surprising as Bcl-2 is usually considered to act upstream of cytochrome c release from the mitochondria, activation of the caspase 9-containing apoptosome, and activation of downstream caspases such as caspase-3 and -7. MCF-7 cells are, however, known as caspase-3-deficient (43), which would be in line with a cell death that is less dependent on caspase activation. Bcl-2 also prevents the release of cytotoxic mitochondrial proteins that function independently of caspases, and it is not excluded that it may also regulate the caspase-independent RIP3-dependent necroptosis pathway (34). Thus, MTDH-mediated induction of Bcl-2 in MCF-7 cells may account for increased resistance against caspase-independent death pathways induced by TRAIL.
It has been reported that overexpression of Bcl-2 conferred protection against TRAIL in breast carcinomas (41), and caspase-8 overexpression could facilitate TRAIL resistance among cancer cells (44). Our results showed that shMTDH could cause the up-regulation of caspase-8 and down-regulation of Bcl-2 in the breast cancer cells. According to these results, it can be inferred that the observed tumor suppression effect by shMTDH in the xenograft model was associated with altered level of Bcl-2 and/or caspase 8 of the tumor cells. Our results, however, indicate that MTDH can also provide protection against the caspase-dependent arm of TRAIL-induced death as depletion of MTDH increased the susceptibility of MDA-MB-231 cells in a Z-VAD-fmk- and caspase-8 siRNA-inhibitable manner.
Taken together, our study shows that MTDH can increase resistance of breast cancer cell lines to TRAIL. Although the protection is partial, MTDH-induced protection is consistently observed in different cell lines with reproducible effects. Enhanced resistance to TRAIL-induced death by MTDH may involve different, partially overlapping mechanisms: Akt activation, up-regulation of Bcl-2 mediated by miR-16, down-regulation of caspase-8, and decreased recruitment of caspase-8 into the DISC. In addition, using a xenograft animal model, we provide a proof of concept for the combination of TRAIL and MTDH inhibition that may represent a promising targeted therapy for breast cancer in the clinic and is definitely worth further exploration.
Acknowledgments
We thank members of the Women's Oncology Laboratory (Shandong University) for valuable input and support throughout this study, Xiulian Sun, Chao Liu, and Yu Wang for helping to generate the rhTRAIL, and Meiyin Zhang for microRNA array data analysis.
This work was supported by National Natural Science Foundation of China (Grants 81072150, 81172529, and 81272903), the Swiss National Science Foundation (Grant 31003A_138065), and Shandong Science and Technology Development Plan (Grant 2012GZC22115).

This article contains supplemental Tables S1–S3 and Figs. S1 and S2.
The microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GSE32960).
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- rhTRAIL
- recombinant human TRAIL
- TRAILR
- TRAIL receptor
- MTDH
- metadherin
- DR
- death receptor
- DISC
- death-inducing signaling complex
- HDRA
- histoculture drug response assay
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- DMSO
- dimethyl sulfoxide
- z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- miRNA
- microRNA
- miR
- microRNA
- shMTDH
- MTDH-targeting shRNA.
REFERENCES
- 1. Wiley S. R., Schooley K., Smolak P. J., Din W. S., Huang C. P., Nicholl J. K., Sutherland G. R., Smith T. D., Rauch C., Smith C. A., et al. (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 [DOI] [PubMed] [Google Scholar]
- 2. Pitti R. M., Marsters S. A., Ruppert S., Donahue C. J., Moore A., Ashkenazi A. (1996) Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271, 12687–12690 [DOI] [PubMed] [Google Scholar]
- 3. Ashkenazi A., Pai R. C., Fong S., Leung S., Lawrence D. A., Marsters S. A., Blackie C., Chang L., McMurtrey A. E., Hebert A., DeForge L., Koumenis I. L., Lewis D., Harris L., Bussiere J., Koeppen H., Shahrokh Z., Schwall R. H. (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walczak H., Miller R. E., Ariail K., Gliniak B., Griffith T. S., Kubin M., Chin W., Jones J., Woodward A., Le T., Smith C., Smolak P., Goodwin R. G., Rauch C. T., Schuh J. C., Lynch D. H. (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5, 157–163 [DOI] [PubMed] [Google Scholar]
- 5. Newsom-Davis T., Prieske S., Walczak H. (2009) Is TRAIL the holy grail of cancer therapy? Apoptosis 14, 607–623 [DOI] [PubMed] [Google Scholar]
- 6. Mahalingam D., Szegezdi E., Keane M., de Jong S., Samali A. (2009) TRAIL receptor signalling and modulation: Are we on the right TRAIL? Cancer Treat. Rev. 35, 280–288 [DOI] [PubMed] [Google Scholar]
- 7. Zhang L., Fang B. (2005) Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 12, 228–237 [DOI] [PubMed] [Google Scholar]
- 8. Pallares J., Llobet D., Santacana M., Eritja N., Velasco A., Cuevas D., Lopez S., Palomar-Asenjo V., Yeramian A., Dolcet X., Matias-Guiu X. (2009) CK2β is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am. J. Pathol. 174, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim E. H., Kim S. U., Shin D. Y., Choi K. S. (2004) Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 23, 446–456 [DOI] [PubMed] [Google Scholar]
- 10. Rahman M., Davis S. R., Pumphrey J. G., Bao J., Nau M. M., Meltzer P. S., Lipkowitz S. (2009) TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 113, 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J., Zhang N., Song L. B., Liao W. T., Jiang L. L., Gong L. Y., Wu J., Yuan J., Zhang H. Z., Zeng M. S., Li M. (2008) Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin. Cancer Res. 14, 3319–3326 [DOI] [PubMed] [Google Scholar]
- 12. Li C., Li R., Song H., Wang D., Feng T., Yu X., Zhao Y., Liu J., Yu X., Wang Y., Geng J. (2011) Significance of AEG-1 expression in correlation with VEGF, microvessel density, and clinicopathological characteristics in triple-negative breast cancer. J. Surg. Oncol. 103, 184–192 [DOI] [PubMed] [Google Scholar]
- 13. Kikuno N., Shiina H., Urakami S., Kawamoto K., Hirata H., Tanaka Y., Place R. F., Pookot D., Majid S., Igawa M., Dahiya R. (2007) Knockdown of astrocyte-elevated gene-1 inhibits prostate cancer progression through upregulation of FOXO3a activity. Oncogene 26, 7647–7655 [DOI] [PubMed] [Google Scholar]
- 14. Lee S. G., Jeon H. Y., Su Z. Z., Richards J. E., Vozhilla N., Sarkar D., Van Maerken T., Fisher P. B. (2009) Astrocyte elevated gene-1 contributes to the pathogenesis of neuroblastoma. Oncogene 28, 2476–2484 [DOI] [PubMed] [Google Scholar]
- 15. Yoo B. K., Emdad L., Su Z. Z., Villanueva A., Chiang D. Y., Mukhopadhyay N. D., Mills A. S., Waxman S., Fisher R. A., Llovet J. M., Fisher P. B., Sarkar D. (2009) Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J. Clin. Invest. 119, 465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X., Zhang N., Li X., Moran M. S., Yuan C., Yan S., Jiang L., Ma T., Haffty B. G., Yang Q. (2011) Identification of novel variants of metadherin in breast cancer. PLoS One 6, e17582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X., Kong X., Huo Q., Guo H., Yan S., Yuan C., Moran M. S., Shao C., Yang Q. (2011) Metadherin enhances the invasiveness of breast cancer cells by inducing epithelial to mesenchymal transition. Cancer Sci. 102, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 18. Li J., Yang L., Song L., Xiong H., Wang L., Yan X., Yuan J., Wu J., Li M. (2009) Astrocyte elevated gene-1 is a proliferation promoter in breast cancer via suppressing transcriptional factor FOXO1. Oncogene 28, 3188–3196 [DOI] [PubMed] [Google Scholar]
- 19. Hu G., Wei Y., Kang Y. (2009) The multifaceted role of MTDH/AEG-1 in cancer progression. Clin. Cancer Res. 15, 5615–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoo B. K., Gredler R., Vozhilla N., Su Z. Z., Chen D., Forcier T., Shah K., Saxena U., Hansen U., Fisher P. B., Sarkar D. (2009) Identification of genes conferring resistance to 5-fluorouracil. Proc. Natl. Acad. Sci. U.S.A. 106, 12938–12943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei Y., Hu G., Kang Y. (2009) Metadherin as a link between metastasis and chemoresistance. Cell Cycle 8, 2132–2133 [PubMed] [Google Scholar]
- 22. Hu G., Chong R. A., Yang Q., Wei Y., Blanco M. A., Li F., Reiss M., Au J. L., Haffty B. G., Kang Y. (2009) MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 15, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bossen C., Tardivel A., Willen L., Fletcher C. A., Perroud M., Beermann F., Rolink A. G., Scott M. L., Mackay F., Schneider P. (2011) Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody responses. Eur. J. Immunol. 41, 787–797 [DOI] [PubMed] [Google Scholar]
- 24. Kim S. H., Kim K., Kwagh J. G., Dicker D. T., Herlyn M., Rustgi A. K., Chen Y., El-Deiry W. S. (2004) Death induction by recombinant native TRAIL and its prevention by a caspase 9 inhibitor in primary human esophageal epithelial cells. J. Biol. Chem. 279, 40044–40052 [DOI] [PubMed] [Google Scholar]
- 25. Schneider P. (2000) Production of recombinant TRAIL and TRAIL receptor: Fc chimeric proteins. Methods Enzymol. 322, 325–345 [DOI] [PubMed] [Google Scholar]
- 26. Yang Q. F., Sakurai T., Yoshimura G., Shan L., Suzuma T., Tamaki T., Umemura T., Kokawa Y., Nakamura Y., Nakamura M., Tang W., Utsunomiya H., Mori I., Kakudo K. (2000) Expression of Bcl-2 but not Bax or p53 correlates with in vitro resistance to a series of anticancer drugs in breast carcinoma. Breast Cancer Res. Treat. 61, 211–216 [DOI] [PubMed] [Google Scholar]
- 27. Chen C., Zhou Z., Liu R., Li Y., Azmi P. B., Seth A. K. (2008) The WW domain containing E3 ubiquitin protein ligase 1 upregulates ErbB2 and EGFR through RING finger protein 11. Oncogene 27, 6845–6855 [DOI] [PubMed] [Google Scholar]
- 28. Shankar S., Davis R., Singh K. P., Kurzrock R., Ross D. D., Srivastava R. K. (2009) Suberoylanilide hydroxamic acid (Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol. Cancer Ther. 8, 1596–1605 [DOI] [PubMed] [Google Scholar]
- 29. Lu M., Strohecker A., Chen F., Kwan T., Bosman J., Jordan V. C., Cryns V. L. (2008) Aspirin sensitizes cancer cells to TRAIL-induced apoptosis by reducing survivin levels. Clin. Cancer Res. 14, 3168–3176 [DOI] [PubMed] [Google Scholar]
- 30. Zhou Z., Liu R., Chen C.(2012) The WWP1 ubiquitin E3 ligase increases TRAIL resistance in breast cancer. Int. J. Cancer 130, 1504–1510 [DOI] [PubMed] [Google Scholar]
- 31. Walczak H., Haas T. L. (2008) Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol. Biol. 414, 221–239 [DOI] [PubMed] [Google Scholar]
- 32. Wang H., Ach R. A., Curry B. (2007) Direct and sensitive miRNA profiling from low-input total RNA. RNA 13, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang H. Y., Luo M., Tereshchenko I. V., Frikker D. M., Cui X., Li J. Y., Hu G., Chu Y., Azaro M. A., Lin Y., Shen L., Yang Q., Kambouris M. E., Gao R., Shih W., Li H. (2005) A genotyping system capable of simultaneously analyzing >1000 single nucleotide polymorphisms in a haploid genome. Genome Res. 15, 276–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galluzzi L., Vitale I., Abrams J. M., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., Dawson T. M., Dawson V. L., El-Deiry W. S., Fulda S., Gottlieb E., Green D. R., Hengartner M. O., Kepp O., Knight R. A., Kumar S., Lipton S. A., Lu X., Madeo F., Malorni W., Mehlen P., Nuñez G., Peter M. E., Piacentini M., Rubinsztein D. C., Shi Y., Simon H. U., Vandenabeele P., White E., Yuan J., Zhivotovsky B., Melino G., Kroemer G. (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu G. S. (2009) TRAIL as a target in anti-cancer therapy. Cancer Lett. 285, 1–5 [DOI] [PubMed] [Google Scholar]
- 36. Maksimovic-Ivanic D., Stosic-Grujicic S., Nicoletti F., Mijatovic S. (2012) Resistance to TRAIL and how to surmount it. Immunol. Res. 52, 157–168 [DOI] [PubMed] [Google Scholar]
- 37. Yoon M. J., Park S. S., Kang Y. J., Kim I. Y., Lee J. A., Lee J. S., Kim E. G., Lee C. W., Choi K. S. (2012) Aurora B confers cancer cell resistance to TRAIL-induced apoptosis via phosphorylation of survivin. Carcinogenesis 33, 492–500 [DOI] [PubMed] [Google Scholar]
- 38. Keane M. M., Ettenberg S. A., Nau M. M., Russell E. K., Lipkowitz S. (1999) Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 59, 734–741 [PubMed] [Google Scholar]
- 39. Lee S. G., Su Z. Z., Emdad L., Sarkar D., Franke T. F., Fisher P. B. (2008) Astrocyte elevated gene-1 activates cell survival pathways through PI3K-Akt signaling. Oncogene 27, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 40. Xu J., Zhou J.-Y., Wei W.-Z., Wu G. S. (2010) Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS One 5, e10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fulda S., Meyer E., Debatin K.-M. (2002) Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 21, 2283–2294 [DOI] [PubMed] [Google Scholar]
- 42. Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mehta K., Devarajan E., Chen J., Multani A., Pathak S. (2002) Multidrug-resistant MCF-7 cells: an identity crisis? J. Natl. Cancer Inst. 94, 1652–1654; author reply 1654 [DOI] [PubMed] [Google Scholar]
- 44. Seol D. W., Li J., Seol M. H., Park S. Y., Talanian R. V., Billiar T. R. (2001) Signaling events triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL): caspase-8 is required for TRAIL-induced apoptosis. Cancer Res. 61, 1138–1143 [PubMed] [Google Scholar]