

Background: The biochemical function of variable N-terminal regions of eukaryotic Dna2 remains unclear.
Results: The N-terminal 45-kDa domain of yeast Dna2 targets the enzyme specifically to a secondary structure flap.
Conclusion: The hairpin binding activity of Dna2 contributes to efficient removal of hairpin flaps together with endonuclease and helicase activities during Okazaki fragment processing.
Significance: The hairpin binding activity of Dna2 is critical for genome stability.
Keywords: DNA Damage, DNA Enzymes, DNA Helicase, DNA Replication, Enzyme Structure, Dna2 Endonuclease, Okazaki Fragment Processing, Double Strand Break, Genome Stability, Resection
Abstract
The removal of initiating primers from the 5′-ends of each Okazaki fragment, required for the generation of contiguous daughter strands, can be catalyzed by the combined action of DNA polymerase δ and Fen1. When the flaps generated by displacement of DNA synthesis activity of polymerase δ become long enough to bind replication protein A or form hairpin structures, the helicase/endonuclease enzyme, Dna2, becomes critical because of its ability to remove replication protein A-coated or secondary structure flaps. In this study, we show that the N-terminal 45-kDa domain of Dna2 binds hairpin structures, allowing the enzyme to target secondary structure flap DNA. We found that this activity was essential for the efficient removal of hairpin flaps by the endonuclease activity of Dna2 with the aid of its helicase activity. Thus, the efficient removal of hairpin structure flaps requires the coordinated action of all three functional domains of Dna2. We also found that deletion of the N-terminal 45-kDa domain of Dna2 led to a partial loss of the intra-S-phase checkpoint function and an increased rate of homologous recombination in yeast. We discuss the potential roles of the N-terminal domain of Dna2 in the maintenance of genomic stability.
Introduction
Dna2 is a highly conserved essential eukaryotic enzyme that contains ATP-dependent helicase and nuclease activities (1–5). Genetic and biochemical data indicate that its single-stranded (ss)2 DNA-specific endonuclease activity is critical in both Okazaki fragment processing (5–7) and double strand break (DSB) repair (8–11). In Saccharomyces cerevisiae, the endonuclease activity of Dna2 is essential for viability (3, 4), whereas its helicase activity is dispensable under some growth conditions (12, 13). Recently, it was shown that Dna2 contributes to the protection of stalled replication forks by inhibiting reversion of replication forks in a manner dependent on the intra-S-phase checkpoint in Schizosaccharomyces pombe (14). The enzymatic properties of Dna2 endonuclease are suited to remove flaps generated from 5′-ends of Okazaki fragments by DNA displacement activity of DNA polymerase (pol) δ (13, 15). It was shown that the coordination of pol δ and flap endonuclease 1 (Fen1) is sufficient to remove initiating primers from 5′-ends of Okazaki fragments (16–18). However, the generation of flaps long enough to bind replication protein A (RPA) or containing sequences to form hairpin structures spontaneously (for example, trinucleotide repeats such as CAG or CTG) can interfere with Fen1 action (7, 19). Under these conditions, Dna2 becomes critical due to its ability to remove RPA-coated or secondary structure flaps (7, 13). Experiments performed with two mutant Dna2 enzymes lacking either endonuclease or helicase activity revealed that those activities are coupled; the helicase activity facilitates the subsequent action of the endonuclease activity by resolving secondary structures in flaps (13).
Genetic data are also in keeping with an important role of Dna2 in the cleavage of long flaps. First, dna2-1 is lethal in combination with a mutation in pol δ (pol3-01) that increases strand displacement synthesis. Furthermore, the deletion of the pol 32 subunit of pol δ, which reduces the strand displacement activity of pol δ in vitro, suppressed growth defects observed with dna2-1 and dna2-2 (20–23). Similar conclusions were derived from studies in S. pombe (24–26). The frequency in production of long flaps can be affected by the interactions of pol δ/Dna2 with other proteins. For example, Pif1 helps create long flaps through its helicase activity in vitro (27) and in vivo (28), and the lethal phenotype of dna2Δ was suppressed by pif1Δ (28, 29).
Formation of long flaps could impose a replication stress to cells, due to their tendency to bind proteins nonspecifically as well as to form hairpins or higher ordered structures that are difficult to process. There are several other lines of evidence that long flaps can be formed in vivo in support of the action of Dna2. The marked increase of unusual duplications or trinucleotide expansions in the absence of Fen1 (30) indicates that cells have the potential to generate long flaps that are deleterious to genome integrity unless removed in a timely fashion. In keeping with this, it was shown that calf thymus pol δ displaces downstream duplex DNAs longer than 200 bp in vitro, revealing its intrinsic ability to form extensively long flaps (31, 32). In vitro reconstitution experiments using yeast enzymes also showed that a portion of flaps grows long, up to 20–30 nt, although the flaps formed in vitro are primarily short, up to 8 nt in length (33). The observation that overexpression of RPA alleviates the requirement of Dna2 helicase activity (13) is also consistent with formation of long flaps in vivo. In order for double-stranded (ds) DNA-destabilizing activity of RPA to substitute for the helicase activity of Dna2, flaps should be long enough at least to form hairpin structures. The requirement of Dna2 endonuclease and helicase activities for the complete removal of long or hairpin flaps supports the idea that the major role of Dna2 is to prevent the formation of excessively long flaps by cleaving them into shorter ones as soon as they occur.
Although well conserved in eukaryotes (1), the sequences of the N-terminal region from the budding and fission yeast Dna2 enzymes lack significant homology, and its precise biochemical function remains unclear. The dna2Δ405N mutant allele of budding yeasts, which lacks the N-terminal 405 amino acid fragment, displayed a temperature-sensitive growth defect (34). In this study, we show that the N-terminal 45-kDa domain binds hairpin structures and helps to target secondary structure flaps. We also show that this activity is required for the removal of flaps containing secondary structures, which is further assisted by the intrinsic helicase activity of Dna2 and RPA. The role of this N-terminal domain of Dna2 in Okazaki fragment processing and genome maintenance is discussed.
MATERIALS AND METHODS
Oligonucleotides and Preparation of Helicase and Nuclease Substrates
All oligonucleotides used to construct DNA substrates were synthesized commercially (Genotech, Daejeon, Korea) and gel-purified prior to use as described previously (2). The preparation of substrates and their labeling at either 5′- or 3′-end were as described previously (2, 15). The structure and position of radioisotopic labels (indicated by asterisks) in substrates are described in each figure. To construct flap-structured substrates with different 5′-tails, both downstream and upstream primers were annealed to the template primer. The sequences of primers used were as follows: downstream primer, 5′-GCG CAT GTG CGT TCC ATT TGC CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73-mer), and upstream primer, 5′-CCG TTA GCA GTT CGC CTT GTG CCT A-3′ (25-mer); the template primer, 5′-GGA AAA CAT TAT TAA TGG CGT CGA GCT AGG CAC AAG GCG AAC TGC TAA CGG-3′ (51-mer). Annealing of the underlined sequence of the downstream primer to the template primer resulted in the formation of a 48-nt flap substrate. The two shorter downstream primers (52- and 40-mer) used lack the first 21 and 33 nt, respectively, of the 73-mer downstream primer. To construct the hairpin-structured substrate, the first 48 nt of the 73-mer downstream primer was substituted by 5′-GCG CAT GTG CGT TCC GTC CGG TTC AAG/underln] CCG CAG CGG CTT GAA CCG GAC-3′. The underlined sequence forms the double-stranded region. For the preparation of flap substrates containing trinucleotide repeats, the first 48 nt of the 73-mer downstream primer was replaced with 16 repeats of an appropriate trinucleotide as indicated in each experiment.
Proteins and Enzymes
Subtilisin was purchased from Sigma. Recombinant wild type and mutant Dna2 proteins were prepared as described previously (2, 7, 13). Construction of the baculovirus that expressed HX-Dna2N (the N-terminal 405 amino acid fragment of Dna2) and purification of the recombinant protein were as described previously (34). RPA was purified from a protease-deficient yeast strain BJ2168 (MATa, ura3-52, trp1-Δ63, leu2-Δ, prb1-1122, pep4-3, prc1-407, gal2) as described previously (35). Taq polymerase and restriction endonucleases were obtained from Enzynomics (Daejeon, Korea). The Klenow fragment of Escherichia coli DNA polymerase was from Stratagene (Santa Clara, CA).
Enzyme Assays
Endonuclease activity of wild type Dna2 and its derivatives was measured in standard reaction mixtures (40 μl) containing 50 mm Tris-HCl (pH 7.8), 2 mm MgCl2, 2 mm dithiothreitol (DTT), 0.25 mg/ml bovine serum albumin (BSA), and 15 fmol of substrate. Other variations are indicated in each figure legend. Reactions were carried out at 37 °C for 5 min unless indicated otherwise. The products were boiled, when necessary, followed by separation in low resolution gels (12% polyacrylamide gel containing 0.5% SDS and 7 m urea) or high resolution denaturing gels as described previously (15). Gels were dried on DEAE-cellulose paper and subjected to autoradiography. Labeled DNA products were quantitated with the use of a phosphorimager. The size markers (indicated by M in each figure) were as described previously (15) and indicated in each figure. To measure the helicase activity of subtilisin-treated Dna2 or examine the influence of helicase activity on the endonuclease activity of Dna2, ATP (2 mm, final concentration) and substrates (15 fmol), as indicated in each experiment, were added. Reactions were preincubated at 37 °C for 5 min and initiated by the addition of enzyme. Reactions were stopped with 6× stop mix (8 μl; 60 mm EDTA (pH 8.0), 40% w/v sucrose, 0.6% SDS, 0.25% bromphenol blue, and 0.25% xylene cyanol), and products were analyzed by 12% native PAGE.
ATP hydrolysis was measured in reaction mixtures (20 μl) containing 50 mm Tris-HCl (pH 7.8), 0.3 mm MgCl2, 2 mm DTT, 0.25 mg/ml BSA, 250 μm cold ATP, 20 nm [γ-32P]ATP, and 50 ng of φX174 DNA. After incubation with indicated levels of Dna2 at 37 °C for 10 min, aliquots (2 μl) were spotted onto a polyethyleneimine-cellulose plate (Mallinckrodt Baker), which was developed with 0.5 m LiCl and 1.0 m formic acid. The products were analyzed using a phosphorimager.
Gel Mobility Shift Assay
Reaction mixtures (40 μl) contained 50 mm Tris-HCl (pH 7.8), 2 mm DTT, 0.25 mg/ml BSA, 125 mm NaCl, and 15 fmol of substrate. The amount of each protein used is indicated in the figure legends. Reactions were preincubated on ice for 10 min, followed by incubation at 37 °C for 5 min. Glycerol and bromphenol blue were added to 10 and 0.05%, respectively, and products were separated for 1 h at 100 V by 5% PAGE in 0.5× TBE. The gel was dried and autoradiographed. The amount of nucleoprotein complex formed was quantified using a phosphorimager.
Dna2 Proteolysis by Subtilisin
Proteolysis of Dna2 (20 μg) was carried out in a buffer (100 μl) containing 50 mm Tris-HCl (pH 7.5), 2 mm DTT, and 0.02% Nonidet P-40. Reactions were preincubated at 37 °C for 5 min, followed by addition of 2 ng of subtilisin. Aliquots (20 μl) were withdrawn at the indicated time and mixed with an equal volume of buffer containing 50 mm Tris-HCl (pH 7.5), 2 mm DTT, 2 mm EDTA, 0.02% Nonidet P-40, 0.5 m NaCl, 0.5 mg/ml BSA, and 0.2 mm PMSF.
Yeast Strains Used in This Study
All strains used are as listed in Table 1. The N-terminal 405 amino acids of Dna2 were deleted in YPH499 to produce YJA2, as described previously (34). YCH6 and YCH7 strains were constructed from MR966 and MR93.28c (kind gifts from Dr. M. Seki), respectively, by deleting the N-terminal 405 amino acids of Dna2 as described previously (34). The YCH5 diploid strain was constructed by mating YCH6 and YCH7 strains and used to measure the inter-chromosomal recombination frequency between the two his1-7 and his1-1 heteroalleles. To measure intra-chromosomal recombination frequencies, the YCH8 and YCH9 strains were constructed from YPH499 and YJA2 by modifying the lys2-801 locus as described previously (36). The pRS306-LYS2-3′ plasmids were constructed by insertion of the 469,566–470,325 region (760-bp) of chromosome II into the unique XhoI site of pRS306. To generate URA3 marker gene (1.17 kb) flanked by a full-length LYS2 and a 760-bp 3′-fragment (see Fig. 8D), the pRS306-LYS2-3′ plasmid was linearized by AflII and inserted into the lys2-801 locus.
TABLE 1.
Yeast strains used in this study
| Strain name | Genotype | Ref. |
|---|---|---|
| MR 966 | MATa ura3-52 leu2-3,112 trp1-289 his1-7 | 54 |
| MR 93.28c | MATa ura3-52 leu2-3,112 trp1-289 his1-1 | 54 |
| YCH4 | MATa/α ura3-52/ura3-52 leu2-3,112/leu2-3,112 trp1-289/trp1-289 his1-7/his1-1 | This study |
| YCH5 | MATa/α ura3-52/ura3-52 leu2-3,112/leu2-3,112 trp1-289/trp1-289 his1-7/his1-1 dna2Δ405N/dna2Δ405N | This study |
| YCH6 | MATa ura3-52 leu2-3,112 trp1-289 his1-7 dna2Δ405N | This study |
| YCH7 | MATa ura3-52 leu2-3,112 trp1-289 his1-1 dna2Δ405N | This study |
| YPH499 | MATa ade2-101 ura3-52 lys2-801 trp1-63 his3-200 leu2-Δ1 GAL+ | 55 |
| YJA2 | MATa ade2-101 ura3-52 lys2-801 trp1-63 his3-200 leu2-Δ1 GAL+ dna2Δ405N | 34 |
| YCH8 | MATa ade2-101 ura3-52 lys2-801 trp1-63 his3-200 leu2-Δ1 GAL+ lys2-801::URA3 | This study |
| YCH9 | MATa ade2-101 ura3-52 lys2-801 trp1-63 his3-200 leu2-Δ1 GAL+ dna2Δ405N lys2-801::URA3 | This study |
FIGURE 8.

Recombination frequencies increased in dna2Δ405N. A, schematic illustration of inter-chromosomal recombination between his1-7 and his1-1 heteroalleles. In this strain (YCH4 and YCH5, see Table 1), mitotic recombination between heteroalleles and his1-1/his1-7 in a diploid could be monitored by the restoration of histidine prototrophy. B, inter-chromosomal recombination frequencies were measured in wild type and dna2Δ405N mutant cells grown at 25 and 34 °C as described under “Materials and Methods.” C, the same experiment as in B was repeated in the presence of 0.002% MMS using wild type and dna2Δ405N cells transformed with pRS314 vector alone or pRS314-DNA2 as indicated (see text for details). D, schematic illustration to show how intra-chromosomal recombination occurs in the test strains, YCH8 (wild type) and YCH9 (dna2Δ405N). In these strains, the recombination between the two tandemly repeated 3′ regions of lys2-801 could be measured by determining the number of cells that grow in media containing 5-FOA. E, intra-chromosomal recombination frequencies obtained in wild type and dna2Δ405N mutant cells grown at 25 and 34 °C.
Synchronization and Budding Analysis of Yeast Cells
Yeast cells in log-phase growth were synchronized to G1-phase with α-factor (3 μg/ml). After a 1-h treatment, a fresh batch of α-factor was added to a final concentration of 6 μg/ml, followed by incubation until most cells became unbudded. The release from α-factor arrest was performed by centrifugation and resuspension of cells in fresh YPD media. Methyl methanesulfonate (MMS, 0.033%) or camptothecin (CPT, 20 μm) was added to the resuspended cells. The cells were then incubated and harvested at the time point indicated for subsequent analyses. The population of budded and unbudded cells was determined by microscopic analyses.
Flow Cytometric Analysis
G1-arrested cells obtained as described above were released into YPD containing 0.033% MMS. Cells (1 ml) were then harvested at each time point as indicated and resuspended in 70% ethanol. MMS-untreated control cells were prepared using the same procedure. Cells were washed by two rounds of centrifugation and resuspension in 0.5 ml of 50 mm sodium/citrate buffer. RNase A was added to the cells to a final concentration of 250 μg/ml, and the mixture was incubated at room temperature overnight, followed by incubation in 37 °C for 1 h in the presence of 1 mg/ml of proteinase K. Cells were collected by centrifugation and resuspended in 1 ml of 50 mm sodium/citrate-containing propidium iodide (16 μg/ml). To disrupt cell aggregates, resuspended cells were subjected to sonication (using Biorupter, Cosmo-Bio) for 10 s at low level power prior to flow cytometric analysis.
Neutral-Neutral Two-dimensional Gel Electrophoresis
Purification of DNA from yeast cells was carried out with cetyltrimethylammonium bromide. Cells (100 ml) grown to A600 = 0.5 were harvested by centrifugation and resuspended in 20 ml of cold water. Cells were cross-linked with two rounds of trioxalen addition (10 μg/ml, final concentration) and UV irradiation, followed by a 10-min incubation at room temperature and subsequent irradiation with 365-nm UV light for 10 min. Cells were harvested and resuspended in 5 ml of spheroplasting buffer (1 m sorbitol, 100 mm EDTA (pH 8.0), 0.1% β-mercaptoethanol). After lyticase (150–200 units/100 ml cell sample) was added and the mixture was incubated at 30 °C for ∼40 min, spheroplast formation was checked by microscopy. Spheroplasts were collected by centrifugation at 4000 rpm for 10 min at 4 °C and vigorously resuspended in 2 ml of water. Solution I (2.5 ml, 2% w/v cetyltrimethylammonium bromide, 1.4 m NaCl, 100 mm Tris-HCl (pH 7.5), and 25 mm EDTA (pH 8.0)) was added, and the mixture was then incubated at 30 °C for 2 h after RNase A was added to a final concentration of 0.4 mg/ml. Proteins were removed by addition of proteinase K (final concentration, 0.8 mg/ml) and incubation overnight at 30 °C. The samples were then centrifuged at 4000 rpm for 10 min at room temperature, and the resulting supernatant and pellet fractions were separately processed as described below. Pellets were resuspended in 2 ml of Solution III (1.4 m NaCl, 10 mm Tris-HCl (pH 7.6), and 1 mm EDTA), and the mixture was incubated 1.5 h at 30 °C. Chloroform/isoamyl alcohol (24:1 v/v; 1 ml) was added and centrifuged at 4000 rpm for 10 min. The upper aqueous phase containing DNA was collected (“pellet” sample). The supernatant fractions obtained from lysis of spheroplasts were added with 2.5 ml of chloroform/isoamyl alcohol (24:1) and centrifuged at 4000 rpm for 10 min. The upper aqueous phase was collected and supplemented with 2 volumes (10 ml) of Solution II (1% w/v cetyltrimethylammonium bromide, 50 mm Tris-HCl (pH 7.6), and 10 mm EDTA). The resulting samples were then incubated >1 h at room temperature to precipitate the DNA, followed by centrifugation at 13,000 rpm for 10 min. The DNA pellet obtained was combined with the “pellet sample” described above. After resuspension, the combined DNA fractions were precipitated with ethanol and sodium acetate and dissolved in 10 mm Tris-HCl (pH 8). The DNA (10–20 μg) was digested with HindIII/EcoRV or XhoI for ARS305 or ARS216, respectively, and subjected to neutral-neutral two-dimensional gel electrophoresis as described previously (37). After electrophoresis, the gels were subjected to depurination, denaturation, and neutralization as described previously (38). DNA in gels was transferred to a nylon membrane and analyzed by Southern blotting. To detect replication intermediates near replication origins, two oligonucleotide sets were used to amplify DNA fragments to prepare labeled probes using a random primer labeling kit (Stratagene) and [α-32P]dCTP as follows: 5′-GAT ACT ACA TTC CGC CAT TTG AAC TCG AC-3′ and 5′-GAT GGA GAA AGA TTG CAA GGG AAA TCT TG-3′ (ARS305), 5′-GAG GAA TGG CTA AAT AAA CAG CAG ATG TG-3′, and 5′-CAA GGG CTT TTA ACC TGA TGT TTG AAA CG-3′ (ARS216).
Measurement of Recombination Frequencies
The strains YCH4 (wild type DNA2) and YCH5 (dna2Δ405N) (see Table 1 for the strains used) were used to measure inter-chromosomal recombination frequencies. These strains contained heteroalleles his1-1/his1-7 in a diploid. Inter-chromosomal homologous recombination (or rare gene conversion) allows the restoration of histidine-auxotrophic cells to histidine prototrophy (see Fig. 8A for an illustration). Cells were spread separately onto SD plates lacking histidine and YPD plates to determine recombination events and the total number of viable cells, respectively. When the frequency of MMS-induced recombination was measured, cells were spread onto SD plates minus histidine and YPD plates containing the indicated levels of MMS. After incubation for 5 days at different temperatures (25, 30, and 34 °C), the number of colonies formed was determined. To assess alterations in intra-chromosomal recombination frequencies in dna2Δ405N cells (see Fig. 8D for illustration of intra-chromosomal recombination events), the number of colonies formed on the plates containing 5-fluoorotic acid (5-FOA) was measured using the YCH8 (wild type control) and YCH9 (dna2Δ405N) strains (Table 1) (constructed as described above). The cells were spread onto SD plates with or without 1 mg/ml of 5-FOA. After incubation for 5 days at different temperatures (25, 30, 32, and 34 °C), the number of colonies formed was determined.
RESULTS
Secondary Structure in DNA Substrates Affects Endonuclease and ATPase Activities of Dna2
We previously showed that the removal of the N-terminal 45-kDa domain of Dna2 by subtilisin significantly activated the DNA-dependent ATPase and endonuclease activities of Dna2 (34). However, this activation was observed only with φX174 single-stranded circular (ssc) DNA as substrate, but not with oligonucleotide-based substrates (supplemental Fig. S1). The DNA-dependent ATPase and endonuclease activities of both wild type Dna2 and Dna2Δ405N mutant enzymes were similar when oligonucleotides were used as substrates for nuclease assays (supplemental Fig. S1A, compare lanes 3–5 and 6–8) or as effector molecules for ATPase assays (supplemental Fig. S1B). In the presence of oligo(dT)25 (closed symbols), wild type Dna2 (closed circle) and Dna2Δ405N (closed square) were virtually indistinguishable in their ATPase activities (supplemental Fig. S1B). In keeping with our previous result (34), φX174 sscDNA stimulated DNA-dependent ATPase activity of Dna2Δ405N more strongly than oligonucleotides (supplemental Fig. S1B, compare open and closed squares).
To explore this difference in more detail, we first examined the rate of digestion of Dna2 with subtilisin and its effect on the enzymatic activities of Dna2 (Fig. 1). Aliquots containing the same amount (0.5 ng) of Dna2 were withdrawn at each time point (1, 2, 5, 10, and 20 min) and scored for ssDNA-dependent ATPase (Fig. 1B) and endonuclease (Fig. 1, C and D) activities. Because Dna2 specifically hydrolyzes ssDNA and preferentially cleaves the end region of the 5′-ssDNA tail (13, 15), the products formed were longer than 52 nt (the size of partial duplex in the φX174 substrate) and heterogeneous in size (bracketed in Fig. 1C). Both ATPase and endonuclease activities of subtilisin-treated Dna2 increased with incubation time (Fig. 1, B and D, closed circle). In contrast, the activities associated with the mock-treated Dna2 were unaffected (Fig. 1, B and D, open circles). The rate of increase in both endonuclease and ATPase activities occurred coincidentally with the increase in formation of the 127-kDa polypeptide (5–10 min). The recombinant Dna2Δ405N lacking the N-terminal 405 amino acid residues behaved similarly to the subtilisin-treated Dna2 (34). For this reason, we decided to use the recombinant Dna2Δ405N in all subsequent experiments.
FIGURE 1.

ATPase and endonuclease activities of Dna2 deleted of the N-terminal 45-kDa domain. A, hydrolysis of the N-terminal region of Dna2 by subtilisin. Proteolysis of Dna2 (100 μg) was carried out in a reaction (0.3 ml) containing 0.2 m potassium phosphate (pH 6.8), 5 mm DTT, and 0.1 μg of subtilisin as described (34). Reaction mixtures were incubated at 37 °C for the indicated time. Aliquots containing 1 μg of Dna2 were withdrawn and subjected to 10% SDS-PAGE followed by silver staining. M denotes marker proteins, and numbers at left indicate molecular size (in kDa). B, ATPase activity of subtilisin-treated Dna2. Closed and open circles represent Dna2 treated with subtilisin and buffer, respectively. Time refers to the incubation period with subtilisin. C, endonuclease activity of proteolyzed Dna2. Schematic structure of the partial duplex φX174 sscDNA substrate used in this assay is shown at the top of the figure. The 32P-labeled 3′-end of the annealed oligonucleotide is indicated by an asterisk. The arrows on the substrate indicate the sites cleaved by Dna2. S is substrate alone. M is the marker indicating the size of a 52-mer. The reaction mixtures were boiled for 3 min and subjected to electrophoresis in a 12% polyacrylamide gel containing 7 m urea. The bracket denotes the migration position of the cleavage products. D, amount of substrate cleaved in C was measured with a phosphorimager, and the results are presented as a graph. The symbols are as indicated in B.
When oligonucleotide-based DNA substrates containing a 15-, 27-, or 48-nt ssDNA flap were used rather than φX174 sscDNA substrate (Fig. 2A), the endonuclease activity of Dna2Δ405N (Δ405N in Fig. 2) was indistinguishable from that of wild type Dna2. This was evident particularly with the 15- and 27-nt 5′-ssDNA flap substrates (Fig. 2A, lanes 1–10); the 48-nt 5′-ssDNA tail was somewhat less (∼2-fold) efficiently cleaved by Dna2Δ405N than by wild type Dna2 (Fig. 2A, lanes 11–15).
FIGURE 2.
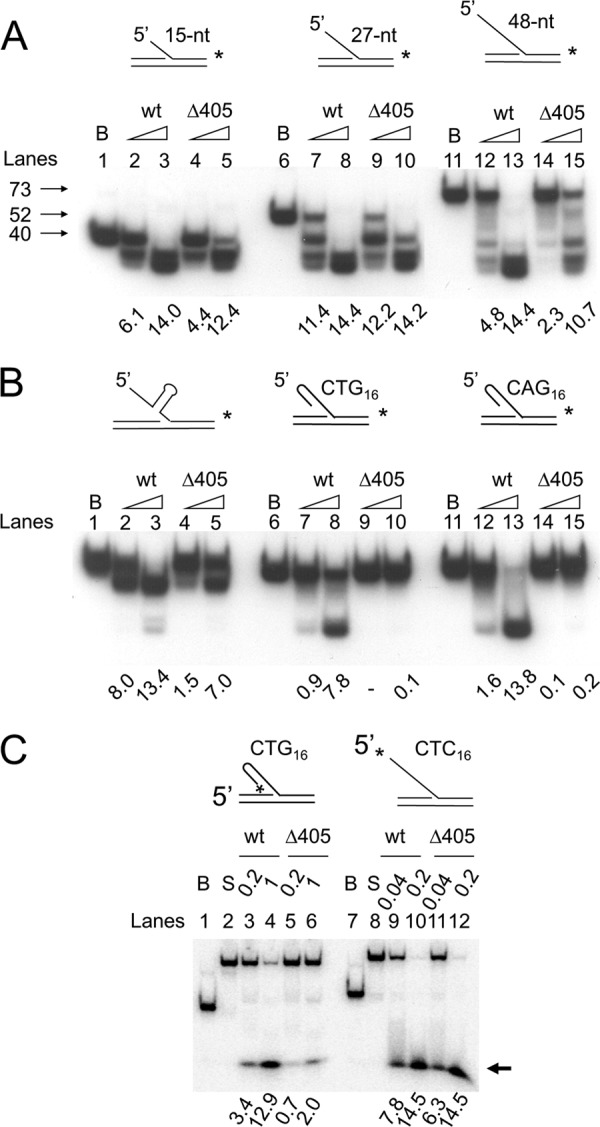
Role of the N-terminal domain in the action of Dna2. Flap substrates were labeled either at the 3′-end (A and B) or 5′-end (C) of the downstream primers. Standard reaction mixtures containing 15 fmol of the indicated substrate were incubated with either Dna2 wild type (wt) or Dna2Δ405N (Δ405) at 37 °C for 5 min. Reaction mixtures were boiled for 3 min and subjected to electrophoresis in a 12% polyacrylamide gel containing 7 m urea. B and S indicate boiled substrate and substrate alone controls, respectively. The amount of substrate cleaved was measured by the use of a phosphorimager as described under “Materials and Methods” and is presented (in fmol) at the bottom of each figure. A and B, increasing amounts (0.1 and 0.5 ng) of wild type Dna2 (wt) or Dna2Δ405N (Δ405) were incubated with each substrate as illustrated at the top of the figure. Arrows indicate the migration position of the labeled strand derived from each substrate. C, amount of wild type and mutant Dna2 enzyme used is as indicated. The reaction mixtures were subjected to the electrophoresis in a 12% native polyacrylamide gel. The arrow indicates the migration position of the cleavage product.
The differences in the response of Dna2 and Dna2Δ405N to φX174 sscDNA- and oligonucleotide-based DNA substrates are likely due to the distinct structural features of the DNA substrates used. For example, φX174 sscDNA contains a large amount of secondary structure, which is absent in the short oligonucleotide DNA substrates. One plausible explanation for the difference in activity is that wild type Dna2 is sequestered by this structure, reducing the effective concentration of the enzyme in the reaction, whereas Dna2Δ405N is not sequestered and thus interacts effectively with ssDNA, leading to increased ATP hydrolysis and cleavage of the ssDNA tail in the substrate.
To address this hypothesis, three additional 48-nt-long 5′-flap substrates containing secondary structures in the flap region were examined. In contrast to 5′-flap substrates of random sequence, the amount of all three substrates hydrolyzed by Dna2Δ405N was reduced dramatically, compared with that observed with wild type Dna2 (Fig. 2B). When a 10-bp hairpin substrate was placed in the middle of the flap, the cleavage activity by Dna2Δ405N at the lower level of enzyme added was reduced markedly (>5-fold) compared with full-length Dna2 (Fig. 2B, lanes 1–5). When 16 repeats of either CTG or CAG, which readily form hairpin structures (19), were present in the flap, the cleavage by Dna2Δ405N was much less efficient than wild type Dna2 (Fig. 2B, lanes 6–15).
Next, we used the 5′-flap substrates labeled at their 5′ termini. The shorter cleavage products formed from these substrates permitted a more accurate measurement of the efficiency of the cleavage reaction catalyzed by wild type and mutant (Dna2Δ405N) enzymes (Fig. 2C). Consistent with the result described above, Dna2Δ405N was much less efficient (<20%) than wild type in cleaving the flap containing 16 CTG repeats (Fig. 2C, lanes 3–6), whereas the cleavage efficiency of a flap DNA with 16 CTC repeats, which do not form hairpin structures, was nearly identical for both enzyme preparations (Fig. 2C, compare lanes 9–12). Based on these results, we conclude that Dna2 devoid of the N-terminal 45-kDa domain is defective in cleaving ssDNA flaps with secondary structure. These results indicate that the N-terminal domain of Dna2 is critical in the interaction of Dna2 with hairpin structures in DNA substrates.
Dna2Δ405N Is Defective in Binding to the Secondary Structures in DNA Substrates
We next examined whether Dna2Δ405N binds to DNA substrates containing secondary structure. Using electrophoretic gel mobility shift assays, we investigated the interaction of wild type Dna2, Dna2Δ405N, and Dna2N (the N-terminal 45-kDa fragment) (Fig. 3A) with flap DNAs. In these experiments, the same molar concentrations (6 and 12 nm) of each protein were used per reaction. Wild type Dna2 formed a complex with the 48-nt-long flap of random sequence slightly more efficiently (10–30%) than Dna2Δ405N (Fig. 3B, compare lanes 2 and 3 with lanes 4 and 5), similar to the cleavage activity observed with these two enzymes (Fig. 2A). One speculation for the reduced efficiencies of Dna2Δ405N in cleavage and binding observed with random 48-nt flap substrates is that the long ssDNA flap sequence might allow the formation of transient or intrinsically weak secondary structure in the flap. Dna2N also formed a complex with the DNA substrate, but less efficiently (30–45%) than the other Dna2 derivatives (Fig. 3B, compare lanes 2–5 with lanes 6 and 7). This indicates that the N-terminal domain itself has intrinsic DNA binding activity. We noted that the nucleoprotein complex formed with Dna2N, despite its small size, migrated more slowly than that formed with Dna2Δ405N (Fig. 3B, compare lanes 4 and 5 with lanes 6 and 7). One speculation is that the unstructured and relatively high content of polar residues in the N-terminal region may contribute to the retardation of the complex during gel electrophoresis. The flap substrate with 16 repeats of CTC that lacks secondary structure supported the formation of complexes with Dna2 and Dna2Δ405N formed with comparable efficiency (Fig. 3B, compare lanes 9 and 10 with lanes 11 and 12). In contrast, in the presence of a flap containing 16 repeats of CTG or CAG, the level of nucleoprotein complex formed with Dna2Δ405N was nearly 10-fold lower than that formed with the wild type protein (Fig. 3B, lanes 16–19 and 23–26). Importantly, Dna2N formed a nucleoprotein complex with these two substrates as efficiently as wild type Dna2 (Fig. 3B, lanes 20 and 21 and lanes 27 and 28). These findings demonstrate that the N-terminal 45-kDa domain of Dna2 contributes importantly to the binding of the enzyme to a flap DNAs with hairpin structures.
FIGURE 3.
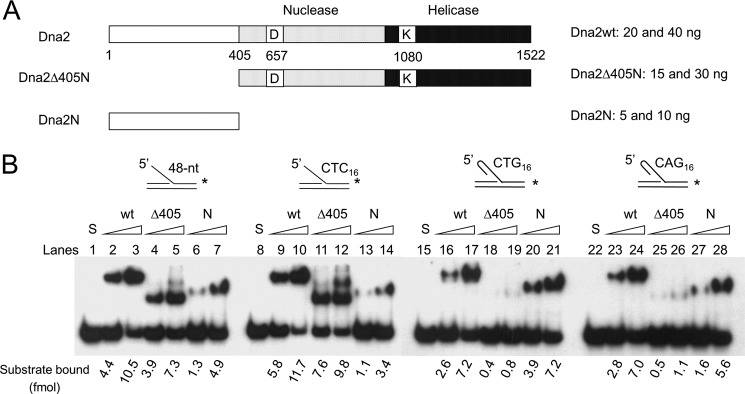
N-terminal domain of Dna2 binds to oligonucleotides possessing secondary structures (in the 5′-flap). A, schematic summary of wild type Dna2 and its derivatives used in gel mobility shift assays; wild type Dna2 (Dna2wt), Dna2 devoid of the N-terminal 405 amino acid residues (Dna2Δ405N), and the N-terminal 405 amino acid fragment (Dna2N). B, substrates used in this experiment were as described in Fig. 2B. Increasing amounts of Dna2 (wt, 20 and 40 ng), Dna2Δ405N (Δ405N, 15 and 30 ng), and the N-terminal fragment of Dna2 (N, 5 and 10 ng) were incubated with each substrate as indicated. Two different levels (110 and 220 fmol) of proteins were used, corresponding to 6 and 12 nm, respectively. Reaction mixtures were assembled and processed as described under “Materials and Methods.” The quantitation of complexes formed was measured by the use of a phosphorimager as described under “Materials and Methods” and is presented (in fmol) at the bottom of the figure.
Deletion of the N-terminal 45-kDa Domain of Dna2 Reduces Its Efficiency to Unwind and Cleave Flap DNAs Containing Secondary Structures
Because the binding of Dna2 to hairpin structures present in flap substrates containing CTG or CAG repeats depends on the N-terminal 45-kDa domain, we predicted that efficient processing of the hairpin flap would require the N-terminal domain of Dna2. As shown in Fig. 4A, Dna2-catalyzed cleavage of the CTG repeat flaps occurred poorly in the absence of ATP, and products were observed only at the highest level (0.5 ng) of Dna2 added (Fig. 4A, lane 4). When ATP (2 mm) or RPA (10 ng) was added, cleavage of hairpin flaps by Dna2 was markedly stimulated (Fig. 4A, lanes 5–7 and 8–10, respectively). Cleavage was most efficient in the presence of both ATP and RPA, and nearly most of the flaps were cleaved at the highest level of Dna2 added (Fig. 4A, lanes 11–13). When Dna2 (0.2 ng) was incubated with a substrate containing a CTG repeat flap in the presence of ATP, 1.7 fmol of cleaved products was formed (Fig. 4B, lane 3), although the same level of Dna2Δ405N hardly produced any product (Fig. 4B, lane 6). The addition of increasing amounts (2 and 10 ng) of RPA stimulated the cleavage activity of both wild type Dna2 and Dna2Δ405N to the same extent with the hairpin flap substrate, although the overall cleavage by Dna2Δ405N was less efficient (Fig. 4B).
FIGURE 4.

Coordination of hairpin binding, helicase, and endonuclease activities is required for efficient removal of the hairpin structure in the flap. A, cleavage of the CTG repeat flap by Dna2 depends on its helicase activity. Increasing amounts (0.1, 0.2, and 0.5 ng) of wild type Dna2 were incubated at 37 °C for 5 min with the (CTG)16-flap DNA substrate (shown in B) in the presence (+) or absence (−) of ATP (2 mm) and/or RPA (10 ng). Reaction products were analyzed in a high resolution gel as described under “Materials and Methods.” M denotes marker (in nucleotides). B, cleavage reactions were carried out with the indicated substrate in the presence of 0.2 ng of either Dna2 or Dna2Δ405. RPA (2 and 10 ng) and ATP (2 mm) additions are indicated. The amount of substrate cleaved was measured by the use of a phosphorimager as described under “Materials and Methods” and is presented (in femtomoles) at the bottom of each figure. C, time course analyses of proteolysis of nuclease-deficient Dna2D657A by subtilisin. Proteolytic products of Dna2D657A (0.5 μg) were subjected to 8% SDS-PAGE and analyzed by Western blot analyses using α-Dna2 (polyclonal antibodies against full-length Dna2, left panel) or α-Dna2N (polyclonal antibodies against the N-terminal 405 amino acid fragment, right panel). An arrowhead indicates the migration position of the 127-kDa proteolytic products devoid of the N-terminal 405 amino acids. D, time course analyses of helicase activities of proteolytic products of nuclease-deficient Dna2D657A. The N-terminal domain of Dna2 is required for the efficient unwinding of hairpin-containing helicase substrates. Dna2D657A was treated with subtilisin as described under “Materials and Methods.” The helicase activity of subtilisin-digested Dna2D657A (endonuclease-deficient) was measured using aliquots (containing 5 ng of the enzyme) withdrawn at each time point (1, 2, 5, and 10 min) of incubation with subtilisin. Helicase reactions were carried out with 15 fmol of substrates containing CTC16-flaps (filled square), T40-flaps (open square), CTG16-flaps (filled triangle), or CAG16-flaps (filled diamond). The structure of each substrate used is as shown to the right of the graph. The amount of labeled flap strands dissociated from each substrate by the helicase activity of proteolyzed Dna2D657A was measured and plotted against time of incubation with subtilisin. The control used was mock-treated Dna2D657A incubated without subtilisin (filled circle).
To further demonstrate the role of the N-terminal 45-kDa domain in recognizing hairpin substrates, we treated Dna2D657A (a full-length Dna2 devoid of endonuclease activity (4)) with subtilisin and examined its ability to unwind hairpin-containing flap substrates. Treatment of this mutant enzyme with subtilisin specifically removed the N-terminal region of Dna2 as it did with the wild type Dna2 (Fig. 4C). When the proteolytic products of Dna2D657A were analyzed by Western blotting using α-Dna2 (polyclonal antibodies against full-length Dna2) and α-Dna2N (polyclonal antibodies against the N-terminal 405 amino acid fragment), the proteolyzed enzyme preparation was not detected by antibodies specific for the N-terminal region (Fig. 4C). The proteolyzed products withdrawn at different times following subtilisin treatment were examined for their ability to unwind DNA substrates containing flaps of either oligo(dT)40 or 16 trinucleotide repeats (CTC, CTG, and CAG) (Fig. 4D). The unwinding of CTC repeat or oligo(dT)40 flap substrates was slightly stimulated (1.6-fold following 2- and 4-min incubation with subtilisin) or unaffected, respectively, by the removal of the N-terminal domain (Fig. 4D). However, subtilisin treatment rapidly (1- or 2-min incubation) reduced the unwinding efficiency of the CTG or CAG flap DNA substrates (Fig. 4D). This finding suggests that the N-terminal 45-kDa domain enables Dna2 to bind to DNA regions containing secondary structures in the flap, which is necessary for efficient unwinding. Taken together, our data demonstrate that the efficient removal of trinucleotide repeat-containing flaps requires all three domains of Dna2 as follows: the N-terminal domain for hairpin-DNA binding activity, the helicase domain for converting it to ssDNA, and the ssDNA-specific endonuclease activity that cleaves the generated ssDNA flap. Thus, removal of trinucleotide repeat-containing flaps requires the coordinated action of the three-independent activities present within the three domains of Dna2.
Unimpeded S-phase Progression Is Observed in dna2Δ405N Following Activation of the Intra-S-phase Checkpoint by MMS
To investigate the biological significance of our in vitro findings, we examined the defects associated with this mutant allele in vivo (7). Based on our biochemical results, we hypothesized that the absence of the N-terminal 45-kDa domain could lead to defects in either Okazaki fragment processing or aberrant resection of DSBs (39). It is reasonable to assume that the two events described above are inseparable as defective Okazaki fragment processing could result in increased DSB formation (36). We consider that cells containing Dna2 devoid of the N-terminal 45-kDa domain would display cell cycle defects when exposed to moderate levels of DNA-damaging agents such as MMS.
To investigate this possibility, cell cycle progression of dna2Δ405N cells was analyzed by flow cytometric analyses in the presence and absence of MMS. When α-factor-arrested cells were released into media without MMS, wild type and dna2Δ405N mutant cells did not show notable differences in cell cycle progression (Fig. 5A, WT and dna2Δ405N, respectively). Moreover, under undisturbed growth conditions, dna2Δ405N cells behaved similarly to the rad53Δsml1Δ double mutant cells (Fig. 5, rad53Δsmil1Δ) that are defective in intra-S-phase checkpoint activation (40). This result indicates that dna2Δ405N mutation did not affect normal cell cycle progression in the absence of DNA damage. In contrast, we observed a remarkable difference in S-phase progression of dna2Δ405N cells compared with wild type cells in the presence of MMS (Fig. 5B). When α-factor-arrested cells were released into media containing 0.033% MMS, wild type cells did not traverse the S-phase and were arrested with 1C DNA contents up to 90 min of incubation (Fig. 5B, WT), indicating that MMS induced intra-S-phase checkpoint response and blocked progression of cells into S-phase. The rad53Δsml1Δ cells entered S-phase efficiently in the presence of MMS (Fig. 5B, rad53Δsml1Δ). Interestingly, dna2Δ405N also entered S-phase in the presence of 0.033% MMS, but the S-phase progression was much slower compared with that of rad53Δsml1Δ (Fig. 5, B and C). Flow cytometric profiles of wild type, dna2Δ405N, and rad53Δsml1Δ at 90 min of incubation in the absence (−MMS) and presence (+MMS) of 0.033% MMS were compared in Fig. 5C. Our results suggest that the intra-S-phase checkpoint may not be fully activated in dna2Δ405N cells, resulting in a partial checkpoint-dependent inhibition of DNA replication.
FIGURE 5.

S-phase progression is unimpeded in dna2Δ405N when intra-S-phase checkpoint is activated by MMS. Cells (wild type (WT), dna2Δ405N, and rad53Δsml1Δ) to be tested were synchronized into G1 with α-factor as described under “Materials and Methods.” G1-arrested cells were then released into media in the absence (A) or presence (B) of 0.033% MMS for 90 min. Samples were taken at 15-min intervals and processed as described under “Materials and Methods.” The cellular DNA contents were analyzed by flow cytometry. Asynch, asynchronous cell preparation. C, 90-min profiles of cell-cycle distribution from A and B merged together. Cells used are indicated in different lines (WT, solid lines; dna2Δ405N, gray lines; and rad53Δsml1Δ, dotted lines). The addition (+) or omission (−) of MMS is indicated.
dna2Δ405N Cells Escape from Intra-S-phase Checkpoint-dependent Inhibition of Bud Formation
To further confirm that S-phase progression in dna2Δ405N cells is unimpeded in the presence of MMS, we determined the population of budded cells because the budding event in S. cerevisiae occurs coincidental with DNA replication (41). In the absence of a DNA-damaging agent, we monitored the changes in distribution of the budded and unbudded cells of wild type (WT) and dna2Δ405N cells and found that they both followed the typical cell cycle oscillation pattern (Fig. 6A). We noted that dna2Δ405N cells formed buds slightly earlier than wild type cells in the absence of DNA- damaging agent (Fig. 6A). However, this difference was not as pronounced as the difference detected in the presence of MMS (compare Fig. 6, A and B). When α-factor-arrested cells were released into media containing 0.033% MMS, bud formation was significantly inhibited in wild type cells. Less than 30% of cells formed buds even after the 120-min incubation. In contrast, a substantial population (>60%) of dna2Δ405N cells formed buds after a 90-min incubation (Fig. 6B). To determine whether the unchecked S-phase progression of dna2Δ405N cells was a phenotype uniquely associated with a defective intra-S-phase checkpoint, we used 20 μm CPT in place of MMS. It is known that CPT-induced lesions, which are converted to DSBs following passage of replication forks, are not detected by the intra-S-phase checkpoint and thus termed “checkpoint-blind lesions” (42). When G1-synchronized cells were released into media containing 20 μm CPT, the rates of bud formation by wild type (WT) and dna2Δ405N cells were indistinguishable (Fig. 6C), indicating that the unhindered S-phase progression phenotype of dna2Δ405N is most likely due to a defective intra-S-phase checkpoint.
FIGURE 6.

Bud formation of dna2Δ405N is not inhibited by MMS treatment. Wild type and dna2Δ405N mutant cells were arrested in G1 with α-factor and released into fresh YPD media containing no drug (A), 0.033% MMS (B), or 20 μm CPT (C). Samples were taken at each time point up to 120 min, and the population of budded cells was determined by microscopy.
Inappropriate Firing of Late Replication Origins in dna2Δ405N Cells
The results shown in Figs. 5 and 6 indicate that dna2Δ405N is defective in the function of the intra-S-phase checkpoint. One major function of the intra-S-phase checkpoint is its regulation of DNA synthesis by inhibiting the firing of late replicating origins (43, 44). Therefore, we investigated the firing of late origins in wild type and dna2Δ405N cells. For this purpose, neutral-neutral two-dimensional gel electrophoresis was used to analyze replication intermediates formed near the late origin, ARS216, in chromosome II (45). For comparison, we also examined the early origin, ARS305, in chromosome III (46). As shown in Fig. 7A, labeled probes were prepared that hybridize to regions ∼4–5 kb away from both origins to detect Y-structured replication intermediates. As described under “Materials and Methods,” G1-arrested cells were released into media containing 20 μm CPT or 0.033% MMS. MMS is known to activate the intra-S-phase checkpoint by interfering with progression of replication fork progression (14, 47, 48), whereas CTP fails to do so (42). DNA was purified from the samples taken at regular (20 or 30 min) intervals after release. When cells were treated with 20 μm CPT, we found that both wild type and dna2Δ405N mutant cells initiated replication from ARS216 even at 20 min after treatment (Fig. 7B). It appears that wild type cells completed replication slightly faster than dna2Δ405N in the presence of CPT (Fig. 7B). We believe that this is most likely due to the compromised function of dna2Δ405N during DNA replication. When treated with 0.033% MMS, however, it is evident that wild type cells did not fire the ARS216 origin in the early stage of S-phase because we failed to detect replication intermediates 20 min after MMS treatment (Fig. 7C). This probably suggests the efficient inhibition of late origin firing by the activation of the intra-S-phase checkpoint by MMS. In contrast, dna2Δ405N cells displayed premature firing of ARS216 under the same condition, indicating that late origins in dna2Δ405N partially escaped this inhibition. When the firing of the early origin ARS305 was examined, we found that deletion of the N-terminal domain of Dna2 did not affect the timing of origin firing (Fig. 7D). This, together with the results described in Figs. 5 and 6, indicates that the in vivo defect associated with the deletion of the N terminus of Dna2 is related to intra-S-phase checkpoint function. However, the mechanism involved in this checkpoint pathway is not clear at present, but it will be addressed under the “Discussion.”
FIGURE 7.

Firing of late replicating origins is not inhibited in dna2Δ405N by the presence of MMS. A, schematic illustration of the relative locations of the two origins, ARS305 (early) and ARS216 (late origin) used to detect replication intermediates and probes. Restriction enzyme sites and sizes of relevant fragments are as shown. The maps were drawn approximately to scale. As described under “Materials and Methods,” G1-arrested cells were released into media containing 20 μm CPT (B) or 0.033% MMS (C and D). Samples were taken at indicated intervals after release and the DNAs purified. The replication intermediates derived from ARS216 (a late origin; B and C) or ARS305 (an early origin; D) were examined by two-dimensional gel electrophoresis as described under “Materials and Methods.”
dna2Δ405N Cells Display Increased Frequency of Mitotic Recombination
The inability of Dna2Δ405N to bind to hairpin DNA structures generated during Okazaki fragment processing or DSB resection could lead to the inefficient processing of such aberrant intermediates. If this were the case, cells expressing Dna2Δ405N could accumulate higher levels of abnormal structures than wild type, resulting in elevated levels of DSBs that require homologous recombination to be repaired. This scenario could be further exacerbated by DNA-damaging agents such as MMS. To investigate this, several tester strains were constructed (see Table 1) to monitor mitotic homologous recombination events in wild type and dna2Δ405N cells.
We first measured inter-chromosomal recombination frequencies using the strains YCH4 (wild type control) and YCH5 (dna2Δ405N) (see Table 1 for genotypes). These two diploid strains, which are isogenic except for the dna2 alleles, contain his1-7/his1-1 heteroalleles. Recombination frequencies were determined based on the restoration of histidine prototrophy that could arise via homologous recombination between the two heteroallelic mutations (Fig. 8A). Histidine prototrophy could also occur by gene conversion, which is also a part of DSB-initiated homologous recombination (Fig. 8A). We first measured the recombination frequencies of cells grown at 25 and 34 °C because dna2Δ405N cells are viable at 35 °C but not at 37 °C (7). At 25 °C, dna2Δ405N cells displayed elevated frequencies in formation of His+ colonies, which were further increased by growth at 34 °C (Fig. 8B). In contrast, wild type cells were not affected by varying the growth temperatures (Fig. 8B). This result is in keeping with the notion that cells expressing Dna2 devoid of the N-terminal 45-kDa domain could accumulate higher levels of aberrant structures than wild type. We next assessed the inter-chromosomal recombination frequencies in the presence of varying concentrations of MMS (0.0005 to 0.002%) using the same cells grown at 30 °C. We found that recombination frequencies increased in both wild type and dna2Δ405N strains in the presence of >0.0005% MMS (Fig. 8C). To examine whether the increased recombination frequencies in dna2Δ405N was due to the defective Dna2, we introduced pRS314-DNA2 into dna2Δ405N cells. The dna2Δ405N cells transformed with pRS314-DNA2 displayed recombination frequencies comparable with that observed in wild type cells (Fig. 8C). This result supports the notion that the deletion of the N-terminal 45-kDa region of Dna2 could lead to increased levels or longer half-life of DSBs.
To confirm our above findings, we investigated whether intra-chromosomal recombination frequencies were increased in dna2Δ405N cells using the strains YCH8 (wild type) and YCH9 (dna2Δ405N). In these tester strains, intra-chromosomal recombination events between two homologous regions result in the loss of URA3, allowing cells to grow in the presence of 5-FOA (Fig. 8D). The frequency of intra-chromosomal recombination was observed in dna2Δ405N-containing YCH9 cells compared with wild type (Fig. 8E). Like inter-chromosomal recombination events, this difference was affected by growth temperatures. Growth of dna2Δ405N cells at 34 °C resulted in a significant increase (14-fold compared with wild type cells) in intra-chromosomal recombination frequencies (Fig. 8E). We also measured the unequal sister-chromatid recombination frequency and found that it was enhanced in dna2Δ405N (supplemental Fig. S2). Collectively, our data suggest that the N-terminal 45-kDa domain of Dna2 plays a critical role in preventing aberrant mitotic recombination by removing the abnormal structures formed during lagging strand DNA synthesis or by facilitating the efficient resection of DSBs.
DISCUSSION
In this study, we show that the N-terminal domain of Dna2 is essential for its binding to the hairpin structures present in single-stranded flaps. We also show that the N-terminal domain of Dna2 facilitates the endonucleolytic removal of the secondary structure flap containing CTG or CAG repeat sequences. Cleavage of hairpin flaps was dramatically enhanced by the addition of RPA and ATP that jointly increase the helicase activity of Dna2. These findings indicate that Dna2 is well suited to cleave secondary structure flaps. To carry out this function efficiently, however, all three domains of Dna2 act in a coordinated and sequential manner; the N-terminal 45-kDa domain of Dna2 targets the enzyme to a hairpin flap, allowing the C-terminally encoded helicase activity to melt the duplex region, and the digestion of unwound DNA by endonuclease activity is located in the central region.
Dna2 was reported to play an important role in homologous recombination by participating in the long range resection of DSBs. In this process, Dna2 acts together with the Sgs1 helicase in a sequential manner. Sgs1 helicase unwinds dsDNA at DSB ends, producing Y-structured DNA. Dna2 cleaves selectively the 5′-overhang ssDNA of the Y-structured DNA, producing a 3′-ss overhang (8). Mre11, one of the first proteins bound to DSB sites, provides a structural platform for the initial recruitment of Dna2 to DSB sites (49, 50). It was noted that the level of Dna2 associated near the DSB sites increased during resection (50). This observation cannot be explained only by the protein-protein interactions between Dna2 and Mre11 (or Sgs1 at the later stage). It is reasonable to assume that the initial resection catalyzed by Mre11 and Sae2 or the unwinding of duplex ends by Sgs1, respectively, would alter the integrity of the structural platform recruiting Dna2. This would lead to the reduction in the further recruitment of Dna2. We suggest that the binding of Dna2 to secondary structures provides an additional mechanism for the recruitment. Once hairpin structures form from long ssDNA regions generated by Sgs1, they could act as binding sites for Dna2. This mechanism may also explain previous observations that the level of Dna2 continued to increase near DSB sites after resection. Consistent with this notion, we found that dna2Δ405N mutant cells displayed defective long range resection activity in the absence of Exo1, a nuclease that acts in conjunction with Sgs1-Dna2 (supplemental Fig. S3). If hairpins formed by Sgs1-dependent DNA unwinding were not processed properly, they could hinder subsequent resections. The biochemical properties of Dna2 revealed here support a role for Dna2 in the removal of stable hairpin structure in ssDNA. Dna2, upon binding to secondary structures, could resolve these structures, which in turn allows RPA binding. RPA binding ultimately leads to the enhanced Dna2-catalyzed cleavage of ssDNA generated from DSB ends by Sgs1. It is worthwhile to note that the N-terminal domain of Dna2 interacts genetically and physically with the N-terminal domain of Rpa1 (51). Therefore, deletion of the N-terminal domain of Dna2 could affect interactions with both RPA and hairpin structures in ssDNA produced by Sgs1, resulting in the impaired long range resection of DSBs.
When replication forks stall upon encountering DNA damage, uncoupling of the replicative helicase and polymerases at the forks can lead to the generation of extensive ssDNA regions, resulting in S-phase checkpoint activation in wild type cells (52). In dna2Δ405N cells, however, formation of extensive secondary structure in ssDNA region is allowed, leading to a net decrease in the amount of RPA-coated ssDNA, a known signal for cell cycle arrest; this would result in the ineffective activation of the S-phase checkpoint. It should be noted that Dna2 does not cleave ssDNA flanked by duplex DNAs, like the ssDNA region generated by the uncoupling of the replicative helicase and polymerases at stalled forks (7). The inability of Dna2 to cleave ssDNA flanked by duplex DNA could provide one conceivable mechanism by which Dna2 plays a role in the maintenance of S-phase checkpoint activation in response to DNA damage. When the fork demise occurs, however, broken forks could be repaired by DSB resection by homologous recombination (53). In this case, long range resection by Dna2 is affected as recently demonstrated (39), which ultimately leads to a reduced level of ssDNA. In both cases, the amount of ssDNA could be reduced to a level that fails to elicit the S-phase checkpoint activation. Thus, we propose that Dna2 can be a major factor or regulator of the amount of ssDNA present in cells.
The inefficient processing in vitro of CTG repeat flaps by Dna2Δ405N described here immediately suggests that the stability of CTG repeats in vivo may be affected by deletion of the N-terminal domain of Dna2. If in vitro defects observed with the N-terminal 45-kDa domain of Dna2 were observed in vivo, both CTG and CAG repeats could be less stable in vivo in a dna2Δ405N mutant strain than in wild type. We tested this possibility by transforming a plasmid containing 174 CTG repeats into wild type and dna2Δ405N mutant cells and found that the 174 CTG repeats in the plasmid were less stable in the mutant dna2Δ405N strain (supplemental Fig. S4). When we tested yeast strains containing a chromosomal insertion of 84 CTG repeats, however, we failed to observe significant differences in their stability between wild type and dna2Δ405N cells (data not shown). The difference in stability between chromosomal and plasmid-born CTG repeats might be attributed to the limited capacity of yeast cells to deal with CTG repeats. If this were the case, it is conceivable that this becomes particularly problematic when CTG repeat sequences are present in a plasmid.
Acknowledgments
We are grateful to Drs. Masayuki Seki (Tohoku University, Japan) and Sang Eun Lee (University of Texas) for strains. We thank Dr. Jerard Hurwitz (Sloan-Kettering Institute) for the critical reading of the manuscript.
This work was supported by a grant from the Korean Ministry of Education, Science and Technology (to Y.-S. S.).

This article contains supplemental Materials and Methods and Figs. S1–S4.
- ss
- single-stranded
- ssc
- single-stranded circular
- pol
- polymerase
- RPA
- replication protein A
- nt
- nucleotide
- MMS
- methyl methanesulfonate
- DSB
- double strand break
- CPT
- camptothecin
- 5-FOA
- 5-fluoorotic acid.
REFERENCES
- 1. Budd M. E., Campbell J. L. (1995) A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc. Natl. Acad. Sci. U.S.A. 92, 7642–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bae S. H., Choi E., Lee K. H., Park J. S., Lee S. H., Seo Y. S. (1998) Dna2 of Saccharomyces cerevisiae possesses a single-stranded DNA-specific endonuclease activity that is able to act on double-stranded DNA in the presence of ATP. J. Biol. Chem. 273, 26880–26890 [DOI] [PubMed] [Google Scholar]
- 3. Budd M. E., Choe W.-C., Campbell J. (2000) The nuclease activity of the yeast Dna2 protein, which is related to the RecB-like nucleases, is essential in vivo. J. Biol. Chem. 275, 16518–16529 [DOI] [PubMed] [Google Scholar]
- 4. Lee K. H., Kim D. W., Bae S. H., Kim J. A., Ryu G. H., Kwon Y. N., Kim K. A., Koo H. S., Seo Y. S. (2000) The endonuclease activity of the yeast Dna2 enzyme is essential in vivo. Nucleic Acids Res. 28, 2873–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kang Y. H., Lee C. H., Seo Y. S. (2010) Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 45, 71–96 [DOI] [PubMed] [Google Scholar]
- 6. Kang H. Y., Choi E., Bae S. H., Lee K. H., Gim B. S., Kim H. D., Park C., MacNeill S. A., Seo Y. S. (2000) Genetic analyses of Schizosaccharomyces pombe dna2+ reveal that Dna2 plays an essential role in Okazaki fragment metabolism. Genetics 155, 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bae S. H., Bae K. H., Kim J. A., Seo Y. S. (2001) RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412, 456–461 [DOI] [PubMed] [Google Scholar]
- 8. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., Kowalczykowski S. C. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 12. Formosa T., Nittis T. (1998) Suppressors of the temperature sensitivity of DNA polymerase α mutations in Saccharomyces cerevisiae. Mol. Gen. Genet. 257, 461–468 [DOI] [PubMed] [Google Scholar]
- 13. Bae S. H., Kim D. W., Kim J., Kim J. H., Kim D. H., Kim H. D., Kang H. Y., Seo Y. S. (2002) Coupling of DNA helicase and endonuclease activities of yeast Dna2 facilitates Okazaki fragment processing. J. Biol. Chem. 277, 26632–26641 [DOI] [PubMed] [Google Scholar]
- 14. Hu J., Sun L., Shen F., Chen Y., Hua Y., Liu Y., Zhang M., Hu Y., Wang Q., Xu W., Sun F., Ji J., Murray J. M., Carr A. M., Kong D. (2012) The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149, 1221–1232 [DOI] [PubMed] [Google Scholar]
- 15. Bae S. H., Seo Y. S. (2000) Characterization of the enzymatic properties of the yeast Dna2 helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem. 275, 38022–38031 [DOI] [PubMed] [Google Scholar]
- 16. Jin Y. H., Ayyagari R., Resnick M. A., Gordenin D. A., Burgers P. M. (2003) Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′–5′-exonuclease activities of pol δ in the creation of a ligatable nick. J. Biol. Chem. 278, 1626–1633 [DOI] [PubMed] [Google Scholar]
- 17. Kao H. I., Bambara R. A. (2003) The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit. Rev. Biochem. Mol. Biol. 38, 433–452 [DOI] [PubMed] [Google Scholar]
- 18. Kao H. I., Veeraraghavan J., Polaczek P., Campbell J. L., Bambara R. A. (2004) On the roles of Saccharomyces cerevisiae Dna2p and Flap endonuclease 1 in Okazaki fragment processing. J. Biol. Chem. 279, 15014–15024 [DOI] [PubMed] [Google Scholar]
- 19. Spiro C., Pelletier R., Rolfsmeier M. L., Dixon M. J., Lahue R. S., Gupta G., Park M. S., Chen X., Mariappan S. V., McMurray C. T. (1999) Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 4, 1079–1085 [DOI] [PubMed] [Google Scholar]
- 20. Burgers P. M., Gerik K. J. (1998) Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem. 273, 19756–19762 [DOI] [PubMed] [Google Scholar]
- 21. Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M. (2004) Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 18, 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johansson E., Garg P., Burgers P. M. (2004) The pol32 subunit of DNA polymerase δ contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J. Biol. Chem. 279, 1907–1915 [DOI] [PubMed] [Google Scholar]
- 23. Budd M. E., Tong A. H., Polaczek P., Peng X., Boone C., Campbell J. L. (2005) A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genet. 6, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reynolds N., Warbrick E., Fantes P. A., MacNeill S. A. (2000) Essential interaction between the fission yeast DNA polymerase δ subunit Cdc27 and Pcn1 (PCNA) mediated through a C-terminal p21(Cip1)-like PCNA-binding motif. EMBO J. 19, 1108–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zuo S., Bermudez V., Zhang G., Kelman Z., Hurwitz J. (2000) Structure and activity associated with multiple forms of Schizosaccharomyces pombe DNA polymerase δ. J. Biol. Chem. 275, 5153–5162 [DOI] [PubMed] [Google Scholar]
- 26. Tanaka H., Ryu G. H., Seo Y. S., MacNeill S. A. (2004) Genetics of lagging strand DNA synthesis and maturation in fission yeast: suppression analysis links the Dna2-Cdc24 complex to DNA polymerase δ. Nucleic Acids Res. 32, 6367–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rossi M. L., Pike J. E., Wang W., Burgers P. M., Campbell J. L., Bambara R. A. (2008) Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J. Biol. Chem. 283, 27483–27493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ryu G. H., Tanaka H., Kim D. H., Kim J. H., Bae S. H., Kwon Y. N., Rhee J. S., MacNeill S. A., Seo Y. S. (2004) Genetic and biochemical analyses of Pfh1 DNA helicase function in fission yeast. Nucleic Acids Res. 32, 4205–4216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Budd M. E., Reis C. C., Smith S., Myung K., Campbell J. L. (2006) Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase δ. Mol. Cell. Biol. 26, 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tishkoff D. X., Filosi N., Gaida G. M., Kolodner R. D. (1997) A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88, 253–263 [DOI] [PubMed] [Google Scholar]
- 31. Podust V. N., Hübscher U. (1993) Lagging strand DNA synthesis by calf thymus DNA polymerases α, β, δ, and ϵ in the presence of auxiliary proteins. Nucleic Acids Res. 21, 841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maga G., Villani G., Tillement V., Stucki M., Locatelli G. A., Frouin I., Spadari S., Hübscher U. (2001) Okazaki fragment processing: modulation of the strand displacement activity of DNA polymerase δ by the concerted action of replication protein A, proliferating cell nuclear antigen, and flap endonuclease-1. Proc. Natl. Acad. Sci. U.S.A. 98, 14298–14303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rossi M. L., Bambara R. A. (2006) Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J. Biol. Chem. 281, 26051–26061 [DOI] [PubMed] [Google Scholar]
- 34. Bae S. H., Kim J. A., Choi E., Lee K. H., Kang H. Y., Kim H. D., Kim J. H., Bae K. H., Cho Y., Park C., Seo Y. S. (2001) Tripartite structure of Saccharomyces cerevisiae Dna2 helicase/endonuclease. Nucleic Acids Res. 29, 3069–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brill S. J., Stillman B. (1989) Yeast replication factor-A functions in the unwinding of the SV40 origin of DNA replication. Nature 342, 92–95 [DOI] [PubMed] [Google Scholar]
- 36. Freudenreich C. H., Kantrow S. M., Zakian V. A. (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856 [DOI] [PubMed] [Google Scholar]
- 37. Friedman K. L., Brewer B. J. (1995) Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 262, 613–627 [DOI] [PubMed] [Google Scholar]
- 38. Wahl G. M., Stern M., Stark G. R. (1979) Efficient transfer of large DNA fragments from agarose gels to diazobenzyloxymethyl-paper and rapid hybridization by using dextran sulfate. Proc. Natl. Acad. Sci. U.S.A. 76, 3683–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen X., Niu H., Chung W. H., Zhu Z., Papusha A., Shim E. Y., Lee S. E., Sung P., Ira G. (2011) Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 18, 1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paulovich A. G., Hartwell L. H. (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82, 841–847 [DOI] [PubMed] [Google Scholar]
- 41. Lew D. J. (2003) The morphogenesis checkpoint: how yeast cells watch their figures. Curr. Opin. Cell Biol. 15, 648–653 [DOI] [PubMed] [Google Scholar]
- 42. Redon C., Pilch D. R., Rogakou E. P., Orr A. H., Lowndes N. F., Bonner W. M. (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 4, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Santocanale C., Diffley J. F. (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395, 615–618 [DOI] [PubMed] [Google Scholar]
- 44. Shirahige K., Hori Y., Shiraishi K., Yamashita M., Takahashi K., Obuse C., Tsurimoto T., Yoshikawa H. (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature 395, 618–621 [DOI] [PubMed] [Google Scholar]
- 45. Yabuki N., Terashima H., Kitada K. (2002) Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7, 781–789 [DOI] [PubMed] [Google Scholar]
- 46. Newlon C. S., Collins I., Dershowitz A., Deshpande A. M., Greenfeder S. A., Ong L. Y., Theis J. F. (1993) Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold Spring Harbor Symp. Quant. Biol. 58, 415–423 [DOI] [PubMed] [Google Scholar]
- 47. Marchetti M. A., Kumar S., Hartsuiker E., Maftahi M., Carr A. M., Freyer G. A., Burhans W. C., Huberman J. A. (2002) A single unbranched S-phase DNA damage and replication fork blockage checkpoint pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 7472–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zegerman P., Diffley J. F. (2010) Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mimitou E. P., Symington L. S. (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shim E. Y., Chung W. H., Nicolette M. L., Zhang Y., Davis M., Zhu Z., Paull T. T., Ira G., Lee S. E. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bae K. H., Kim H. S., Bae S. H., Kang H. Y., Brill S., Seo Y. S. (2003) Bimodal interaction between replication protein A and Dna2 is critical for Dna2 function both in vivo and in vitro. Nucleic Acids Res. 31, 3006–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Segurado M., Tercero J. A. (2009) The S-phase checkpoint: targeting the replication fork. Biol. Cell 101, 617–627 [DOI] [PubMed] [Google Scholar]
- 53. Lopes M., Cotta-Ramusino C., Pellicioli A., Liberi G., Plevani P., Muzi-Falconi M., Newlon C. S., Foiani M. (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412, 557–561 [DOI] [PubMed] [Google Scholar]
- 54. Onoda F., Takeda M., Seki M., Maeda D., Tajima J., Ui A., Yagi H., Enomoto T. (2004) SMC6 is required for MMS-induced interchromosomal and sister chromatid recombinations in Saccharomyces cerevisiae. DNA Repair 3, 429–439 [DOI] [PubMed] [Google Scholar]
- 55. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]