

Background: Neuronatin was identified as a novel substrate of Lafora disease ubiquitin ligase malin; however, its role in disease biology is unknown.
Results: Neuronatin causes increased intracellular Ca2+ and ER stress and accumulated as insoluble aggregates in LD brain and skin biopsy samples.
Conclusion: Neuronatin-induced aberrant Ca+2 signaling might trigger LD pathogenesis.
Significance: These findings provide a new insight in understanding pathogenesis of LD.
Keywords: Calcium Signaling, Endoplasmic Reticulum Stress, Glycogen Storage Disease, Neurodegenerative Diseases, Protein Aggregation
Abstract
Lafora disease (LD) is a teenage-onset inherited progressive myoclonus epilepsy characterized by the accumulations of intracellular inclusions called Lafora bodies and caused by mutations in protein phosphatase laforin or ubiquitin ligase malin. But how the loss of function of either laforin or malin causes disease pathogenesis is poorly understood. Recently, neuronatin was identified as a novel substrate of malin that regulates glycogen synthesis. Here we demonstrate that the level of neuronatin is significantly up-regulated in the skin biopsy sample of LD patients having mutations in both malin and laforin. Neuronatin is highly expressed in human fetal brain with gradual decrease in expression in developing and adult brain. However, in adult brain, neuronatin is predominantly expressed in parvalbumin-positive GABAergic interneurons and localized in their processes. The level of neuronatin is increased and accumulated as insoluble aggregates in the cortical area of LD brain biopsy samples, and there is also a dramatic loss of parvalbumin-positive GABAergic interneurons. Ectopic expression of neuronatin in cultured neuronal cells results in increased intracellular Ca2+, endoplasmic reticulum stress, proteasomal dysfunction, and cell death that can be partially rescued by malin. These findings suggest that the neuronatin-induced aberrant Ca2+ signaling and endoplasmic reticulum stress might underlie LD pathogenesis.
Introduction
Lafora disease (LD)4 is an autosomal recessive progressive myoclonus epilepsy, usually manifest during teenage, and the patient dies within a decade of disease onset. The disease is clinically characterized by progressive increase in generalized tonic-clonic seizures, myoclonic and visual seizures, dementia, psychoses, muscle wasting, leading the patient to a vegetative state and ultimately death (1–5). Pathological features of LD include gradual increase in accumulation of insoluble polyglucosan bodies (commonly known as Lafora bodies) and in neuronal loss. Lafora bodies are not only observed in brain but also in other non-neuronal tissues like liver, heart, skeletal muscle, and skin (1, 3, 6).
LD is caused by mutations in the EPM2A or EPM2B (NHLRC1) genes encoding laforin (a protein phosphatase) and malin (a E3 ubiquitin ligase), respectively (2, 7–9). Patients with mutations in either laforin or malin are clinically and pathologically indistinguishable (2, 10), indicating that both of these proteins work together in some common signaling pathways, and defect in those pathways could lead to disease manifestation. Emerging evidence indicates that both laforin and malin could play an important role in regulation of glycogen metabolism (11–15), in autophagy (16–18), and in Wnt signaling pathway (19, 20). Knock-out mice for both laforin and malin also exhibits progressive accumulation of Lafora bodies in various tissues including brain, defects in autophagic degradation pathway, and widespread neurodegeneration (17, 18, 21–24). Current finding in LD mice models point toward the role of abnormal accumulation of Lafora bodies (18, 23, 25) and impairment of intracellular protein degradation pathways (17, 18, 26) in the disease pathogenesis. Lafora bodies in the brain are also associated with a number of cellular factors including the components of ubiquitin-proteasome system (UPS) and autophagy, indicating further the involvement of these protein degradation pathways in disease progression (17, 26). But how exactly Lafora bodies induce neurodegeneration is not clear at present, and the possible mechanism of dysfunction of protein degradation pathways in LD is also poorly understood.
Neuronatin, a small proteolipid family membrane protein, is highly expressed in embryonic brain and suggested to be involved in brain development (27, 28). It exists in two major isoforms, α and β, consisting of 81 and 54 amino acids, respectively (29, 30). In adult mice brain, it is expressed at very low levels and largely restricted to limbic structures (31). Neuronatin is also expressed in several other peripheral tissues including pancreas, adipose tissues, and skin (32–34). Although the physiological function of neuronatin is poorly understood, existing literature indicates its involvement in glucose-mediated insulin secretion from pancreas, in adipogenesis, and in metabolic regulation (31, 32, 35–37). Interestingly, neuronatin has significant homology with phospholamban and PMP1 that function as regulatory subunit of ion channel and shown to promote neural lineage in embryonic stem cells through increasing the intracellular Ca2+ by antagonizing serco/endoplasmic reticulum Ca2+-ATPase-2 (SERCA2) (27, 38). Recently, we have reported that malin interacts with neuronatin and promotes its proteasome-mediated degradation. Malin also negatively regulates neuronatin-stimulated glycogen synthesis (39). Here, we demonstrate that neuronatin level is increased in LD skin biopsy samples irrespective of mutation in either malin or laforin and accumulated as insoluble aggregates in LD brain biopsy samples. We also provide evidence that neuronatin-mediated aberrant Ca2+ signaling might be an important early event leading to neurodegeneration in LD by inducing endoplasmic reticulum (ER) and proteasomal stress.
EXPERIMENTAL PROCEDURES
Materials
Cell culture media, MG132, dbcAMP, thapsigargin, tunicamycin, and mouse monoclonal calbindin antibody were purchased from Sigma. Lipofectamine 2000, OptiMEM, Fura-2/AM, and mouse monoclonal V5 antibody were purchased from Invitrogen. Fetal bovine serum and antibiotics penicillin/streptomycin were obtained from GIBCO, and Bradford reagent and prestained protein molecular mass markers were from Bio-Rad. Mouse monoclonal anti-myc, anti-GAPDH, anti-CHOP, rabbit polyclonal anti-IRE-α, anti-β-tubulin, and goat polyclonal anti-Hsc70/Hsp70 and anti-Grp78 were purchased from Santa Cruz Biotechnology. Rabbit polyclonal anti-ubiquitin, mouse monoclonal anti-GFP, and rabbit polyclonal anti-neuronatin were purchased from Dako, Roche Applied Science, and Abcam, respectively. Rabbit polyclonal parvalbumin antibody was obtained from Swant. HRP- and fluorophore-conjugated secondary antibodies, Novared kit, and Vectashield mounting media with 4′6′-diamidino-2-phenylindole (DAPI) were purchased from Vector Laboratories. The construction of malin-V5, neuronatin β-myc plasmid, and the source of pd1EGFP and C26S malin plasmids were described elsewhere (39–41). Mouse-specific neuronatin siRNA (a pool of three target-specific 20–25 nucleotide) and control (scramble sequences) were purchased from Santa Cruz Biotechnology.
The serial paraffin sections of axillary skin and brain biopsy samples, paraffin section of control brain with different ages (39 weeks, 11, 15, and 50 years) were obtained from the archives of Human Brain Tissue Repository, National Institute of Mental Health and Neurosciences, Bangalore, India, according to the ethical guidelines of Indian Council of Medical Research, with the consent of the subject to utilize the material for research purposes, protecting the confidentiality of the subject. We used three clinically and genetically confirmed LD (malin mutants; L279P and delF216-D233 and laforin mutant; N148Y) and three normal skin biopsy samples (samples serial numbers 1718, 1762, and 1848) in this study. Two clinically confirmed cases of LD brain biopsy samples (LD1, 623; LD2, 544) were used in our study. LD1 and LD2 patients were of 17 and 23 years old, respectively.
Cell Culture, Transfection, and Immunoblotting
Neuro 2a cells were routinely cultured in DMEM with 10% fetal bovine serum and antibiotics penicillin/streptomycin. Cells were plated at subconfluent density into 6-well tissue cultured plates and transfected after 24 h with Lipofectamine 2000 according to the manufacturer's instruction. Transfection efficiency was ∼50–60%. After 36 or 48 h of transfection, cells were collected, washed twice in ice-cold phosphate-buffered saline (PBS), pelleted by centrifugation, and lysed under ice for 30 min using Nonidet P-40 lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, complete EDTA-free protease inhibitor mixture). Cell lysates were collected after brief sonication and centrifugation at 20,000 × g for 15 min. Protein concentrations were measured using the Bradford method. Total cell lysates were resolved through SDS-polyacrylamide gel electrophoresis and processed for immunoblotting as described elsewhere (42). Neuronatin and V5 antibody were used at 1:5000 dilution; myc, IRE-α, Grp78, CHOP, ubiquitin, and β-tubulin antibody at 1:1000 dilution; GAPDH at 1:15,000 dilution.
Immunofluorescence Staining and Counting of Apoptotic Cells
Neuro 2a cells grown in 2-well tissue cultured chamber slides were transiently transfected with various plasmids, differentiated with dbcAMP, and 72 h after transfection, cells were processed for immunofluorescence staining. In some experiment cells were treated with MG132 and subjected to immunofluorescence staining. Briefly, cells were washed with PBS three times, fixed with 4% paraformaldehyde in PBS for 30 min. After fixation, cell were washed three times again with PBS and permeabilized with 0.3% TritonX-100 in PBS for 10 min and subsequently blocked with 3% BSA in PBS for 1 h. Cells were incubated with various primary antibodies overnight at 4 °C. After three washings with PBS, cells were incubated with fluorophore-conjugated secondary antibody for 1 h. Cells were finally washed with PBS and mounted in medium containing DAPI and imaged using Axioplan fluorescence microscope/Apotome (Zeiss). Anti-myc, anti-neuronatin, and anti-V5 were used at 1:1000 dilution.
Transfected cells exhibiting apoptotic bodies (fragmented nuclei) were manually counted at a magnification of ×20. Fields were randomly selected, and ∼200 cells were scored per experiment. Each experiment was repeated at least three times, and counts were performed in a blinded manner.
Measurement of Intracellular Ca2+ Level
Neuro 2a cells were plated into 6-well tissue culture plates and transfected with various plasmids and neuronatin siRNA. Thapsigargin (1 μm) was added in one of the wells for 12 h to serve as the positive control. Thirty-six hours after transfection, cells were washed with PBS and loaded with 5 μm Fura-2/AM for 30 min. Cells were then washed and treated with 1× Hanks' balanced salt solution for 45 min, washed, collected, and transferred to a 96-well black bottom plate, and the fluorescence was measured using a fluorescence plate reader having an excitation filter of 340 and 380 nm and an emission filter of 510 nm. The raw data were obtained, and the intracellular calcium concentration was calculated from the ratio of fluorescence emissions at excitation wavelengths of 340 and 380 nm (F340/F380) according to the equation described earlier (43).
Immunohistochemical Techniques and Periodic Acid Schiff (PAS) Staining
The paraffin-embedded axillary skin and brain sections were deparaffinized with xylene, rehydrated with a gradually decreasing percentage of alcohol, and subjected to antigen retrieval in a pressure cooker. The sections were treated with 3% H2O2 in methanol, permeabilized in 0.3% Triton X-100, and blocked with 5% goat serum containing 0.1% Triton X-100. The sections were incubated with primary antibodies to neuronatin (1:1000 dilution), ubiquitin (1:200 dilution), and Hsc70/Hsp70 (1:250 dilution), respectively, overnight. Color development was carried out using the Nova red kit. One section from each case was subjected to PAS staining to confirm the presence of Lafora bodies according to previously described method (44). In some experiment, control human brain section was subjected to double immunofluorescence staining using either parvalbumin and neuronatin antibodies or neuronatin and calbindin antibodies.
Proteasome Activity Assays
Neuro 2a cells were seeded onto 6-well tissue-cultured plates and after overnight of platting cells were transfected with various plasmids and treated with dbcAMP. Collected cells were processed for proteasome activity assays as previously described (41). The substrates Suc-Leu-Leu-Val-Tyr-MCA (methyl cumaryl amide) and Z-Leu-Leu-Glu-MCA were used to measure the chymotrypsin-like and post-glutamyl peptidyl hydrolytic-like protease activity, respectively.
Statistical Analysis
Data were analyzed by one-way analysis of variance using SigmaStat software. Values are presented as mean ± S.D. The Bonferroni post hoc test was conducted to compare individual means where analysis of variance indicated statistical differences. p < 0.05 was considered statistically significant.
RESULTS
Skin Biopsy Samples of LD Patients Having Mutations in Either Laforin or Malin Exhibit Increased Level of Neuronatin
We have recently identified neuronatin as a novel interacting partner of malin. Malin induces ubiquitination and proteasome-mediated degradation of neuronatin (39). We further explored the significance of interaction of malin with neuronatin in the context of LD pathogenesis. First, we studied and compared the level of neuronatin in skin biopsy sample of LD patients along with age-matched control subjects. It is important to note that the Lafora bodies are frequently observed in the apocrine sweat gland of skin (34), and skin biopsies are routinely carried out to diagnose LD (1). Because neuronatin is abundantly expressed in sweat gland of human skin, we took advantage of easily accessible LD skin biopsy samples to study the level of neuronatin. In control subjects, neuronatin was expressed in myoepithelial cells of the sweat glands and localized mostly in the membrane with diffuse cytoplasmic staining pattern. However, in the malin mutants LD skin biopsy sample, neuronatin staining was strongly increased not only in the membrane and cytoplasm but also in the nucleus of the myoepithelial cells of sweat gland (Fig. 1). A similar increase and redistribution of neuronatin into nucleus were also detected in the LD skin biopsy samples having laforin mutation. These finding suggests that functional laforin-malin complex might be essential in the regulation of neuronatin level.
FIGURE 1.

Neuronatin level is increased in axillary skin biopsy samples collected from malin and laforin mutant LD patients. Skin biopsy samples obtained from three controls (sample numbers 1718, 1762, and 1848) and three LD patients (one laforin and two malin mutants) were stained immunohistochemically using neuronatin antibody. Each LD skin sample showed PAS-positive Lafora bodies. Hsc70/Hsp70 antibody was used as positive control that detects Lafora bodies. In negative control (NC), sample was processed without primary antibody. Scale bar, 20 μm.
Age-dependent Expression and Localization of Neuronatin in Human Brain
Our next goal was to characterize the expression of neuronatin in the LD brain and its involvement in LD pathogenesis. Neuronatin expression is developmentally regulated in rodent brain. However, its expression pattern and localization in human brain are not well known. Therefore, we next attempted to study the expression and localization of neuronatin in human brain of different ages. Fig. 2A shows that the expression of neuronatin is very high in the cortical neurons of 39-week-old brain. However, its expression was drastically reduced in the cortical neurons of 15- and 50-year-old brain. Immunoblot analysis of neuronatin also confirmed similar findings (Fig. 2B). Interestingly, a selective population of neurons in 15- and 50-year-old brain expressed high level of neuronatin and was localized not only in the cell soma but also in the neuronal processes. Further search revealed that the parvalbumin-positive (PV+ve) GABAergic interneurons selectively expressed high level of neuronatin in the brains of 15- and 50-year-old individuals (Fig. 2C), whereas neuronatin was not expressed in calbindin-positive GABAergic neurons. Extensive studies in adult mouse brain also showed high levels of expression of neuronatin in PV+ve GABAergic interneurons in the cortex, hippocampus, amygdala, and hypothalamus.
FIGURE 2.

Age-dependent changes in the expression and localization of neuronatin in human brain. A, brain sections of different ages were immunohistochemically stained with neuronatin antibody. Arrow indicates the cell expressing a high level of neuronatin, and the arrowhead points to the localization of neuronatin in neuritic puncta. NC, negative control. Scale bar, 40 μm. B, human brain lysates (cortex area) were subjected to immunoblot analysis using neuronatin antibody. C, double immunofluorescence staining of brain section (cortex area from 15-year-old control) shows localization of neuronatin with PV+ve GABAergic interneurons. Nuclei were counterstained with DAPI. FITC-conjugated secondary antibody was used to detect PV or calbindin, and Alexa Fluor 594-labeled secondary was used to recognize neuronatin. Scale bar, 20 μm.
LD Brain Exhibits Increased Accumulation of Neuronatin and Loss of PV+ve GABAergic Interneurons
The symptoms of LD usually appear early in adolescence. Because neuronatin is widely expressed in developing brain including a subpopulation of GABAergic neurons, we further explored its level and localization in cortical areas of LD brain. To our surprise, we noticed frequent accumulation of neuronatin as insoluble aggregates in cortical areas (Fig. 3). Some neurons and their processes also showed a very high level of accumulation of neuronatin compared with age-matched control brain. Ubiquitin immunostaining also revealed accumulation of insoluble inclusions in the LD brain. Ubiquitin and neuronatin-positive Lafora bodies were strikingly similar in their appearance. Most Lafora bodies were strongly labeled with ubiquitin (Fig. 4). In addition, ubiquitin-positive multiple inclusions were also sometimes observed around the perinuclear region (Fig. 4). Because neuronatin is expressed in PV+ve GABAergic neurons, we were curious to know the status of these interneurons in LD brain biopsy samples. Immunohistochemical staining revealed approximately 75–85% reduction in the numbers of the PV+ve GABAergic neuron in the cortical areas of both the LD brain biopsy samples (Fig. 5). Existing PV+ve GABAergic neurons in LD brain biopsy samples also showed degeneration in their processes (Fig. 5).
FIGURE 3.

Immunohistochemical staining of neuronatin and ubiquitin in control (15 years old) and two different LD brain biopsy samples (LD1 and LD2 of 17 and 23 years old, respectively). Lower panel of neuronatin staining shows higher magnification images. Arrow indicates neuronatin or ubiquitin-positive inclusions. Arrowhead points to the cell or neurites exhibiting high level of neuronatin in LD brain. PAS staining was conducted to confirm the presence of Lafora bodies (indicated by arrows) in LD brain samples. In negative control (NC), LD brain section was processed without neuronatin antibody. Scale bars, 40 μm in smaller magnification images and 20 μm in higher magnification images. Bottom panel shows higher magnification images of localization of neuronatin or ubiquitin into Lafora bodies.
FIGURE 4.

A, immunohistochemical staining of ubiquitin in LD brain biopsy samples showing ubiquitin-positive perinuclear aggregates. B, co-localization of ubiquitin with PAS-positive Lafora body (indicated by arrow).
FIGURE 5.

Immunohistochemical staining of PV+ve GABAergic interneurons in control (15 years old) and two different LD brain biopsy samples (LD1, 17 years and LD2, 23 years old). Arrow indicates PV+ve neurons. Scale bar, 20 μm. For counting PV-immunostained cells in cortex area, two serial sections in each sample (two controls and two LD) were used. At least three 20× images of same size (500 μm × 400 μm) in each section were used for counting PV-immunostained cells. The numbers of PV+ve neurons in control brain was 9.7 ± 2.68, whereas in LD brain it was 1.8 ± 0.53 (2.2 in LD1 and 1.45 in LD2). LD1 showed approximately 78% reduction of PV-stained cells whereas LD2 showed ∼85% reduction compared with control.
Neuronatin Is Accumulated in Perinuclear Region in Response to Proteasomal Inhibition, and Its Overexpression Causes Increased Intracellular Ca2+ and Apoptosis, Which Are Partially Restored by Malin
Because neuronatin is accumulated and forms inclusions in LD brain, we next explored its possible role in neurodegeneration using cultured neuronal cells. Neuronatin was expressed in Neuro 2a cells and localized in the membrane and cytosol. Treatment with a proteasome inhibitor caused its accumulation in perinuclear region in the cell indicating that it is rapidly degraded via UPS (Fig. 6A). Immnoblot analysis also confirmed the increased level of neuronatin in proteasome inhibitor-treated cells. Neuronatin was also accumulated in insoluble fraction (Fig. 6A). The half-life of neuronatin was also very short (approximately 1 h), which was extended upon inhibition of proteasome function (Fig. 6B). Recently, neuronatin was shown to increase the intracellular Ca2+ levels by antagonizing the SERCA2 pump located in the ER. We have also observed that the overexpression or knockdown of neuronatin in Neuro 2a cells caused an increase or decrease in intracellular Ca2+, respectively (Fig. 6C). Thapsigargin, a noncompetitive inhibitor of SERCA pump, was used as positive control. Interestingly, co-expression of wild type malin significantly restored the neuronatin-induced increased intracellular Ca2+ (Fig. 6D). Malin also rescued neuronatin-induced apoptotic cell death (Fig. 6E). Although wild type malin had no effect on intracellular Ca2+ levels, its LD-associated C26S mutant caused significant increase in intracellular Ca2+ and also apoptosis. This is not surprising because the C26S mutant of malin is shown to form aggregates in cell culture and induce apoptosis (40).
FIGURE 6.

Neuronatin is degraded by proteasome, and its overexpression leads to increased intracellular Ca2+ and apoptosis that can be suppressed by malin. A, Neuro 2a cells were plated onto 6-well tissue-cultured plates or two-well chamber slides and on the following day cells were treated with MG132 (10 μm for 6 h) and processed for immunofluorescence staining or subjected to preparation of soluble and insoluble fractions followed by immunoblot analysis using neuronatin antibody. Arrows indicate accumulation of neuronatin in perinuclear region. Scale bar, 20 μm. B, cells were seeded in a 6-well tissue culture plate and, on the following day, they were chased with 25 μg/ml cycloheximide for different time periods in the presence or absence of 10 μm MG132. Collected cells at each time point were then processed for immunoblotting using antibodies against neuronatin and GAPDH. Quantitation of band intensities was performed using NIH ImageJ analysis software, and values were normalized against GAPDH. Values represent the mean ± S.D. (error bars) of three independent experiments. MG132 treatment caused significant increase (a indicates p < 0.001) in half-life of neuronatin. C, Neuro 2a cells were plated onto 6-well tissue-cultured plates and 24 h later cells were transiently transfected with either neuronatin β plasmid (Nnat OE, 2 μg/well) or neuronatin and control siRNA (30 pmol/well) for 36 h. In some experiment, cells were treated with thapsigargin (1 μm for 12 h). The cells were then processed for either measurement of intracellular Ca2+ or immunoblot analysis using neuronatin, myc (to detect overexpressed neuronatin), and GAPDH antibodies. D, cells were transfected with plasmid encoding empty pcDNA3.1 (control) or neuronatin β plasmid alone or along with wild-type or C26S mutant of malin (2 μg of each plasmid/well of 6-well plate). Thirty-six hours after transfection, cells were subjected to the analysis of intracellular Ca2+ levels. In C and D, values are mean ± S.D. of three independent experiments each performed in triplicate. The a indicates p < 0.01 compared with control, and b indicates p < 0.01 compared with neuronatin-transfected group. E, neuronatin-induced cell death is protected by malin and aggravated by LD-associated C26S mutant of malin. Cells were transfected with LacZ or neuronatin β plasmid independently or along with malin plasmids as in D for 72 h. Cells were differentiated with dbcAMP (5 mm for 48 h) and then processed for immunofluorescence staining to identify neuronatin or neuronatin- and malin-transfected cells. Nuclei were stained with DAPI to identify the apoptotic cell (fragmented nuclei). Values are mean ± S.D. of three independent experiments with minimum of 200 transfected cells counted for each experiment. The a indicates p < 0.001 in comparison with LacZ transfected control, and b shows p < 0.01 compared with neuronatin-transfected group.
Malin Suppresses Neuronatin-induced ER Stress and Proteasomal Dysfunction
Increased intracellular free Ca2+ as well as accumulation of misfolded proteins are known to generate ER stress in the cell. Because neuronatin increases the cytosolic Ca2+, we further tested its involvement on the ER stress. As shown in Fig. 7, ectopic expression of neuronatin in Neuro 2a cells resulted in ER stress in concentration-dependent manner as evident from the increased expression of Grp78, CHOP, and IRE-α. Transient co-expression of malin significantly suppressed neuronatin-induced ER stress (Fig. 7B). The C26S mutant of malin was also found to generate ER stress and amplified neuronatin-induced ER stress. We have also observed proteasomal dysfunction in neuronatin overexpressed cells as shown from the decrease in proteasome activity and increased accumulation of ubiquitinated proteins (Fig. 8, A and B). Neuronatin overexpression also caused increased accumulation of d1EGFP, a model substrate of proteasome with half-life of about 1 h (Fig. 8, C and D). Co-expression of malin along with neuronatin partially restored neuronatin-induced proteasomal inhibition (Fig. 8E). The C26S mutant of malin also inhibited cellular proteasomal function that was enhanced further in the presence of neuronatin. These in vitro studies indicate that increased accumulation of neuronatin is toxic to the neuron, which is mediated by increased intracellular free Ca2+ followed by ER stress and proteasomal dysfunction. LD-associated mutant malin also could be implicated in the ER stress generation, proteasomal dysfunction, and neurodegeneration.
FIGURE 7.
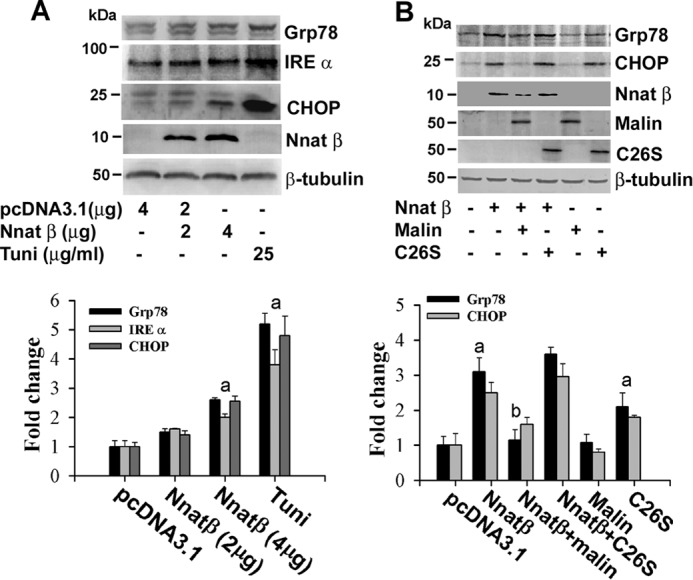
Neuronatin induces ER stress, which is suppressed by malin and enhanced by C26S mutant of malin. A, Neuro 2a cells were transfected with different concentrations of neuronatin plasmid for 48 h. Cells were treated with dbcAMP (5 mm) for 24 h before collection and then subjected to immunoblot analysis using various antibodies as indicated in the figure. Tunicamycin (Tuni, 25 μg/ml) was treated for 6 h. Band intensities of Grp78, IRE-α, and CHOP were quantified using NIH ImageJ analysis software, normalized against β-tubulin, and expressed as -fold change. B, cells were transfected with plasmid encoding neuronatin or malin independently or together for 48 h, treated with dbcAMP as in A, and then processed for immunoblotting using various antibodies shown in the figure. Grp78 and CHOP blots were quantified, normalized against β-tubulin, and expressed as -fold change. Values are mean ± S.D. of four different experiments. The a represents p < 0.05 compared with the respective empty pcDNA3.1-transfected group, and b represents p < 0.01 compared with neuronatin-transfected group.
FIGURE 8.
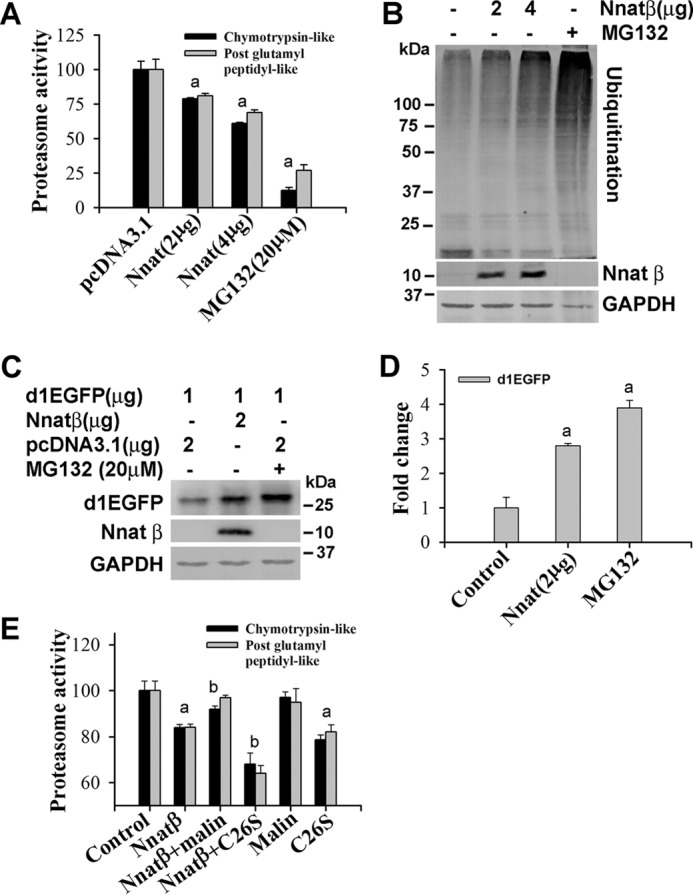
Neuronatin-induced proteasomal dysfunction is partially recovered by malin. A and B, Neuro 2a cells were transfected with different concentrations of plasmid encoding neuronatin and differentiated as described in Fig. 6A. Forty-eight hours after transfection, cells were either processed for proteasome activity assays (A) or immunoblot analysis using ubiquitin, myc (to detect neuronatin β) and GAPDH antibodies (B). Values are mean ± S.D. (error bars) of three independent experiments each performed in triplicate. The a indicates p < 0.01 compared with control. C, cells were transiently transfected with plasmid encoding pd1EGFP alone or along with neuronatin β for 48 h, and the collected cells were then processed for immunoblotting using antibody against GFP. D, the band intensities of d1EGFP from three independent experiments were quantified using NIH ImageJ analysis software. Proteasome inhibitor MG132 (20 μm for 6 h) was used as positive control. The a indicates p < 0.001 compared with control. E, cells were transfected with various plasmids and differentiated as described in Fig. 6D. Cells were collected and subjected to proteasome activity assay. Values are mean ± S.D. of three different experiments each performed in triplicate. The a indicates p < 0.01 compared with the empty pcDNA3.1-transfected group (control), and b represents p < 0.01 compared with the neuronatin-transfected group.
DISCUSSION
We had demonstrated earlier that malin interacts with neuronatin and promotes its degradation through proteasome (39). Here, we provide substantial evidence that indicates the crucial role of neuronatin in LD pathogenesis. First, we show that the level of neuronatin is significantly increased in the sweat gland of skin of LD patients having mutations not only in malin but also in laforin in contrast to control samples. The neuronatin level is also increased and often aggregated and sometimes associated with Lafora bodies in cortical neurons of LD brain biopsy samples. Ectopic expression of neuronatin in the neuronal cell leads to increase in intracellular Ca2+ and induction of apoptotic cell death. Increased accumulation of neuronatin in the skin samples of either malin or laforin mutant patients implies that these two protein complexes are involved in the turnover of neuronatin. Laforin and malin are shown to function as a complex (14, 45, 46), and in the malin knock-out mice, laforin is increased in insoluble fraction (21, 22) and therefore might be biologically inactive. Similarly, in the absence of laforin, malin also could be nonfunctional because malin is shown to be an aggregation-prone protein and can be stabilized upon binding with laforin (40).
Neuronatin is highly expressed in neonatal brain and possibly involved in neural induction and neuronal differentiation during brain development (27, 38). In adult mice, it is also found to be selectively expressed in various limbic structures (31) of brain and several non-neural tissues including skin (33, 34). Existing literature suggests the involvement of neuronatin in the regulation of glycogen and lipid metabolism, and its mutation is associated with obesity (31, 32, 37, 39). The primary and secondary structure of neuronatin is also strikingly similar with phospholamban (an ER-resident Ca2+ regulator found in cardiac muscle), and recent reports have demonstrated that it is indeed involved in increasing the intracellular Ca2+ by antagonizing SERCA2 pump in the ER (27, 38). We have also detected a significant increase in intracellular Ca2+ in neuronatin-transfected neuroblastoma cells. Because malin regulates the neuronatin level, we speculate that malin might also be associated with certain aspects of brain development and regulation of metabolism.
Although the biological role of neuronatin in the skin is not clear, it is possible that a high level of neuronatin in LD skin might be involved with abnormal function of sweat gland and Lafora body formation. In the developing LD brain, the neuronatin level is increased and might be progressively accumulated as an insoluble aggregates in postmitotic neurons. Neuronatin seems to be an unfolded and aggregate prone protein that is recognized and rapidly cleared by UPS. Blockade of cellular proteasome function lead to its aberrant accumulation and aggregation. Therefore, lack of function of the malin-laforin complex could result in inefficient clearance of unfolded neuronatin leading to its accumulation and aggregation particularly in the neurons. Increased accumulation of neuronatin or its aggregates could potentially induce neurodegeneration by increasing intracellular Ca2+ overload followed by ER and proteasomal stress. We have also shown here that overexpression of malin partially recovered neuronatin-induced ER stress and proteasomal dysfunction and rescued the neuronatin-induced cell death. Our findings are consistent with other reports suggesting a role for malin-laforin complex in the clearance of misfolded proteins and involvement of ER stress in LD pathogenesis (46, 47).
Interestingly, neuronatin is highly expressed in PV+ve GABAergic interneuron, and these inhibitory interneurons dramatically degenerated in LD brain. PV is a Ca2+-buffering protein in cytosol, and expression of neuronatin in PV-containing GABAergic interneurons indicates that neuronatin might be playing a pivotal role in Ca2+-mediated GABAergic neurotransmission and signaling pathways. Excessive accumulation of neuronatin could potentially lead to the degeneration of these inhibitory interneurons in LD brain by disturbing Ca2+ homeostasis and inducing ER stress. Our findings are very similar with a recent report that demonstrates progressive degeneration of PV+ve GABAergic interneuron in the hippocampus of malin knock-out mice (23). The loss of PV+ve inhibitory interneurons in LD brain could potentially explain the rapidly progressing seizures and myoclonus in this disease.
Although the Lafora body is one of the common pathological hallmarks of LD, its involvement in disease pathogenesis is not clearly understood. In laforin-deficient mice, degenerating neurons sometimes lacked Lafora bodies (24), whereas depletion of protein targeting to glycogen in the same mice results in disappearance of Lafora bodies and rescue of neurodegeneration and seizures (25). Emerging literature now indicates that the defect in UPS and autophagy might be causative for the evolution of LD (17, 18, 26, 46, 47). Lafora bodies are found to be associated with various components of UPS and autophagy and could potentially induce the defect in these intracellular protein degradation pathways (17, 26). Accumulation of unfolded neuronatin along with LD-associated malin or laforin mutant proteins in neurons could also potentially disturb the protein quality control system. Taken together, our finding suggests that the neuronatin-induced aberrant Ca2+ signaling and ER stress might be an important contributor in LD pathogenesis. We also indicate that LD can now join to the list of neurodegenerative disorders involving accumulation of misfolded proteins.
This work was supported in part by a core grant from the Department of Biotechnology to National Brain Research Centre, Government of India, and Extramural Grant BT/PR13590/MED/30/286/2009 from the Department of Biotechnology, Government of India.
- LD
- Lafora disease
- dbcAMP
- dibutyryl-adenosine-3′,5′ cyclic monophosphate
- ER
- endoplasmic reticulum
- PAS
- Periodic acid Schiff
- PV+ve
- parvalbumin-positive
- SERCA2
- serco/endoplasmic reticulum Ca2+-ATPase-2
- UPS
- ubiquitin proteasome system
- Z
- benzoyloxycarbonyl
- MCA
- methyl cumaryl amide.
REFERENCES
- 1. Acharya J. N., Satishchandra P., Asha T., Shankar S. K. (1993) Lafora's disease in South India: a clinical, electrophysiologic, and pathologic study. Epilepsia 34, 476–487 [DOI] [PubMed] [Google Scholar]
- 2. Singh S., Ganesh S. (2009) Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum. Mutat. 30, 715–723 [DOI] [PubMed] [Google Scholar]
- 3. Delgado-Escueta A. V. (2007) Advances in Lafora progressive myoclonus epilepsy. Curr. Neurol. Neurosci. Rep. 7, 428–433 [DOI] [PubMed] [Google Scholar]
- 4. Minassian B. A. (2001) Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 25, 21–29 [DOI] [PubMed] [Google Scholar]
- 5. Ganesh S., Puri R., Singh S., Mittal S., Dubey D. (2006) Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 51, 1–8 [DOI] [PubMed] [Google Scholar]
- 6. Carpenter S., Karpati G. (1981) Sweat gland duct cells in Lafora disease: diagnosis by skin biopsy. Neurology 31, 1564–1568 [DOI] [PubMed] [Google Scholar]
- 7. Chan E. M., Young E. J., Ianzano L., Munteanu I., Zhao X., Christopoulos C. C., Avanzini G., Elia M., Ackerley C. A., Jovic N. J., Bohlega S., Andermann E., Rouleau G. A., Delgado-Escueta A. V., Minassian B. A., Scherer S. W. (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127 [DOI] [PubMed] [Google Scholar]
- 8. Chan E. M., Omer S., Ahmed M., Bridges L. R., Bennett C., Scherer S. W., Minassian B. A. (2004) Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology 63, 565–567 [DOI] [PubMed] [Google Scholar]
- 9. Minassian B. A., Lee J. R., Herbrick J. A., Huizenga J., Soder S., Mungall A. J., Dunham I., Gardner R., Fong C. Y., Carpenter S., Jardim L., Satishchandra P., Andermann E., Snead O. C., 3rd, Lopes-Cendes I., Tsui L. C., Delgado-Escueta A. V., Rouleau G. A., Scherer S. W. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 20, 171–174 [DOI] [PubMed] [Google Scholar]
- 10. Singh S., Sethi I., Francheschetti S., Riggio C., Avanzini G., Yamakawa K., Delgado-Escueta A. V., Ganesh S. (2006) Novel NHLRC1 mutations and genotype-phenotype correlations in patients with Lafora's progressive myoclonic epilepsy. J. Med. Genet. 43, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lohi H., Ianzano L., Zhao X. C., Chan E. M., Turnbull J., Scherer S. W., Ackerley C. A., Minassian B. A. (2005) Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 14, 2727–2736 [DOI] [PubMed] [Google Scholar]
- 12. Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Ros S., Fernandez-Sanchez M. E., Garcia-Fojeda B., Criado Garcia O., Vilchez D., Dominguez J., Garcia-Rocha M., Sanchez-Piris M., Aguado C., Knecht E., Serratosa J., Guinovart J. J., Sanz P., Rodriguez de Córdoba S. (2008) Regulation of glycogen synthesis by the Laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 17, 667–678 [DOI] [PubMed] [Google Scholar]
- 13. Worby C. A., Gentry M. S., Dixon J. E. (2008) Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medraño-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez de Córdoba S., Guinovart J. J. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413 [DOI] [PubMed] [Google Scholar]
- 15. Cheng A., Zhang M., Gentry M. S., Worby C. A., Dixon J. E., Saltiel A. R. (2007) A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease. Genes Dev. 21, 2399–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aguado C., Sarkar S., Korolchuk V. I., Criado O., Vernia S., Boya P., Sanz P., de Córdoba S. R., Knecht E., Rubinsztein D. C. (2010) Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 19, 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puri R., Suzuki T., Yamakawa K., Ganesh S. (2012) Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 21, 175–184 [DOI] [PubMed] [Google Scholar]
- 18. Criado O., Aguado C., Gayarre J., Duran-Trio L., Garcia-Cabrero A. M., Vernia S., San Millán B., Heredia M., Romá-Mateo C., Mouron S., Juana-López L., Domínguez M., Navarro C., Serratosa J. M., Sanchez M., Sanz P., Bovolenta P., Knecht E., Rodriguez de Cordoba S. (2012) Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 21, 1521–1533 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y., Liu Y., Wu C., Zhang H., Zheng X., Zheng Z., Geiger T. L., Nuovo G. J., Liu Y., Zheng P. (2006) Epm2a suppresses tumor growth in an immunocompromised host by inhibiting Wnt signaling. Cancer Cell 10, 179–190 [DOI] [PubMed] [Google Scholar]
- 20. Sharma J., Mulherkar S., Mukherjee D., Jana N. R. (2012) Malin regulates Wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 287, 6830–6839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DePaoli-Roach A. A., Tagliabracci V. S., Segvich D. M., Meyer C. M., Irimia J. M., Roach P. J. (2010) Genetic depletion of the malin E3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 285, 25372–25381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turnbull J., Wang P., Girard J. M., Ruggieri A., Wang T. J., Draginov A. G., Kameka A. P., Pencea N., Zhao X., Ackerley C. A., Minassian B. A. (2010) Glycogen hyperphosphorylation underlies Lafora body formation. Ann. Neurol. 68, 925–933 [DOI] [PubMed] [Google Scholar]
- 23. Valles-Ortega J., Duran J., Garcia-Rocha M., Bosch C., Saez I., Pujadas L., Serafin A., Cañas X., Soriano E., Delgado-García J. M., Gruart A., Guinovart J. J. (2011) Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol. Med. 3, 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ganesh S., Delgado-Escueta A. V., Sakamoto T., Avila M. R., Machado-Salas J., Hoshii Y., Akagi T., Gomi H., Suzuki T., Amano K., Agarwala K. L., Hasegawa Y., Bai D. S., Ishihara T., Hashikawa T., Itohara S., Cornford E. M., Niki H., Yamakawa K. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 11, 1251–1262 [DOI] [PubMed] [Google Scholar]
- 25. Turnbull J., DePaoli-Roach A. A., Zhao X., Cortez M. A., Pencea N., Tiberia E., Piliguian M., Roach P. J., Wang P., Ackerley C. A., Minassian B. A. (2011) PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 7, e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao S. N., Maity R., Sharma J., Dey P., Shankar S. K., Satishchandra P., Jana N. R. (2010) Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 19, 4726–4734 [DOI] [PubMed] [Google Scholar]
- 27. Dou D., Joseph R. (1996) Cloning of human neuronatin gene and its localization to chromosome-20q 11.2-12: the deduced protein is a novel “proteolipid.” Brain Res. 723, 8–22 [DOI] [PubMed] [Google Scholar]
- 28. Joseph R., Dou D., Tsang W. (1994) Molecular cloning of a novel mRNA (neuronatin) that is highly expressed in neonatal mammalian brain. Biochem. Biophys. Res. Commun. 201, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 29. Joseph R., Dou D., Tsang W. (1995) Neuronatin mRNA: alternatively spliced forms of a novel brain-specific mammalian developmental gene. Brain Res. 690, 92–98 [DOI] [PubMed] [Google Scholar]
- 30. Dou D., Joseph R. (1996) Structure and organization of the human neuronatin gene. Genomics 33, 292–297 [DOI] [PubMed] [Google Scholar]
- 31. Vrang N., Meyre D., Froguel P., Jelsing J., Tang-Christensen M., Vatin V., Mikkelsen J. D., Thirstrup K., Larsen L. K., Cullberg K. B., Fahrenkrug J., Jacobson P., Sjöström L., Carlsson L. M., Liu Y., Liu X., Deng H. W., Larsen P. J. (2010) The imprinted gene neuronatin is regulated by metabolic status and associated with obesity. Obesity 18, 1289–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mzhavia N., Yu S., Ikeda S., Chu T. T., Goldberg I., Dansky H. M. (2008) Neuronatin: a new inflammation gene expressed on the aortic endothelium of diabetic mice. Diabetes 57, 2774–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arava Y., Adamsky K., Ezerzer C., Ablamunits V., Walker M. D. (1999) Specific gene expression in pancreatic beta-cells: cloning and characterization of differentially expressed genes. Diabetes 48, 552–556 [DOI] [PubMed] [Google Scholar]
- 34. Dugu L., Takahara M., Tsuji G., Iwashita Y., Liu X., Furue M. (2010) Abundant expression of neuronatin in normal eccrine, apocrine and sebaceous glands and their neoplasms. J. Dermatol. 37, 846–848 [DOI] [PubMed] [Google Scholar]
- 35. Chu K., Tsai M. J. (2005) Neuronatin, a downstream target of BETA2/NeuroD1 in the pancreas, is involved in glucose-mediated insulin secretion. Diabetes 54, 1064–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suh Y. H., Kim W. H., Moon C., Hong Y. H., Eun S. Y., Lim J. H., Choi J. S., Song J., Jung M. H. (2005) Ectopic expression of Neuronatin potentiates adipogenesis through enhanced phosphorylation of cAMP-response element-binding protein in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 337, 481–489 [DOI] [PubMed] [Google Scholar]
- 37. Joe M. K., Lee H. J., Suh Y. H., Han K. L., Lim J. H., Song J., Seong J. K., Jung M. H. (2008) Crucial roles of neuronatin in insulin secretion and high glucose-induced apoptosis in pancreatic beta-cells. Cell. Signal. 20, 907–915 [DOI] [PubMed] [Google Scholar]
- 38. Lin H. H., Bell E., Uwanogho D., Perfect L. W., Noristani H., Bates T. J., Snetkov V., Price J., Sun Y. M. (2010) Neuronatin promotes neural lineage in ESCs via Ca2+ signaling. Stem Cells 28, 1950–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sharma J., Rao S. N., Shankar S. K., Satishchandra P., Jana N. R. (2011) Lafora disease ubiquitin ligase malin promotes proteasomal degradation of neuronatin and regulates glycogen synthesis. Neurobiol. Dis. 44, 133–141 [DOI] [PubMed] [Google Scholar]
- 40. Rao S. N., Sharma J., Maity R., Jana N. R. (2010) Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 285, 1404–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jana N. R., Dikshit P., Goswami A., Nukina N. (2004) Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J. Biol. Chem. 279, 11680–11685 [DOI] [PubMed] [Google Scholar]
- 42. Mishra A., Godavarthi S. K., Maheshwari M., Goswami A., Jana N. R. (2009) The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284, 10537–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 44. Sinha S., Satishchandra P., Gayathri N., Yasha T. C., Shankar S. K. (2007) Progressive myoclonic epilepsy: a clinical, electrophysiological and pathological study from South India. J. Neurol. Sci. 252, 16–23 [DOI] [PubMed] [Google Scholar]
- 45. Gentry M. S., Worby C. A., Dixon J. E. (2005) Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. U.S.A. 102, 8501–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garyali P., Siwach P., Singh P. K., Puri R., Mittal S., Sengupta S., Parihar R., Ganesh S. (2009) The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 18, 688–700 [DOI] [PubMed] [Google Scholar]
- 47. Vernia S., Rubio T., Heredia M., Rodríguez de Córdoba S., Sanz P. (2009) Increased endoplasmic reticulum stress and decreased proteasomal function in Lafora disease models lacking the phosphatase laforin. PLoS One 4, e5907. [DOI] [PMC free article] [PubMed] [Google Scholar]