

Background: Plant aminoaldehyde dehydrogenases (AMADHs) detoxify ω-aminoaldehydes from several metabolic pathways.
Results: Two of five new AMADHs exhibit unusual kinetic properties. A thiohemiacetal intermediate was trapped in a crystal structure.
Conclusion: Five critical residues can modulate substrate specificity, and a new substrate was identified.
Significance: The present findings allow sequence-based predictions of AMADH substrate specificity linked with the production of individual osmoprotectants in plants.
Keywords: Enzyme Kinetics, Enzyme Structure, Gene Expression, NAD, Plant Biochemistry, ALDH10 Family, Thiohemiacetal Intermediate, Aminoaldehyde Dehydrogenase, Betaine Aldehyde, Substrate Specificity
Abstract
Plant ALDH10 family members are aminoaldehyde dehydrogenases (AMADHs), which oxidize ω-aminoaldehydes to the corresponding acids. They have been linked to polyamine catabolism, osmoprotection, secondary metabolism (fragrance), and carnitine biosynthesis. Plants commonly contain two AMADH isoenzymes. We previously studied the substrate specificity of two AMADH isoforms from peas (PsAMADHs). Here, two isoenzymes from tomato (Solanum lycopersicum), SlAMADHs, and three AMADHs from maize (Zea mays), ZmAMADHs, were kinetically investigated to obtain further clues to the catalytic mechanism and the substrate specificity. We also solved the high resolution crystal structures of SlAMADH1 and ZmAMADH1a because these enzymes stand out from the others regarding their activity. From the structural and kinetic analysis, we can state that five residues at positions 163, 288, 289, 444, and 454 (PsAMADHs numbering) can, directly or not, significantly modulate AMADH substrate specificity. In the SlAMADH1 structure, a PEG aldehyde derived from the precipitant forms a thiohemiacetal intermediate, never observed so far. Its absence in the SlAMADH1-E260A structure suggests that Glu-260 can activate the catalytic cysteine as a nucleophile. We show that the five AMADHs studied here are capable of oxidizing 3-dimethylsulfoniopropionaldehyde to the cryo- and osmoprotectant 3-dimethylsulfoniopropionate. For the first time, we also show that 3-acetamidopropionaldehyde, the third aminoaldehyde besides 3-aminopropionaldehyde and 4-aminobutyraldehyde, is generally oxidized by AMADHs, meaning that these enzymes are unique in metabolizing and detoxifying aldehyde products of polyamine degradation to nontoxic amino acids. Finally, gene expression profiles in maize indicate that AMADHs might be important for controlling ω-aminoaldehyde levels during early stages of the seed development.
Introduction
In plants, polyamines like spermine, spermidine, and putrescine are involved in various developmental processes and stress responses (1). Polyamines are degraded by copper diamine oxidases (EC 1.4.3.22, formerly 1.4.3.6) and FAD-containing polyamine oxidases (PAOs5; EC 1.5.3.14 and 1.5.3.17), leading to the production of reactive and cytotoxic ω-aminoaldehydes, such as 3-aminopropionaldehyde (APAL) or 4-aminobutyraldehyde (ABAL; in equilibrium with Δ1-pyrroline) plus hydrogen peroxide (2, 3). The oxidation of ω-aminoaldehydes by aminoaldehyde dehydrogenase (AMADH) ends up with the formation of the nontoxic metabolites β-alanine and γ-aminobutyric acid (GABA), respectively (4, 5).
Studying the physiological aspects of plant AMADHs (EC 1.2.1.19) has become attractive for economic reasons. It has been shown that an AMADH gene mutation leads to the acetylation of free ABAL (or its cyclic form Δ1-pyrroline) and accumulation of 2-acetyl-Δ1-pyrroline, a potent flavor component conferring a fragrance to several rice varieties like jasmine and basmati or to soybean (6, 7). Plant AMADHs have also been studied for their role in stress responses and in the production of the osmoprotectants glycine betaine (GB) and dimethylsulfoniopropionate (DMSP) by the oxidation of betaine aldehyde (BAL) and 3-dimethylsulfoniopropionaldehyde (DMSPAL), respectively (8, 9, 10).
AMADHs are classified in different aldehyde dehydrogenase (ALDH) families because of the established criteria stating that ALDH sequences have to be 40% identical to create a family. Because plant AMADHs display between 35 and 39% sequence identity with bacterial, fungal, fish, or mammalian AMADHs, they have recently been classified in the separate family ALDH10 (11). Several enzymes of the ALDH9 family covering mammalian AMADHs (12) and of the ALDH10 family from various species have been independently shown to oxidize a wide range of aminoaldehydes and other aldehydes (Fig. 1). Therefore, they have been also called 4-aminobutyraldehyde dehydrogenases (EC 1.2.1.19), 4-guanidinobutyraldehyde dehydrogenases (EC 1.2.1.54), betaine aldehyde dehydrogenases (BADHs; EC 1.2.1.8), and 4-trimethylaminobutyraldehyde dehydrogenases (EC 1.2.1.47).
FIGURE 1.

An overview of possible natural substrates oxidized by plant members of the ALDH10 family (AMADHs). APAL is converted to β-alanine, ABAL to GABA, ACAPAL to 3-acetamidopropionate, GBAL to 4-guanidinobutyrate, TMABAL to γ-butyrobetaine, BAL to glycine betaine, and DMSPAL to DMSP. A nomenclature derived from well characterized substrates has been frequently used for individual enzymes: 4-aminobutyraldehyde dehydrogenase (EC 1.2.1.19), 4-trimethylaminobutyraldehyde dehydrogenase (EC 1.2.1.47), 4-guanidinobutyraldehyde dehydrogenase (EC 1.2.1.54), and BADH (EC 1.2.1.8).
Plant AMADHs share around 70–80% sequence identity. One complete AMADH sequence is usually known for many plant species. However, ongoing genome analyses have gradually shown the presence of two genes coding for two homologous proteins of at least 80% sequence identity. Interestingly, most plant AMADHs carry the C-terminal peroxisomal targeting signal S(A)KL and are found in peroxisomes (13, 14). We have previously identified two pea AMADHs (Pisum sativum; PsAMADH1 and PsAMADH2) and analyzed their crystal structures (4). Plant AMADHs are dimeric and possess a 14-Å-long substrate channel in each monomer. There are three strictly conserved residues essential for the catalysis, Asn-162, Cys-294, and Glu-260, which lie in PWNYP, GQI(V)CSATSR, and ELGGKSP consensus motifs. The three catalytic residues (Asn, Cys, and Glu) lie at the substrate channel bottom and together form the active site. The catalytic mechanism follows the well described sequential binding model valid for the ALDH superfamily (15, 16). Aminoaldehyde substrates undergo a nucleophilic attack by the catalytic cysteine, leading to a thioester formation (i.e. a covalent intermediate) and the subsequent hydride transfer to NAD+. The conserved glutamate residue functions as a general base activating a water molecule. Such a molecule performs a nucleophilic attack at the thioester acyl-sulfur bond, resulting in the release of the amino acid.
Using site-directed mutagenesis performed on PsAMADH2 (17), we showed that two totally conserved aspartate residues located at the entrance of the substrate channel (Asp-110 and -113) are essential for high affinity binding of ω-aminoaldehydes (APAL is the best substrate). We also noticed that the activity on several aminoaldehyde substrates is affected by Trp-288. In order to predict the substrate specificity differences in AMADH pairs from a single species and between species and because only a few plant AMADH pairs were reported and studied (4, 6, 18), we cloned and biochemically characterized five ALDH10 members (19) (Table 1): two tomato (Solanum lycopersicum) isoenzymes SlAMADH1 and SlAMADH2 and three maize (Zea mays) isoenzymes ZmAMADH1a, ZmAMADH1b, and ZmAMADH2. Both tomato AMADHs share 80% sequence identity. ZmAMADH1a and -1b differ in 13 amino acids only (97% sequence identity). ZmAMADH2 and both ZmAMADH1a/1b are 75% sequence identical.
TABLE 1.
Enzyme nomenclature and GenBankTM accession numbers of the five studied ALDH10 family members (AMADHs)
The nomenclature of Z. mays ALDH10 family has been recently published (19). Because both tomato enzymes, SlAMADH1 and SlAMADH2, are not clustering (see “Phylogenetic Analysis”) too close to the sequences with established ALDH10 nomenclature (11), we named them ALDH10A12 and ALDH10A13.
Based on kinetic results, we decided to co-crystallize SlAMADH1 and ZmAMADH1a with NAD+. The dimeric structures were determined at 1.90 and 1.95 Å resolution, respectively. Although both soaking and co-crystallization with substrates were attempted and despite using the active-site base mutant SlAMADH1-E260A for this purpose, obtaining AMADH in such a complex was unsuccessful. However, ethylene glycol and polyethylene glycol (PEG) aldehyde molecules from the crystallization solution were found to mimic a substrate bound at the substrate binding site in ZmAMADH1a and SlAMADH1 structures, respectively. Moreover, we obtained the first crystal structure of a thiohemiacetal intermediate in AMADH family; a PEG aldehyde bound covalently to the catalytic cysteine forms an intermediate in one subunit of the SlAMADH1 structure in which only ADP moiety of NAD+ is ordered. The whole NAD+ is ordered in ZmAMADH1a and SlAMADH1-E260A structures and adopts the common conformation observed in ALDH structures. Combining structural analysis and kinetics, we can state that five residues at positions 163, 288, 289, 444, and 454 (PsAMADH numbering) can significantly modulate AMADH substrate specificity. Trp-288 and Tyr-163 are essential for the high affinity to ω-aminoaldehyde substrates. The absence of this Trp in addition to the presence of a Thr (instead of Cys in other enzymes) in SlAMADH1 leads to a broader substrate specificity due to an enlargement of its substrate channel. We can predict that AMADHs containing Ile-444 have only a negligible activity with BAL. Conversely, those containing an Asn at position 289 may display BAL activity. For the first time, we identify a new and common substrate for plant AMADHs.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of AMADHs from Tomato and Maize
The total RNA from apical meristems and leaves of 7-day-old tomato seedlings (S. lycopersicum cv. Amateur) and 5-day-old maize seedlings (Z. mays cv. Sachara) was extracted using the Midi spin column kit (Macherey-Nagel, Düren, Germany), and the cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen). SlAMADH1 and -2 and ZmAMADH1a, -1b, and -2 cDNA sequences were originally obtained in this work and submitted to the EMBL/GenBankTM database. The SlAMADH1 ORF (1515 bp; EMBL/GenBankTM accession number AY796114) was amplified using a primer pair with a BamHI (5′-CAGGGATCCGGCAAATCGTAATGTACCA-3′) and XhoI site (5′-CGTCTCGAGCTAATTCTTTGAAGGTGACTTAT-3′). For the SlAMADH2 ORF (1518 bp; FJ228482), an upstream primer with the EcoRI site (5′-CATGAATTCGGCGATTCCTAATATACGGAT-3′) and a downstream primer with the KpnI site (5′-AGTGGTACCTTACAGCTTTGAAGGAGACT-3′) were used. All three maize genes were cloned with primer pairs containing EcoRI and XhoI sites. ZmAMADH1a ORF (1518 bp; GQ184593) was amplified using the following primers: 5′-ATTGAATTCTGCCTCGCCAGCGATGGTCCCG-3′ and 5′-CGTCTCGAGTTCGTCTTGCCAGTTTACAGC-3′. ZmAMADH1b ORF (1518 bp; JN635700) was amplified using the primer pair 5′-ATTGAATTCTGCCTCGCAAGCGATGGTACCG-3′ and 5′-CGTCTCGAGTTCGTCTTGCCAGTTTACAGC-3′, and ZmAMADH2 ORF (1521 bp; GQ184594) was cloned using the primer pair 5′-ATAGAATTCAGCGCCGCCGCAGACGAT-3′ and 5′-ATACTCGAGTCACAGCTTAGATGGAGGCTGGT-3′.
The amplified sequences were inserted into a pCDFDuet vector (Novagen, La Jolla, CA), sequenced, and transformed into T7 express Escherichia coli cells for expression as N-terminal His6 tag proteins. Enzymes were produced and purified on a column filled with Co(II)-charged IDA-Sepharose 6B (Sigma-Aldrich) as described previously (20). The complete purification from a 600-ml culture yielded about 20 mg of recombinant SlAMADHs. Specific activity values measured with 1 mm APAL as a substrate were 3.1 units mg−1 for SlAMADH1, 3.6 units mg−1 for SlAMADH2, 2.6 units mg−1 for ZmAMADH1a, 1.9 units mg−1 for ZmAMADH1b, 4.1 units mg−1 for ZmAMADH2.
For crystallization, the plasmid was transformed into E. coli BL21 pLysS (Novagen). The cells were grown in 2YT medium at 37 °C to an A600 = 0.6, the temperature was reduced to 20 °C, and expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside overnight. The cells were suspended in buffer A (50 mm Tris-HCl buffer, pH 8, 20 mm imidazole, and 500 mm NaCl) and disrupted by sonication. The lysate was cleared by centrifugation at 20,000 × g at 4 °C for 30 min. The supernatant was filtered through a 0.22-μm membrane (Millipore) and loaded on a 5-ml nickel-nitriloacetic acid-agarose column (GE Healthcare). After washing with 10% buffer B (50 mm Tris-HCl, pH 8, 300 mm imidazole, and 500 mm NaCl), elution was performed with 100% buffer B. Fractions containing AMADH were pooled, and the buffer exchange was performed on a HiLoad 26/60 Superdex 200 prep grade (GE Healthcare), using 50 mm Tris-HCl, pH 8, 150 mm NaCl. Fractions were analyzed by SDS-PAGE, and those containing AMADH were pooled, concentrated using Vivaspin-10kDa (Sartorius) at 14 mg/ml, and stored at −20 °C. The identity of the purified proteins was confirmed by peptide mass fingerprinting on a Microflex LRF20 MALDI-TOF mass spectrometer (Bruker Daltonik, Bremen, Germany) after SDS-PAGE and in-gel digestion (21).
Substrates, Activity Assays, and pH Optimum Determination
Elementary aliphatic aldehydes, pyridine carboxaldehydes, BAL chloride together with N,N-dimethyl-4-aminobutyraldehyde, APAL, and ABAL diethylacetals were purchased from Sigma-Aldrich. DMSPAL and diethylacetals of 4-amino-2-hydroxybutyraldehyde, 4-guanidinobutyraldehyde (GBAL), 4-guanidino-2-hydroxybutyraldehyde, and 3-guanidinopropionaldehyde were all synthetic preparations (9, 22). Diethyl acetal iodides of N,N,N-trimethyl-3-aminopropionaldehyde and TMABAL were synthesized according to a protocol described for the latter compound (23). 3-Acetamidopropionaldehyde (ACAPAL) diethylacetal was synthesized as described (24); pyridine, acetic acid, and acetic anhydride were removed from the reaction mixture in a lyophilizer; 1H NMR (300 MHz, DMSO-d6), δ (ppm): 1.10 (t, J = 7.04 Hz, 6 H) 1.63 (q, J = 6.65 Hz, 2 H) 1.77 (s, 3 H) 3.03 (q, J = 6.71 Hz, 2 H) 3.29–3.62 (m, 4 H) 4.48 (t, J = 5.58 Hz, 1 H) 7.80 (br. s., 1 H). 13C NMR (75 MHz, DMSO-d6), δ (ppm): 15.8, 23.1, 33.9, 35.3, 61.2, 101.0, 169.5. If necessary, free aminoaldehydes were prepared by heating their acetals in a plugged test tube with 0.2 m HCl for 10 min (10). NAD+ and NAD analogs were purchased from Sigma-Aldrich.
Activity was measured spectrophotometrically by monitoring the formation of NADH (ϵ340 = 6.62 mm−1 cm−1) at 37 °C. The reaction mixture in a cuvette contained 0.15 m Tris-HCl buffer, pH 9.0, 1 mm NAD+, and appropriate amounts of AMADH. The enzyme reaction was started by the addition of APAL (or another aminoaldehyde) at a final 1 mm concentration. NAD+ analogs were assayed at 0.5 mm concentration with 1 mm APAL as described previously (4). Tris-HCl buffers (150 mm) in the pH range from 7.0 to 9.0 and 150 mm glycine-NaOH buffers in the pH range from 8.8 to 10.6 were used to determine pH optima using 1 mm APAL and 0.5 mm NAD+. Kinetic constants were determined with GraphPad Prism 5.0 data analysis software (GraphPad Software, Inc., La Jolla, CA). In those cases where no substrate inhibition was observed, the data were fitted to the Michaelis-Menten equation. The data were analyzed by a nonlinear regression using the Michaelis-Menten equation that includes terms in the numerator and denominator to account for partial substrate inhibition (25),
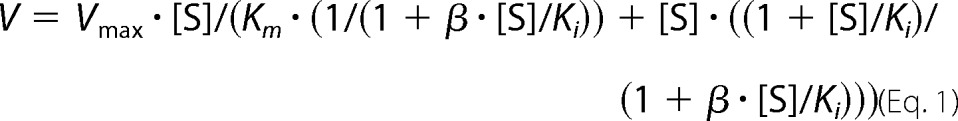 |
where v represents the determined initial velocity, Vmax is the maximal velocity, [S] is the concentration of the substrate, Km is the substrate concentration at half-maximal velocity, Ki is the substrate inhibition constant, and β is the factor that describes the effect of substrate inhibition on Vmax.
Plant AMADHs share a similar pH optimum The pH optima of ZmAMADH1a, ZmAMADH2, and SlAMADH2 fall within the pH range of 9.4–9.8 (i.e. similarly to that of pea or rice AMADHs) (4, 6). The pH optima of SlAMADH1 and ZmAMADH1b are slightly more basic (pH ∼10.2).
Crystallization and Data Collection
Crystallization conditions using commercial kits (Classics, PEGs II, and JCSG I suites from Qiagen) were screened in sitting-drop vapor diffusion experiments using a Cartesian nanodrop robot (Genomic Solutions) on IMAGIF (CNRS, Gif sur Yvette, France). SlAMADH1 with 5 mm NAD+ was crystallized over reservoirs containing 23% PEG 1000, 0.1 m HEPES, pH 7.5. ZmAMADH1a with 5 mm NAD+ was crystallized over reservoirs containing 16% PEG 4000, 0.1 m HEPES, pH 7.5, and 10% isopropyl alcohol. The E260A mutant of SlAMADH1 with 5 mm NAD+ was crystallized over reservoirs containing 15% PEG 1500, 0.1 m imidazole, pH 7.0, and 10% glycerol. Crystals were transferred to a cryoprotectant solution (mother liquor supplemented with 25% PEG 400 for SlAMADH1 or 20% glycerol for the E260A mutant and ZmAMADH1a) and flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K on the PROXIMA 1 beamline (SOLEIL Synchrotron, Saint-Aubin, France). Diffraction intensities were integrated with the program XDS (26). Data collection and processing statistics are given in Table 2.
TABLE 2.
Data collection and refinement statistics
| SlAMADH1 | E260A mutant of SlAMADH1 | ZmAMADH1a | |
|---|---|---|---|
| Protein Data Bank code | 4I9B | 4I8Q | 4I8P |
| Space group | P212121 | I212121 | C121 |
| Asymmetric unit | 1 dimer | 1 monomer | 1 dimer |
| Unit cell (Å) | |||
| a | 84.15 | 94.04 | 181.19 |
| b | 94.79 | 118.07 | 49.89 |
| c | 118.17 | 128.21 | 154.53 |
| α (degrees) | 90.0 | 90.0 | 90.0 |
| β (degrees) | 90.0 | 90.0 | 124.15 |
| γ (degrees) | 90.0 | 90.0 | 90.0 |
| Resolution (Å) | 43.9-1.90 | 50.0-2.65 | 45.0-1.95 |
| Observed reflections | 405,447 (45,398)a | 142,957 (22,300) | 338,559 (52,896) |
| Unique reflections | 75,000 (11,743) | 21,922 (3441) | 83,412 (13,238) |
| Completeness (%) | 99.2 (97.2) | 99.2 (98.5) | 99.1 (98.2) |
| I/σ (I) | 12.2 (1.93) | 15.3 (2.36) | 10.71 (2.29) |
| Rsym (%) | 9.8 (76.3) | 12.0 (78.4) | 10.9 (62.2) |
| Rcryst (%) | 17.4 | 20.8 | 17.6 |
| Rfree (%) | 21.2 | 28.2 | 21.0 |
| Root mean square deviation | |||
| Bond lengths (Å) | 0.010 | 0.010 | 0.010 |
| Bond angles (degrees) | 1.12 | 1.23 | 1.07 |
| Mean B value (Å2) | |||
| Protein | 26.0 | 45.1 | 27.6 |
| NAD+ | 37.4 | 23.3 | |
| ADP part | 31.5 | ||
| PEG aldehyde intermediate | 35.7 | ||
| Solvent | 35.3 | 44.2 | 34.8 |
a Numbers in parentheses represent values in the highest resolution shell: 1.90–2.01 Å (SlAMADH1), 2.65–2.81 Å (E260A mutant of SlAMADH1), and 1.95–2.07 Å (ZmAMADH1a).
Molecular Replacement, Refinement, and Final Model
The structures of SlAMADH1 and of ZmAMADH1a were solved by molecular replacement with Phaser (27) using the dimer structure of PsAMADH2 (Protein Data Bank code 3IWJ) as a search model. The structure of the SlAMADH1-E260A mutant was also solved with Phaser using a monomer of the WT structure as a search model. Model refinement was performed with BUSTER-TNT (28). Electron density maps were evaluated using COOT (29) for manual refinement. In the SlAMADH1 structure, the maps revealed clear density for the ADP moiety of the NAD+ coenzyme. In contrast, the side chain of the active-site base Glu-260 is disordered. For this reason, we set the occupancy of each atom forming the carboxylate at 0. In the ZmAMADH1a and SlAMADH1-E260A structures, the electron density of the whole NAD+ molecule was observed. Glu-262 (equivalent to Glu-260) is well defined in the maize structure. Refinement details of the three structures are shown in Table 2. Molecular graphics images were generated using PyMOL (Schroedinger, LLC, New York).
qPCR Analysis
The total RNA from 10-day-old and 2 month-old tomato plants (cv. Amateur) for reverse transcription was isolated using the RNAqueous kit and Plant RNA Isolation Aid solutions from Ambion (Austin, TX). Isolated RNA was treated twice by the TURBO DNase-free kit (Ambion) to remove all traces of genomic DNA contamination and to minimize bias in qPCR data. First-strand cDNA was synthesized by RevertAid H Minus Moloney murine leukemia virus reverse transcriptase and oligo(dT) primers (Fermentas, Vilnius, Lithuania). Diluted cDNA samples were used as templates in real-time PCRs containing TaqMan Gene Expression Master Mix (Applied Biosystems), both primers at 300 nm concentrations, and 250 nm TaqMan 6-FAM MGB probe. For the SlAMADH1 gene, the primer pair 5′-TGGACGTGATCTAGGGAAATGG-3′ plus 5′-CCAATGGTTCAGCAGAAGTATACTCT-3′ and a probe (5′-AATTTCTTGAACATTAAGCAGG-MGB-3′) were used. For the SlAMADH2 gene, the following primer pair and probe were used: 5′-GTAGTGGTTTTGGACGTGAGCTT-3′, 5′-TCGTCCGGAGTCACATACTGAGT-3′, and 5′-FAM-AATGGAGTCTCGAGAACT-3′, respectively. Expression of ZmAMADH1 genes was analyzed using the following primer pairs and probes. 5′-CCAGAGGTTATCTGAGGAGATTGAC-3′, 5′-CCAAATCCACTGCGCTTGTT-3′, and 5′-FAM-TGGGTAAACTGCTCGCAACCCTGC-MGB-3′, respectively, were used for ZmAMADH1a; 5′-TGAGCGCTGCCAGAGATTATC-3′, 5′-AGCAGGGCTGTGAGCAGTTT-3′, and 5′-FAM-ATGCTGGAATTATCTGGG-MGB-3′, respectively, were used for ZmAMADH1b; and 5′-AGGTGTGAGCGCATTTCAAAG-3′, 5′-AAAACCGCTCCGCTTGTTC-3′, and 5′-FAM-TTCCCAACCATGCTTCGTTCAAGCTC-Tamra-3′, respectively, were used for ZmAMADH2.
The TaqMan probes together with the corresponding primers were designed by Applied Biosystems customer service. RNA from every biological replicate was at least transcribed in two independent reactions, and each cDNA sample was run in at least two technical replications on a StepOne-Plus real-time PCR system using a default program (Applied Biosystems). For each primer pair, plasmid DNA was used as a template to generate a calibration curve for determining the PCR efficiency. Cycle threshold values were normalized with respect to the elongation factor 1α of tomato (SlEF1α; primer pair 5′-GGTCATCATCATGAACCATCC-3′ and 5′-CATACCAGCATCACCGTTCTT-3′) or maize (ZmEF1α; primer pair 5′-TGATACCCACCAAGCCTATGGT-3′ and 5′-CATGTCGCGGACAGCAAAC-3′) and the efficiency of amplification.
Phylogenetic Analysis
Amino acid alignment was performed with MUSCLE version 3.8 (30), and then the maximum likelihood phylogeny with bootstrap analysis was performed with PhyML version 3.0 (31), using an LG amino acid replacement matrix. AMADH sequences were obtained from the EMBL/GenBankTM database, TGI database (tentative consensus numbers), or Phytozome version 8.0 database: Amaranthus hypochondriacus 1 and 2 (AF017150/O04895); Avicennia marina 1/2 (mangrove, AF170094, and AB043540); Arabidopsis thaliana 1/2 (AY093071/AF370333); Beta vulgaris (sugar beet, AB221006); Brassica napus 1/2 (rapeseed, AY351634/TC111063); Glycine max 1/2 (soybean, AB333793/AB333794); Gossypium hirsutum (cotton, AY461804); Hordeum vulgare 1/2 (barley, AB063179/AB063178); Lycium barbarum (wolfberry, FJ514799); Leymus chinensis 1/2 (wild rye, AB183715/AB183716); Medicago truncatula 1/2 (barrel medic, TC151999/BT053176); Oryza sativa 1/2 (rice, AB001348/AB096083); Physcomitrella patens (moss, EDQ78577); Picea sitchensis (spruce, BT070941); Pinus sylvestris (pine, TC133801); P. sativum 1/2 (pea, AJ315852/AJ315853); Populus trichocarpa 1/2 (poplar, XM_002322111/DR448910); Prunus persica 1/2 (peach, PPA004563M/PPA022568M); S. lycopersicum 1/2 (tomato, this work); Solanum tuberosum 1/2 (potato, PGSC0003DMC400055759/PGSC0003DMC400042549); Sorghum bicolor (sorghum, XM_002444312); Spinacia oleracea (spinach, M31480); Triticum aestivum 1/2 (wheat, AY050316/TC389888 + AK332255); Z. mays 1a/1b/2 (maize, this work); Vitis vinifera 1/2 (grapevine, XM_002283654/XM_002281948); and Zoysia tenuifolia 1/2 (velvetgrass, AB161712/AB161702).
Accession Numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the following accession codes: 4I9B for SlAMADH1 in complex with NAD+ and a thiohemiacetal intermediate, 4I8Q for the Glu-260 mutant of SlAMADH1 in complex with NAD+, and 4I8P for ZmAMADH1a in complex with NAD+.
RESULTS AND DISCUSSION
Crystal Structures of SlAMADH1, ZmAMADH1a, and SlAMADH1-E260A
Because SlAMADH1 and ZmAMADH1a differ in terms of activity from the other new studied AMADHs from tomato (S. lycopersicum) and maize (Z. mays) as well as those from peas (see “Substrate Catalysis and Specificity”), their crystal structures were solved at high resolution (Table 2). We also crystallized SlAMADH1-E260A mutant to trap an intermediate or a bound substrate, but these attempts were unsuccessful. However, we have been able to trap an intermediate in the SlAMADH1 structure, thanks to the PEG solution used as precipitant in the crystallization condition.
The asymmetric unit of SlAMADH1 and ZmAMADH1a crystals contains a dimer, which is the active form in solution in accordance with results from gel permeation chromatography (molecular mass estimates between 106 and 112 kDa). The asymmetric unit of SlAMADH1-E260A is composed of one molecule, which forms a dimer by the crystallographic symmetry. The dimers of tomato, maize, and pea (P. sativum) AMADHs are very similar, sharing the same oligomerization interface, folding, and topology. A major difference is that SlAMADH1 is shorter by three residues in the loop from residue 63 to 70 when compared with ZmAMADH1a and PsAMADHs.
All monomers are identical, with root mean square deviations for 494 Cα atoms between 0.6 and 0.8 Å. They adopt the characteristic ALDH fold, consisting of an NAD(P)+ coenzyme binding domain (residues 1–131, 152–261, and 456–480), a catalytic domain (residues 262–455), and an oligomerization domain (132–151 and 481 at the C terminus). They all contain a monovalent cation present in a conserved intrasubunit cavity close to the coenzyme binding site. The cation cavity in SlAMADH1 (and also in ZmAMADH1a) is formed by the three equivalent carbonyl groups of Ile-31 (Val-30), Asp-99 (Asp-101), and Leu-189 (Leu-191) and the carboxyl group of Asp-99 (Asp-101). The nature of the protein ligands and the distances of around 2.4–2.6 Å are typical for a bound sodium ion (32). This ion probably allows maintenance of the protein structural integrity and/or stabilizes the position of a loop involved in NAD+ binding (15).
The oligomerization domains connect both monomers by protruding over the surface of the neighboring subunits around substrate channel entrances, whereas the NAD+ binding sites reside on the opposite side of the dimer. The catalytic cysteine lies between the NAD+ and the substrate binding sites. All enzymes were co-crystallized with NAD+. Whereas the whole well defined NAD+ molecule adopts an extended conformation typical for oxidized NAD+ (15) in ZmAMADH1a and SlAMADH1-E260A structures, the nicotinamide riboside (NiR) moiety is disordered in the SlAMADH1 structure, as frequently observed in structures of NAD(P)+-dependent ALDHs, including PsAMADH2 (4, 33, 34). With the bound coenzyme fully observed in electron density maps, the catalytic cysteine points toward the substrate channel (attacking conformation), allowing the nicotinamide ring to interact with the active-site base Glu. In contrast, when the NiR moiety is mobile, the catalytic cysteine adopts the resting conformation by pointing toward the NAD+ cavity and prevents the NiR moiety from being in the vicinity of the active-site base glutamate. In this state, mixed conformations of NAD+ and NADH coenzyme are observed.
Coenzyme Binding Site
The coenzyme binding site is highly conserved in the plant AMADH family. The adenine moiety of NAD+ inserted in a hydrophobic pocket flanked by two helices (αD and αE) makes no polar contact with the enzyme, so that the free amino group is exposed to the solvent. The O2B and O3B oxygen atoms of the ribose and both α- and β-phosphate oxygen atoms establish identical hydrogen bonds with the enzyme as described for PsAMADH2 (4). All five AMADHs display similar reaction rates and affinity for NAD+ (Km values are in the range of 10−5 μm; Tables 3 and 4). These enzymes are NAD+-specific because they possess a conserved glutamate (Glu-188 in PsAMADH2 numbering), preventing the binding of the 2′-phosphate group of NADP+, as described previously (4). Thus, their activity with NADP+ is very low (Fig. 2).
TABLE 3.
Kinetic parameters of ALDH10 family from tomato for selected substrates and the coenzyme NAD+
All Km and kcat values are given in μm and s−1, respectively, and kcat/Km ratios are expressed in m−1 s−1. Activities were measured in 0.15 m Tris-HCl buffer, pH 9.0. Specific activity values with 1 mm APAL were 3.1 units mg−1 for SlAMADH1 and 3.6 units mg−1 for SlAMADH2. Kinetic constants were measured using a saturating NAD+ concentration of 500 μm. The Km value for NAD+ was measured at a fixed 1 mm concentration of APAL. The data were further analyzed by a nonlinear regression using either a simpler equation for substrate inhibition, v = Vmax·[S]/(Km + [S]·(1 + [S]/Ki)), or an equation for partial substrate inhibition, v = Vmax·[S]/(Km·(1/(1 + β·[S]/Ki)) + [S]·((1 + [S]/Ki)/(1 + β·[S]/Ki))), where v is the determined initial velocity, Vmax is the maximal velocity, [S] is the concentration of the substrate, Km is the substrate concentration at half-maximal velocity, Ki is the substrate inhibition constant, and β is the factor that describes the effect of substrate inhibition on Vmax. In cases where substrate inhibition was observed, Ki values are given in μm. −, not determined (very weak substrates); None, no substrate inhibition observed in the measured range.
| Ligand | SlAMADH1 |
SlAMADH2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Ki | β | Km | kcat | kcat/Km | Ki | β | |
| μm | s−1 | m−1 s−1 | μm | μm | s−1 | m−1 s−1 | μm | |||
| NAD+ | 72 ± 7 | − | − | − | − | 89 ± 3 | − | − | − | − |
| TMABAL | 17 ± 1.4 | 3.6 ± 0.2 | 2.1 × 105 | 90 ± 14 | 0.22 | 141 ± 17 | 2.0 ± 0.1 | 1.4 × 104 | 1560 ± 108 | 0.04 |
| GBAL | 85 ± 13 | 4.5 ± 0.4 | 5.3 × 104 | 581 ± 74 | − | 22 ± 3 | 3.9 ± 0.3 | 1.8 × 105 | 211 ± 40 | − |
| APAL | 41 ± 5 | 6.0 ± 0.4 | 1.5 × 105 | 914 ± 84 | − | 9 ± 1.4 | 8.2 ± 0.7 | 9.1 × 105 | 111 ± 18 | 0.36 |
| ABAL | 278 ± 19 | 5.2 ± 0.4 | 1.9 × 104 | None | − | 54 ± 5 | 2.5 ± 0.1 | 4.6 × 104 | 858 ± 81 | − |
| BAL | 2051 ± 83 | 1.0 ± 0.02 | 4.6 × 102 | None | − | − | − | − | − | − |
| 3-PCAL | 316 ± 14 | 2.2 ± 0.03 | 6.9 × 103 | None | − | − | − | − | − | − |
| 4-PCAL | 50 ± 4 | 1.4 ± 0.03 | 2.8 × 104 | None | − | − | − | − | − | − |
TABLE 4.
Kinetic parameters of ALDH10 family from maize for selected substrates and the coenzyme NAD+
All Km and kcat values are given in μm and s−1, respectively, and kcat/Km ratios are expressed in m−1 s−1. Activities were measured in 0.15 m Tris-HCl buffer, pH 9.0. Specific activity values with 1 mm APAL were 2.6 units mg−1 for ZmAMADH1a, 1.9 units mg−1 for ZmAMADH1b, and 4.1 units mg−1 for ZmAMADH2. Kinetic constants were measured using a saturating NAD+ concentration of 500 μm. The Km value for NAD+ was measured at a fixed 1 mm concentration of APAL. The data were further analyzed by a nonlinear regression using either a simpler equation for substrate inhibition, v = Vmax·[S]/(Km + [S]·(1 + [S]/Ki)), or an equation for partial substrate inhibition, v = Vmax·[S]/(Km·(1/(1 + β·[S]/Ki)) + [S]·((1 + [S]/Ki)/(1 + β·[S]/Ki))), where v is the determined initial velocity, Vmax is the maximal velocity, [S] is the concentration of the substrate, Km is the substrate concentration at half-maximal velocity, Ki is the substrate inhibition constant, and β is the factor that describes the effect of substrate inhibition on Vmax. In cases where substrate inhibition was observed, Ki values are given in μm. −, not determined (very weak substrates); None, no substrate inhibition observed in the measured range.
| Ligand | ZmAMADH1a |
ZmAMADH1b |
ZmAMADH2 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Ki | β | Km | kcat | kcat/Km | Ki | β | Km | kcat | kcat/Km | Ki | β | |
| μm | s−1 | m−1 s−1 | μm | μm | s−1 | m−1 s−1 | μm | μm | s−1 | m−1 s−1 | μm | ||||
| NAD+ | 91 ± 4 | − | − | − | − | 79 ± 6 | − | − | − | − | 86 ± 3 | − | − | − | |
| TMABAL | 6 ± 0.8 | 5.1 ± 0.3 | 8.4 × 105 | 66 ± 7 | 0.1 | 10 ± 2 | 5.7 ± 0.5 | 5.7 × 105 | 89 ± 11 | − | 16 ± 1.1 | 10.8 ± 0.7 | 6.8 × 105 | 38 ± 8 | 0.06 |
| GBAL | 3 ± 0.7 | 1.6 ± 0.2 | 5.2 × 105 | 21 ± 3 | 0.04 | 5 ± 1 | 1.7 ± 0.2 | 3.5 × 105 | 18 ± 4 | 0.04 | 11 ± 1.2 | 5.0 ± 0.4 | 4.5 × 105 | 29 ± 4 | 0.07 |
| APAL | 9 ± 2 | 11.8 ± 0.5 | 1.3 × 106 | 182 ± 31 | 0.09 | 11 ± 2 | 11.1 ± 0.4 | 1.0 × 106 | 109 ± 8 | 0.1 | 98 ± 8 | 10.3 ± 0.5 | 1.1 × 105 | 505 ± 28 | 0.07 |
| ABAL | 28 ± 3 | 1.2 ± 0.1 | 4.2 × 104 | 1214 ± 102 | − | 26 ± 4 | 1.7 ± 0.2 | 6.5 × 104 | 108 ± 17 | − | 59 ± 2 | 4.3 ± 0.1 | 7.2 × 104 | 652 ± 72 | 0.11 |
| BAL | 14 ± 1.6 | 0.6 ± 0.02 | 4.5 × 104 | None | − | 29 ± 4 | 0.07 ± 0.01 | 2.3 × 103 | None | − | − | − | − | − | − |
| 3-PCAL | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 4-PCAL | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
FIGURE 2.

Relative activities of tomato (A) and maize (B) AMADHs with NAD+ analogs. Coenzyme efficiency was measured with 1 mm APAL as a substrate and 500 μm NAD+ analogs, in 0.15 m Tris-HCl buffer, pH 9.0. The rate of the NAD+-mediated reaction was arbitrarily taken as 100%. Error bars, S.D.
A screening test using NAD+ analogs with substitutions at the nicotinamide ring in the presence of APAL as a substrate shows that these compounds are less efficient as coenzymes (Fig. 2). 3-Acetylpyridine-NAD+ is the best electron acceptor and leads to ∼45% activity for ZmAMADH1a and ZmAMADH1b (as well as for both PsAMADHs) and ∼75% activity for both SlAMADHs and ZmAMADH2 compared with that with NAD+. Thio-NAD+ drastically decreases AMADH activity (between 4.5 and 25%), especially that of ZmAMADH2 and PsAMADH1. 3-Pyridinealdehyde-NAD+ is generally not accepted as a coenzyme for our studied AMADHs. To our knowledge, there is so far only one detailed analysis of the coenzyme binding site using various NAD+ analogs performed on human ALDH1 (ALDH1A1), ALDH2, and AMADH (ALDH9A1) (35). The human ALDHs (hALDHs) significantly differ in their activity with nicotinamide analogs, and these differences are rather difficult to interpret in terms of interactions with the conserved catalytic residues. For example, thio-NAD+, which is inactive with hALDH2 (reduced Vmax and unaffected Km), is a good coenzyme of hAMADH and a weak coenzyme for hALDH1. Similarly, 3-pyridinealdehyde-NAD+, which is a coenzyme for hALDH1 and hALDH2, is inactive for hAMADH as well as for plant AMADHs. Indeed, 3-pyridinealdehyde-NAD+ acts as a strong competitive inhibitor of hAMADH and might form a thiohemiacetal linkage with the catalytic cysteine, preventing its reduction (36).
Substrate Catalysis and Specificity
A large screening study performed using 1 mm substrates, such as natural ω-aminoaldehydes, including APAL, ABAL, GBAL, TMABAL, and BAL (Fig. 1), proves that all five plant AMADHs from tomato and maize most readily oxidize propionaldehyde and butyraldehyde natural and synthetic aminoaldehydes (Fig. 3). The highest reaction rates are obtained with APAL for all maize enzymes and SlAMADH2. Only SlAMADH1 displays the highest activity for ABAL. Methyl and guanidine derivatives of APAL and ABAL are also well oxidized, with 10–60% rates on average (relative to that of the best substrate), whereas hydroxylated derivatives are oxidized more slowly. Taken together, the three ZmAMADHs and SlAMADH2 resemble both PsAMADHs with respect to their substrate preference. Unexpectedly, SlAMADH1 exhibits much broader substrate specificity than SlAMADH2 and the other studied AMADHs. This enzyme oxidizes much better not only the guanidine derivatives 3-guanidinopropionaldehyde and GBAL but also 3- and 4-pyridine carboxaldehydes (3- and 4-PCAL; ∼40% of the rate with ABAL) and several n-alkyl aldehydes like valeraldehyde and capronaldehyde (∼15% of the rate with ABAL). One more unexpected result is that ZmAMADH1a shows a high activity with BAL, comparable with ABAL oxidation (∼16% compared with APAL). To shed light on these surprising results, we focused on SlAMADH1 and ZmAMADH1a for a crystallographic study.
FIGURE 3.

Substrate specificity of ALDH10 isoenzymes from tomato (top) and maize (bottom). The activity of each AMADH isoenzyme with the best substrate was arbitrarily taken as 100%. The measurements were performed with 1 mm substrates in 0.15 m Tris-HCl buffer, pH 9.0, containing 1 mm NAD+. The following substrates were tested: APAL, N,N,N-trimethyl-3-aminopropionaldehyde (TMAPAL), 3-guanidinopropionaldehyde (GPAL), ABAL, N,N-dimethyl-4-aminobutyraldehyde (DMABAL), TMABAL, 4-amino-2-hydroxybutyraldehyde (AHBAL), GBAL, 4-guanidino-2-hydroxybutyraldehyde (GHBAL), BAL, acetaldehyde (C2), propionaldehyde (C3), butyraldehyde (C4), valeraldehyde (C5), capronaldehyde (C6), enanthaldehyde (C7), and pyridine carboxaldehydes (PCAL). Specific activity values with 1 mm APAL were 3.1 units mg−1 for SlAMADH1, 3.6 units mg−1 for SlAMADH2, 2.6 units mg−1 for ZmAMADH1a, 1.9 units mg−1 for ZmAMADH1b, and 4.1 units mg−1 for ZmAMADH2. Error bars, S.D.
SlAMADH1 structure reveals well defined electron density maps in the substrate binding site of both subunits despite the absence of a co-crystallized or soaked substrate. The only compound considered as a potential ligand comes from the PEG solution used as a precipitant for crystallization. A di(hydroxyethyl)ether molecule was straightforward modeled. A continuous electron density between the SG atom of the catalytic cysteine Cys-295 and the C1 atom of the di(hydroxyethyl)ether molecule suggests a 1.7-Å-long covalent bond in subunit A (Fig. 4A). The density shape at C1 is not flat and satisfies an sp3 hybridization expected for a thiohemiacetal intermediate. The oxygen atom of the hydroxyl group at C1 is hydrogen-bonded to the Cys-295 amide nitrogen (2.9 Å) and to the ND2 atom of Asn-162 (2.8 Å), two groups formerly postulated to be involved in the formation of the oxyanion hole and to stabilize the thiohemiacetal and thioacyl intermediates (37) (Fig. 4B). So far, the only reported intermediate in the ALDH family corresponding to the E268A glyceraldehyde-3-phosphate dehydrogenase mutant (38) is a thioester intermediate due to the flat density observed at the C1 atom, corresponding to a sp2 hybridization.
FIGURE 4.

SlAMADH1 substrate channel with a bound intermediate. A, a close-up view of the active site in subunit A showing a continuous electron density from the Fo − Fc omit map contoured at 3σ between the catalytic Cys-295 and the PEG aldehyde, which forms a thiohemiacetal intermediate. B, a covalent bond is formed between the SG atom of Cys-295 and the C1 atom of the PEG aldehyde. Hydrogen bonds between the intermediate and enzyme residues (Cys-295 and Asn-162) are shown as dashed lines. The angle geometry of this intermediate is shown. C, a close-up view of the active site in subunit B showing a discontinuous electron density map between the catalytic cysteine and the PEG aldehyde. Residues are labeled.
The existence of the covalent bond and the disordered Glu-260 side chain strongly suggests that the enzyme is turning over in the crystal with a highly mobile Glu-260. Kinetic measurements with PEG solutions (PEG 1000 and 1500) used for crystallization confirm the presence of aldehydes and show an activity of about a 1% rate compared with that with capronaldehyde (data not shown). The presence of an aldehyde in PEG solution was also mentioned in an hALDH2 crystallographic study, where we believe that the soaked crotonaldehyde substrate forms an intermediate (Protein Data Bank code 1O01) (15). In consequence, instead of a di(hydroxyethyl)ether molecule, we modeled a 2-(hydroxyethoxy)acetaldehyde molecule in two conformations in subunit B of the SlAMADH1 structure because there was no continuous electron density proving the absence of a covalent bond (Fig. 4C). Indeed, the catalytic cysteine points toward the coenzyme cavity. However, we cannot rule out the presence of a carboxylate product because the NiR moiety of the coenzyme molecule is disordered in both subunits, indicating the presence of an NAD+/NADH mixture in the crystals as previously shown in the hALDH2 study (15). The extended conformation typical for oxidized NAD+ in the SlAMADH1-E260A structure proves that no turnover occurs in the crystal, although PEG was used as a precipitant. We verified that this mutant is inactive with the aldehyde in PEG solutions and capronaldehyde, at pH 7.0 and in the same buffer used in the crystallization solution, explaining the unsuccessful attempts at obtaining intermediate or bound substrates by soaking methods. At this pH, the catalytic cysteine (pKa ∼8.0) (39) should carry a thiol group and needs to be deprotonated to the nucleophilic thiolate to function properly. Because an intermediate was trapped in the WT enzyme, the acylation reaction is slow enough to observe the intermediate accumulation in the crystal (i.e. the nucleophilic activation is faster). Therefore, the absence of an intermediate formation in the mutant structure suggests that Glu-260 was required to activate the catalytic cysteine in the WT structure. The same function has been shown for the equivalent glutamate in hALDH2 (40, 41). However, under physiological conditions occurring in plant peroxisomes (a basic pH), we believe that the role of this glutamate is rather for proton abstraction from the hydrolytic water.
The presence of either intermediate or product/substrate in the active site does not induce conformational changes. The finding of a thiohemiacetal intermediate in only one subunit (subunit A) and the presence of either a substrate or carboxylate product in the other subunit would imply that in the context of dimer and the ALDH10 family, the hydride transfer (from NAD+ to NADH followed by thioester formation) occurs in each subunit in turn. Otherwise, the covalent bond (between the catalytic cysteine Cys-295 in attacking conformation and a substrate molecule) would be randomly observed in both subunits. At the moment, we cannot conclude whether this property would be unique for the ALDH10 family or also apply for other ALDHs. Indeed, in the only other known case of the Streptococcus E268A-glyceraldehyde-3-phosphate dehydrogenase mutant (38), a thioester intermediate in all four subunits of the tetramer was observed due to a very slow deacylation rate allowing the accumulation of the intermediate in each subunit.
Role of Trp-288 and Tyr-163 in Catalysis of Aliphatic and Aromatic Aminoaldehydes
A comparison of the substrate channel residues in tomato, maize, and pea AMADHs shows that almost all residues are evolutionarily highly conserved (Table 5). Noticeably, two residues at positions 288 and 453 (PsAMADH numbering) are less preserved. Trp-288 could be substituted by Ala, Phe, or Ser, and Cys-453 could be substituted by Ser or Thr.
TABLE 5.
A comparison of active-site residues in ALDH10 family from several plant species
Shown are active-site residues and C-terminal motifs of AMADHs from pea (PsAMADH) (4), tomato (SlAMADH), and maize (ZmAMADH). The numbering used follows the sequence of PsAMADH2, of which the substrate channel was previously analyzed by site-directed mutagenesis (17). Rice (OsAMADHs, UniProtKB numbers O24174 and Q84LK3) and barley enzymes (HvAMADHs, UniProtKB numbers Q94IC0 and Q94IC1) analyzed by other groups (6, 18) are shown for comparison and indicated by an asterisk. Enzymes accepting BAL as an efficient substrate are labeled with a cross.
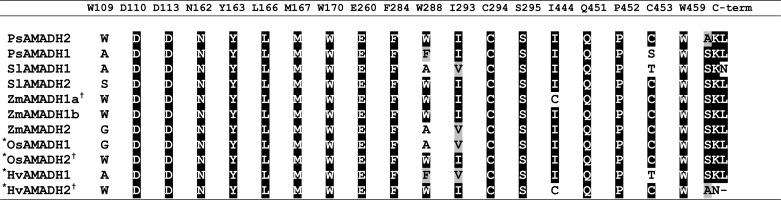
The five studied AMADHs exhibit the highest kcat value with APAL (Tables 3 and 4). However, based on the kcat/Km ratio, APAL is the best substrate only for three of them (SlAMADH2, ZmAMADH1a, and ZmAMADH1b) as well as for PsAMADH2 (4), which all possess Trp-288 (Trp-289 in SlAMADH numbering, Trp-290 or -291 in ZmAMADHs). SlAMADH1 and ZmAMADH2, which contain an Ala at this position, prefer TMABAL as the best substrate, accented by the increased Km values for APAL and ABAL. PsAMADH1 containing a phenylalanine also prefers TMABAL (4). Saturation curves for the five enzymes show that a strong excess substrate inhibition appears at relatively low substrate concentrations (Fig. 5, A–E). The inhibition appears at higher substrate concentrations for SlAMADH1 and ZmAMADH2 lacking Trp-288. This is in line with the behavior of the PsAMADH2-W288A mutant showing Km values for APAL and ABAL 2–3-fold higher than those for the WT enzyme (17). The preference of TMABAL for Trp-288-absent AMADHs appears either directly due to the increase in the substrate channel diameter and the loss of possible π-electron stacking interaction with the electrophilic protonated amino group of the substrate or indirectly by affecting the position of the Tyr-163 side chain (see “BADH Activity of ZmAMADH1a and SlAMADH1; Role of Cys-446 and Asn-290”). Similar kinetic results have been reported for two rice (OsAMADHs) and barley (HvAMADHs) isoenzymes (6, 18). OsAMADH2 and HvAMADH2, which contain the Trp-288 residue, display significantly lower Km values for APAL or ABAL than OsAMADH1 and HvAMADH1 lacking this tryptophan.
FIGURE 5.

Broad substrate specificity of plant AMADHs (ALDH10 family). A–E, saturation curves for activity determination of ZmAMADHs and SlAMADHs. Data were measured in 0.15 m Tris-HCl buffer, pH 9.0, with APAL, ABAL, TMABAL, GBAL, BAL, 3-PCAL, and 4-PCAL as substrates in the presence of 1 mm NAD+. F, relative activity values for all studied AMADHs with ACAPAL and DMSPAL. The data were measured in 0.15 m Tris-HCl, pH 9.0, using 1 mm substrate and in the presence of 1 mm NAD+. The activity with APAL was taken as 100%. Error bars, S.D.
The five studied AMADHs oxidize 3- and 4-PCAL, representing heterocyclic aminoaldehydes with a tertiary nitrogen atom (Fig. 3). Interestingly, the product of 3-PCAL oxidation, named nicotinic acid, is methylated to the alkaloid trigonelline in plants, which acts among others as an osmoprotectant. AMADHs lacking the Trp-288 residue, such as SlAMADH1, ZmAMADH2, and PsAMADH1, should be more efficient, as demonstrated by the PsAMADH2-W288A mutant showing a 4 times higher rate with 3-PCAL than the WT enzyme (17). However, the activity of ZmAMADH2 and PsAMADH1 is not as high as that of SlAMADH1, which displays similar Km and kcat values for 3- and 4-PCAL as for the best aminoaldehyde substrates (Tables 3 and 4). Therefore, it becomes clear that such a change in SlAMADH1 kinetics does not only result from the Trp-288 absence.
Broad Substrate Specificity of SlAMADH1; Role of Thr-454
SlAMADH1 is unique among the studied enzymes in that it has a threonine at position 454 instead of a highly conserved cysteine or serine (Cys-453 in PsAMADHs). The substrate channel volume differs between SlAMADH1 and PsAMADH2, as is shown in Fig. 6A. A structural comparison of the substrate binding site between SlAMADH1 and known plant AMADH structures reveals a displacement of four residues (from Thr-454 to Asp-457), which open the channel cavity in SlAMADH1. Thr-454, the most affected residue, is shifted by 1.8 Å away from the active site, allowing its side chain to make a hydrogen bond with the conserved Gln-485 NE1 atom from the oligomerization domain of the other subunit (Fig. 6B). Thr-454 is thus directly involved in the dimer interface interaction. The position of the Thr-454 side chain leads to a large and less polar channel interior, resulting in high rates of oxidation of aliphatic and aromatic aldehydes by SlAMADH1 compared with SlAMADH2 and both PsAMADHs (Figs. 3 and 5, D and E). Indeed, in PsAMADH1/2, the side chains of Ser-453/Cys-453 make the channel interior smaller by pointing inward and polar for substrate interaction (Fig. 6C). Therefore, the combination of the two residues, Ala-289 and Thr-454, distinguishes SlAMADH1 for its broad substrate specificity. The role of Thr-454 is emphasized by the PsAMADH2-C453A mutant, which does not display any significant differences from the WT enzyme (17).
FIGURE 6.

Substrate channels of SlAMADH1, ZmAMADH1a, and PsAMADH2. A, a comparison between the substrate channel volumes of SlAMADH1 (in red) and PsAMADH2 (in blue mesh; Protein Data Bank code 3IWJ). The enlarged volume of SlAMADH1 comes from the absence of Trp-109 and Trp-288 and a displacement at position 453; B and C, a transversal volume section of substrate channel of SlAMADH1 (in red) and PsAMADH2 (in blue), both with a docked ABAL ligand. These images show the enlarged middle section of the substrate channel of SlAMADH1, which occurs due to the presence of Ala-289 and Thr-454 residues compared with the presence of Trp-288 and Cys-453 in PsAMADH2. Thr-454 is hydrogen-bonded to Gln-485 from the other subunit and thus is involved in the dimer interface. Amino acid residues are shown as sticks and labeled. The total surface of the cavities was calculated using Hollow with 0.5-Å grid spacing and a 1.4-Å interior probe. The ABAL molecule (in black) is shown for illustration. It was docked into the active site using AutoDock, and its carbonyl oxygen atom establishes a hydrogen bond to the catalytic cysteine Cys-295/Cys-294, whereas the amino group is hydrogen-bonded to Tyr-163/Tyr-163; D and E, a substrate channel structural comparison of ZmAMADH1a (in purple) and SlAMADH1 (in orange) with PsAMADH2 (in blue, used as the reference for residue numbering shown in parentheses). Cys-446 in ZmAMADH1a allows a conserved Trp to move away from Tyr-165, thus opening the channel for a bulkier BAL molecule. Asn-290 in SlAMADH1 could have a similar role on the opposite side, allowing Tyr-163 to move toward Asn-290, thus opening the channel. Residues labeled in red are not totally conserved in plant AMADHs.
BADH Activity of ZmAMADH1a and SlAMADH1; Role of Cys-446 and Asn-290
BAL, formed by choline monooxygenase (42) in plants, is oxidized by AMADH/BADH to the osmoprotectant GB. Under stress conditions, high levels of GB have been found in leaves of diverse families of dicots and monocots, including barley, wheat, and rye (43). In contrast, maize cultivars synthesize considerably lower levels of GB, and naturally occurring GB-deficient inbred lines have also been identified (44). Moreover, certain maize varieties with tomato and pea do not accumulate GB due to the loss of a functional CMO (choline monooxygenase) gene (45–47), correlated with the very low activity of ZmAMADH1b, ZmAMADH2, SlAMADH2, and PsAMADHs with BAL (below 1.5% compared with that with APAL; Fig. 3) (4). Interestingly, ZmAMADH1a exhibits a good activity with BAL (Km = 14 μm, kcat = 0.6 s−1). SlAMADH1 oxidizes BAL significantly only at high concentrations, as indicated by the corresponding Km value (Tables 3 and 4).
Based on AMADH structures, four strictly conserved residues, Tyr-163, Trp-170, Trp-288, and Trp-459 (Tyr-165, Trp-172, Trp-290, and Trp-461 in ZmAMADH1a), seem to be arranged suitably for cation-π interactions with the trimethylammonium group of BAL, which becomes sandwiched between Tyr-163 and Trp-459 upon binding. A structural comparison of the four plant AMADH structures (ZmAMADH1a, SlAMADH1, and PsAMADH1/2) reveals a difference between the position of Trp-461 in ZmAMADH1a and those of the equivalent tryptophans in the other three structures. Indeed, its side chain is shifted by 1.6 Å (corresponding to a rotation of 23° along the Cβ-Cγ axis) away from Tyr-163, leading to an enlargement of the substrate channel favorable for binding of the bulky trimethylammonium group (Fig. 6D). Ile-444 in PsAMADHs (Ile-445 in SlAMADH1) prevents Trp-459 from adopting the observed position in ZmAMADH1a due to a steric hindrance. Thus, the equivalent cysteine (Cys-446) in ZmAMADH1a, located in the second sphere of the substrate interaction, is responsible for the high activity with BAL, as shown in spinach AMADH/BADH (SoBADH; Protein Data Bank code 4A0M) (48). Notably, the concerned tryptophan and cysteine residues adopt the same position in ZmAMADH1a and SoBADH structures. HvAMADH2, which also contains a cysteine, exhibits a good BAL activity (18). Moreover, AMADHs from members of the Amaranthaceae family (amaranth, spinach, and sugarbeet), from several monocots like barley or wheat, and from several grasses like sorghum or velvetgrass, known for their high BADH activity, possess such a cysteine (or an alanine) (49, 50, 10, 51). The low or negligible BADH activity of ZmAMADH1b, ZmAMADH2, SlAMADH2, and PsAMADHs correlates with the presence of Ile-444 (Table 5). However, despite the presence of Ile-445 in SlAMADH1, its activity with BAL resembles that of the mutant Y163A from PsAMADH2 (17). Here the structural comparison shows that Tyr-163, in SlAMADH1, has no hydrogen bond involving its hydroxyl group (conversely, there is a hydrogen bond with the Thr-289 side chain in PsAMADH, which is replaced by Asn-290 in SlAMADH1). Asn-290 does not interact with Tyr-163, which could move toward Asn-290 and away from the channel substrate. The flexibility of Tyr-163 in SlAMADH1 would allow bulkier BAL to pass to the catalytic cysteine. Thus, Asn-290 in SlAMADH1, located in the second sphere of substrate interaction, might have a similar role like that of Cys-446 in ZmAMADH1a, in terms of the channel widening, acting on the opposite side on the substrate channel. Another interesting different movement of Tyr-163 is observed in SlAMADH1 and PsAMADH2 structures compared with the two others. Its hydroxyl group can move 1.1 Å (corresponding to a rotation of 9° along the Cα-Cβ axis) toward Ala-289 and the equivalent Phe-288 residue, respectively, which is only possible due to the absence of Trp-288 at this position (Fig. 6E). Consequently, the nature of the residue at position 288 will have a significant effect on the interaction between the Tyr-163 side chain and the substrate.
Interestingly, human ALDH9 (ALDH9A1, also called E3 isoenzyme) has been shown to prefer TMABAL (Km = 1.4 μm) over ABAL (Km = 8–14 μm) and BAL (Km = 120 μm) (23). The low Km value for ABAL indicates that this enzyme is involved in GABA formation (reviewed in Ref. 53). So far, APAL and GBAL have not been tested with this enzyme. The other well studied ALDH9 from rat liver also shows a high preference for TMABAL. Fish ALDH9 from cod liver (Protein Data Bank codes 1A4S and 1BPW) (54) shows a higher kcat/Km ratio for BAL over aliphatic and aromatic aldehydes, and this is why the enzyme is called BADH (55). To our knowledge, other ω-aminoaldehydes have not yet been tested, but the active site preserves aromatic residues responsible for AMADH activity, typical for the ALDH10 family, namely Tyr-167 (Tyr-163 in ALDH10), Trp-174 (Trp-170), and Phe-466 (Trp-459). The variable Trp-288 in ALDH10 is replaced by Leu-291.
ACAPAL and DMSPAL Dehydrogenase Activities
Besides the two natural compounds APAL and ABAL, known to be among the best substrates of all plant AMADHs studied so far, we show for the first time that ACAPAL may also be generally oxidized (Fig. 5F). Therefore, these enzymes are unique in metabolizing and detoxifying characteristic aldehyde products of polyamine degradation to nontoxic acids. SlAMADH1 shows 85% activity with ACAPAL compared with that with APAL, whereas SlAMADH2 displays only 30% activity. For ZmAMADHs, the activity is between 43 and 55%, and for PsAMADHs, it is between 28 and 35%. With respect to ACAPAL oxidation by AMADH isoenzymes in a single species, the activity of the Trp-288-absent AMADH isoenzyme is higher. APAL mainly originates from the “polyamine back-conversion pathway” via the oxidation of spermine (to spermidine) and spermidine (to putrescine) on the exo-side of N4-nitrogen by plant PAOs (56, 57). Likewise, the oxidation of N1-acetylspermine and N1-acetylspermidine ends up with ACAPAL. APAL or ACAPAL is oxidized by AMADH to β-alanine and 3-acetamidopropionate, respectively. β-Alanine can subsequently be methylated to the osmoprotectant β-alanine betaine (58). It is notable that three of five Arabidopsis PAOs are localized in peroxisomes (57, 59, 60). Another part of polyamine catabolism is localized outside peroxisomes because the existence of apoplastic and symplastic PAOs has also been reported (56, 57). These PAOs also oxidize polyamines on the endo-side of N4-nitrogen, producing 1,3-propanediamine (from spermine) and ABAL (from spermidine). Because plant PAOs seem to have no activity or negligible activity with putrescine (57, 59), ABAL apparently originates from putrescine oxidation by plant copper diamine oxidases. It is then oxidized by AMADHs to GABA.
DMSPAL accumulates in many marine algae and a few higher plant species, including the Asteraceae and Poaceae families (9, 61). So far, DMSPAL has been shown to be oxidized by sugar beet and amaranth AMADHs/BADHs (10). Here we show that all studied AMADHs are capable of oxidizing it to the cryo- and osmoprotectant DMSP sharing similar properties with betaines (8, 61) (Fig. 5F). Indeed, SlAMADH1 and SlAMADH2 oxidize this compound at a 36 and 10% rate, respectively, compared with that with their best substrate. Likewise, ZmAMADHs display rates between 8 and 16% compared with those with APAL and both PsAMADHs at a 22% rate. The universal ability of AMADHs to convert DMSPAL seems related to its stable positive charge at the dimethylsulfonio group and its structural similarity with APAL, which is protonated at the primary amino group under physiological conditions. As well as ω-aminoaldehydes, DMSPAL is trapped into a nucleophilic cage formed by the carboxylic and aromatic residues at the active site of AMADH.
AMADH Gene Expression and Subcellular Localization
qPCR analysis shows that the expression of both tomato AMADH genes is relatively different. In contrast to the SlAMADH1 gene, the SlAMADH2 gene is highly and ubiquitously expressed in all analyzed tissues (Fig. 7). Whereas SlAMADH2 is well expressed in roots at 10 days after germination and at 6 weeks, SlAMADH1 is not expressed at all and expressed at a low extent (by 2 orders of magnitude), respectively. Both genes are expressed in cotyledons at 10 days after germination and later in flower organs (petals, carpel, and stamens) and senescent leaves. These data indicate that SlAMADH2 is playing a major role in all tomato organs in detoxifying toxic ω-aminoaldehydes. The observed high activity of both SlAMADHs with APAL and ABAL is in a good agreement with previous data obtained from in vivo experiments on tomatoes, where the formation of β-alanine and GABA was observed as a result of polyamine degradation (62).
FIGURE 7.
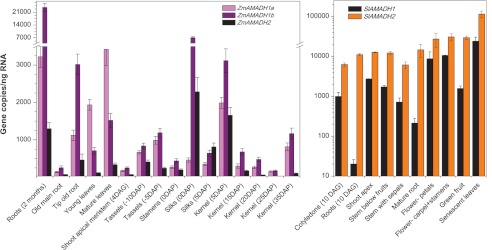
Expression profiles of two SlAMADH and three ZmAMADH genes in various organs as evaluated by qPCR. Transcript abundance is expressed as gene copy number in 1 ng of total RNA amplified by qPCR, normalized to EF1α, and recalculated as primer pair efficiency. Tomato samples, including roots and cotyledons, were collected at 10 days after germination (DAG); shoot apices (stem plus leaves), stems below flowers (including sepals), stems below fruits, petals, reproductive organs, green fruit, roots, and senescent leaves were collected from 6-week-old plants grown on a soil/peat mixture (2:1, v/v). Maize samples were collected from 3-month-old plants unless indicated otherwise. Tassels, silks, and kernels were collected at several days before, during, and after pollination (DAP). Error bars, S.D.
Because of the incomplete knowledge of tomato genome, it was impossible to localize the tomato genes, whereas those for maize could be mapped. The paralogs ZmAMADH1a and ZmAMADH1b are located on chromosomes 4 and 1, respectively. The gene of ZmAMADH2 is located on chromosome 10. The three maize genes are expressed differently (Fig. 7). A major difference concerns the ZmAMADH2 gene, which is markedly less expressed than the others. Transcript abundance of ZmAMADH2 is higher in silks at 0 and 5 days after pollination and in the kernel at 5 days after pollination. ZmAMADH1a (coding for the AMADH isoform with higher BADH activity) is highly expressed in young and mature leaves as well as in roots of younger plants. Transcript abundance of ZmAMADH1b is weaker in leaves but higher in roots compared with that of ZmAMADH1a. ZmAMADH1b is also highly expressed in silks at 0 days after pollination and in the kernel at 5 days after pollination. Both ZmAMADH1 genes are well expressed in tassels. These data indicate that AMADHs might be important for controlling ω-aminoaldehyde levels during early stages of the seed development.
Recently, several studies have shown that AMADH2 gene mutation, naturally occurring in several rice varieties, is responsible for their fragrance (6, 63, 64). AMADH2 was found to be transcribed constitutively in all tissues tested except for roots. The fragrance appears due to high abundance and subsequent acetylation of free ABAL and accumulation of 2-acetyl-Δ1-pyrroline. Traditional aromatic cultivars often have undesirable properties, including poor crop yield, susceptibility to pests and diseases, and strong shedding. Although their causes have not been analyzed so far, based on our results, these will be a combination of accumulation of toxic aminoaldehydes and lower content of zwitterionic products (β-alanine betaine, GABA, and others) having osmo-, cryo-, and heat-protecting properties.
Most plant AMADHs, including rice AMADHs and HvAMADH1, carry the SKL C-terminal peroxisomal targeting signal (PTS-1) (14, 65). All of our studied AMADHs except for SlAMADH1 contain PTS-1. Indeed, SlAMADH1 ends with a tripeptide SKN, which is not a peroxisomal targeting signal, making SlAMADH1 probably localized in the cytosol. The loss of the peroxisomal signal in one of the two AMADH isoforms has been observed in many plant species. For example, in Arabidopsis, one isoform (with SKL signal) is localized in peroxisomes, whereas the other (KSPN motif at the C terminus) is targeted to leucoplasts (66). Similarly, in barley, one isoform was demonstrated to be localized in peroxisomes, whereas the second appears to reside in the cytosol (65). The exact subcellular localization of SlAMADH1 remains unclear.
The phylogenetic analysis of numerous AMADH/BADH sequences from monocots and dicots shows that there are two homologous sequences per species among flowering plants and an additional one for maize. Thus, we believe this number can be higher in species with genome duplications. Among monocots, including maize, rice, and barley, two orthologous AMADH subgroups can be identified (Fig. 8). One subgroup contains a cluster of the Triticeae (Poaceae family) predictable as wide substrate-specific AMADHs (due to the presence of Phe-288 and Thr-454 in PsAMADH numbering; Fig. 8, highlighted in yellow). Moreover, the presence of Asn-289 implies that Tyr-163 is mobile, affecting the substrate binding. A similar cluster is found in dicots from the Solanaceae family. The majority of members in the other monocot subgroup display significant BADH activity due to the presence of an Ala-444 or Cys-444 residue favoring BAL binding (48). They also possess the Trp-288 residue. Interestingly, higher BAL activity correlates in part with nonperoxisomal targeting. For example, HvAMADH2, wild rye AMADH2, and wheat AMADH1 miss the SKL targeting signal and carry a C-terminal KAPAN motif instead. In the dicots, such a clustering only exists among closely related species. Here again, sequences of AMADHs with BADH activity from the Amaranthaceae, Acanthaceae, and Salicaceae families carry the KSP C-terminal signal as well as Ala-444 and Trp-288. Such a variable targeting indicates that AMADH genes might be differently responsive to, for example, stress conditions in various plants. The differential subcellular localization of AMADHs may arise from the interconnection of several metabolic pathways. Several PAOs are targeted to peroxisomes, where they generate APAL and ACAPAL from polyamines, which are detoxified by peroxisomal AMADHs. Other PAOs as well as copper diamine oxidases are localized in the cytoplasm. Copper diamine oxidases produce ABAL from putrescine, which is oxidized to GABA. In species accumulating GB, AMADHs are often found in chloroplasts (as well as choline monooxygenase), where they convert BAL to GB.
FIGURE 8.

Phylogenetic analysis of the plant ALDH10 family (AMADHs). The unrooted phylogenetic consensus tree shows two distinct groups of AMADHs, one from dicots (solid line) and the other from monocots (dashed line). PsAMADH numbering is used to point out crucial residues modulating substrate specificity. AMADHs exhibiting a significant BADH activity are highlighted in green. AMADHs predictable as broad substrate specific enzymes are highlighted in yellow due to the presence of Thr-454. The presence of Asn-289 implies that Tyr-163 is mobile, affecting the substrate binding. The enzymes from maize (ZmAMADHs) and tomato (SlAMADHs) analyzed in this work are shown in boldface type, as are the previously characterized reference enzymes from peas (PsAMADHs) (4), rice (OsAMADHs) (6), and barley (HvAMADHs) (18). The internal labels give bootstrap frequencies for each clade. See “Experimental Procedures” for the corresponding GenBankTM accession numbers.
CONCLUSIONS
In this work, we continued studies on substrate specificity of plant AMADHs. After previous analysis of two PsAMADHs, we now cloned five more genes: two from tomato and three from maize. We produced and purified the corresponding enzymes (SlAMADH1, SlAMADH2, ZmAMADH1a, ZMAMADH1b, and ZmAMADH2) to characterize them kinetically. Because two of them stand out from the others in terms of substrate specificity, SlAMADH1 having the broadest range of substrates and ZmAMADH1a having high BADH activity, we determined their crystal structures. From structural comparisons, we observe that SlAMADH1 is distinguished by its large and less polar substrate channel due to residues at positions 289 and 454 (288 and 453 in PsAMADHs), which are responsible for this large specificity. We can also predict that AMADHs containing Ile-444 (PsAMADH numbering) have a negligible BADH activity, and those containing Asn-289 may display some BADH activity. The wide substrate specificity of SlAMADH1 allowed us to trap a thiohemiacetal intermediate in the structure, never observed before, which, in addition to the SlAMADH1-E260A structure, suggests a bifunctional role of the active-site base glutamate. In addition to its known role for the activation of the hydrolytic water, it can activate the protonated catalytic cysteine.
Based on results of the entire study, we can now state that a tryptophan (Trp-288 in PsAMADHs), which is found in more than three-quarters of AMADH sequences, together with the conserved cluster of Tyr-163, Trp-170, and Trp-459, is responsible for the high affinity to ω-aminoaldehydes with APAL as the best substrate. AMADHs devoid of this tryptophan prefer TMABAL either directly due to the increase in the substrate channel diameter (and consequent loss of possible π-electron stacking interaction with the electrophilic protonated amino group of the substrate) or indirectly by affecting the position of the Tyr-163 side chain involved in substrate interaction. For the first time, we identified ACAPAL as a possible common substrate for all plant AMADHs. We also showed that the enzymes are capable of oxidizing DMSPAL to form the cryo- and osmoprotectant DMSP.
It becomes now obvious that AMADHs, depending on the plant species, are capable of oxidizing a wide range of naturally occurring ω-aminoaldehydes, resulting in the production of various amino acids that may directly or after a further conversion function as osmoprotectants. Thus, in plants, AMADHs control levels of ω-aminoaldehydes, which are produced under normal and stress conditions. Current transgenic studies on the overexpression of AMADH/BADH in plants conferring increased abiotic stress tolerance are aimed especially at GB production. Related engineering of GB synthesis in species with nonfunctional choline monooxygenase is also targeted rather narrowly. In the context of the present work, the wide substrate specificity of plant AMADHs indicates that there is a possibility of altered production of β-alanine betaine, GABA, or γ-butyrobetaine, which is worthy of consideration for the development and analysis of stress-tolerant transgenic crops. The latter compound may also be involved in the biosynthetic pathway of l-carnitine in plants, which shares similar features with pathways in mammals and fungi. In addition to its role in the mitochondrial transport of fatty acids, l-carnitine was also shown to confer tolerance to abiotic stress (52, 67).
Acknowledgments
We are grateful to Beatriz Guimaraes for help in data collection on PROXIMA 1 at the SOLEIL synchrotron and to Dr. Jacques Snégaroff (Institut Jean-Pierre Bourgin, INRA Versailles) and Prof. Thomas D. Hurley (Indiana University School of Medicine) for helpful discussion during manuscript preparation. This work benefited from IMAGIF platform facilities at the Centre de Recherche de Gif-sur-Yvette (FRC3115, CNRS, France) for crystallization work at the Structural Biology and Proteomic Unit in the Laboratoire d'Enzymologie et Biochimie Structurales.
This work was supported by Czech Science Foundation Grants P501/11/1591 and 522/08/0555 and OP RD&I Grant ED0007/01/01 (Centre of the Region Haná for Biotechnological and Agricultural Research) and supported in part by CNRS (to S. M. and A.V.).
The atomic coordinates and structure factors (codes 4I9B, 4I8Q, and 4I8P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PAO
- polyamine oxidase
- ABAL
- 4-aminobutyraldehyde
- ACAPAL
- 3-acetamidopropionaldehyde
- ALDH
- aldehyde dehydrogenase
- hALDH
- human ALDH
- AMADH
- aminoaldehyde dehydrogenase
- APAL
- 3-aminopropionaldehyde
- BADH
- betaine aldehyde dehydrogenase
- BAL
- betaine aldehyde
- DMSP
- 3-dimethylsulfoniopropionate
- DMSPAL
- 3-dimethylsulfoniopropionaldehyde
- GBAL
- 4-guanidinobutyraldehyde
- PCAL
- pyridine carboxaldehyde
- TMABAL
- N,N,N-trimethyl-4-aminobutyraldehyde
- GB
- glycine betaine
- NiR
- nicotinamide riboside
- qPCR
- quantitative PCR
- GABA
- γ-aminobutyric acid.
REFERENCES
- 1. Bouchereau A., Aziz A., Larher F., Martin-Tanguy J. (1999) Polyamines and environmental challenges. Recent development. Plant Sci. 140, 103–125 [Google Scholar]
- 2. Šebela M., Frébort I., Petı̌valský M., Peč P. (2002) Copper/topa quinone-containing amine oxidases. Recent research developments. in Studies in Natural Products Chemistry, Vol. 26, Bioactive Natural Products, Part G (Atta-ur-Rahman, ed) pp. 1259–1299, Elsevier, Amsterdam [Google Scholar]
- 3. Li W., Yuan X. M., Ivanova S., Tracey K. J., Eaton J. W., Brunk U. T. (2003) 3-Aminopropanal, formed during cerebral ischaemia, is a potent lysosomotropic neurotoxin. Biochem. J. 371, 429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tylichová M., Kopečný D., Moréra S., Briozzo P., Lenobel R., Snégaroff J., Šebela M. (2010) Structural and functional characterization of plant aminoaldehyde dehydrogenase from Pisum sativum with a broad specificity for natural and synthetic aminoaldehydes. J. Mol. Biol. 396, 870–882 [DOI] [PubMed] [Google Scholar]
- 5. Bouché N., Fromm H. (2004) GABA in plants. Just a metabolite? Trends Plant Sci. 9, 110–115 [DOI] [PubMed] [Google Scholar]
- 6. Bradbury L. M., Gillies S. A., Brushett D. J., Waters D. L., Henry R. J. (2008) Inactivation of an aminoaldehyde dehydrogenase is responsible for fragrance in rice. Plant Mol. Biol. 68, 439–449 [DOI] [PubMed] [Google Scholar]
- 7. Arikit S., Yoshihashi T., Wanchana S., Uyen T. T., Huong N. T., Wongpornchai S., Vanavichit A. (2011) Deficiency in the amino aldehyde dehydrogenase encoded by GmAMADH2, the homologue of rice Os2AP, enhances 2-acetyl-1-pyrroline biosynthesis in soybeans (Glycine max L.). Plant Biotechnol. J. 9, 75–87 [DOI] [PubMed] [Google Scholar]
- 8. Rhodes D., Hanson A. D. (1993) Quaternary ammonium and tertiary sulfonium compounds in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 44, 357–384 [Google Scholar]
- 9. James F., Paquet L., Sparace S. A., Gage D. A., Hanson A. D. (1995) Evidence implicating dimethylsulfoniopropionaldehyde as an intermediate in dimethylsulfoniopropionate biosynthesis. Plant Physiol. 108, 1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trossat C., Rathinasabapathi B., Hanson A. D. (1997) Transgenically expressed betaine aldehyde dehydrogenase efficiently catalyzes oxidation of dimethylsulfoniopropionaldehyde and ω-aminoaldehydes. Plant Physiol. 113, 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kirch H. H., Bartels D., Wei Y., Schnable P. S., Wood A. J. (2004) The ALDH gene superfamily of Arabidopsis. Trends Plant Sci. 9, 371–377 [DOI] [PubMed] [Google Scholar]
- 12. Sophos N. A., Vasiliou V. (2003) Aldehyde dehydrogenase gene superfamily. The 2002 update. Chem. Biol. Interact. 143, 5–22 [DOI] [PubMed] [Google Scholar]
- 13. Nakamura T., Yokota S., Muramoto Y., Tsutsui K., Oguri Y., Fukui K., Takabe T. (1997) Expression of a betaine aldehyde dehydrogenase gene in rice, a glycinebetaine nonaccumulator, and possible localization of its protein in peroxisomes. Plant J. 11, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 14. Reumann S., Babujee L., Ma C., Wienkoop S., Siemsen T., Antonicelli G. E., Rasche N., Lüder F., Weckwerth W., Jahn O. (2007) Proteome analysis of Arabidopsis leaf peroxisomes reveals novel targeting peptides, metabolic pathways, and defense mechanisms. Plant Cell 19, 3170–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez-Miller S. J., Hurley T. D. (2003) Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 42, 7100–7109 [DOI] [PubMed] [Google Scholar]
- 16. Wymore T., Hempel J., Cho S. S., Mackerell A. D., Jr., Nicholas H. B., Jr., Deerfield D. W., 2nd (2004) Molecular recognition of aldehydes by aldehyde dehydrogenase and mechanism of nucleophile activation. Proteins 57, 758–771 [DOI] [PubMed] [Google Scholar]
- 17. Kopečný D., Tylichová M., Snegaroff J., Popelková H., Šebela M. (2011) Carboxylate and aromatic active-site residues are determinants of high-affinity binding of ω-aminoaldehydes to plant aminoaldehyde dehydrogenases. FEBS J. 278, 3130–3139 [DOI] [PubMed] [Google Scholar]
- 18. Fujiwara T., Hori K., Ozaki K., Yokota Y., Mitsuya S., Ichiyanagi T., Hattori T., Takabe T. (2008) Enzymatic characterization of peroxisomal and cytosolic betaine aldehyde dehydrogenases in barley. Physiol. Plant 134, 22–30 [DOI] [PubMed] [Google Scholar]
- 19. Brocker C., Vasiliou M., Carpenter S., Carpenter C., Zhang Y., Wang X., Kotchoni S. O., Wood A. J., Kirch H. H., Kopečný D., Nebert D. W, Vasiliou V. (2013) Aldehyde dehydrogenase (ALDH) superfamily in plants. Gene nomenclature and comparative genomics. Planta 237, 189–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tylichová M., Briozzo P., Kopečný D., Ferrero J., Moréra S., Joly N., Snégaroff J., Šebela M. (2008) Purification, crystallization and preliminary crystallographic study of a recombinant plant aminoaldehyde dehydrogenase from Pisum sativum. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 88–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Šebela M., Štosová T., Havlis J., Wielsch N., Thomas H., Zdráhal Z., Shevchenko A. (2006) Thermostable trypsin conjugates for high-throughput proteomics. Synthesis and performance evaluation. Proteomics 6, 2959–2963 [DOI] [PubMed] [Google Scholar]
- 22. Sebela M., Brauner F., Radová A., Jacobsen S., Havlis J., Galuszka P., Pec P. (2000) Characterisation of a homogeneous plant aminoaldehyde dehydrogenase. Biochim. Biophys. Acta. 1480, 329–341 [DOI] [PubMed] [Google Scholar]
- 23. Vaz F. M., Fouchier S. W., Ofman R., Sommer M., Wanders R. J. (2000) Molecular and biochemical characterization of rat γ-trimethylaminobutyraldehyde dehydrogenase and evidence for the involvement of human aldehyde dehydrogenase 9 in carnitine biosynthesis. J. Biol. Chem. 275, 7390–7394 [DOI] [PubMed] [Google Scholar]
- 24. Wood P. L., Khan M. A., Moskal J. R. (2007) The concept of “aldehyde load” in neurodegenerative mechanisms. Cytotoxicity of the polyamine degradation products hydrogen peroxide, acrolein, 3-aminopropanal, 3-acetamidopropanal and 4-aminobutanal in a retinal ganglion cell line. Brain Res. 1145, 150–156 [DOI] [PubMed] [Google Scholar]
- 25. Holt A., Degenhardt O. S., Berry P. D., Kapty J. S., Mithani S., Smith D. J., Di Paolo M. L. (2007) The effects of buffer cations on interactions between mammalian copper-containing amine oxidases and their substrates. J. Neural Transm. 114, 733–741 [DOI] [PubMed] [Google Scholar]
- 26. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30. Edgar R. C. (2004) MUSCLE. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guindon S., Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 32. Zheng H., Chruszcz M., Lasota P., Lebioda L., Minor W. (2008) Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 102, 1765–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamb A. L., Newcomer M. E. (1999) The structure of retinal dehydrogenase type II at 2.7 Å resolution. Implications for retinal specificity. Biochemistry 38, 6003–6011 [DOI] [PubMed] [Google Scholar]
- 34. González-Segura L., Rudiño-Piñera E., Muñoz-Clares R. A., Horjales E. (2009) The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa provides new insight into the reaction mechanism and shows a novel binding mode of the 2′-phosphate of NADP+ and a novel cation binding site. J. Mol. Biol. 385, 542–557 [DOI] [PubMed] [Google Scholar]
- 35. Izaguirre G., Pietruszko R., Cho S., MacKerell A., Jr. (2001) Human aldehyde dehydrogenase catalytic activity and structural interactions with coenzyme analogs. J. Biomol. Struct. Dyn. 19, 429–447 [DOI] [PubMed] [Google Scholar]
- 36. Hill E. J., Chou T. H., Shih M. C., Park J. H. (1975) Covalent binding of 3-pyridinealdehyde nicotinamide adenine dinucleotide and substrate to glyceraldehyde 3-phosphate dehydrogenase. J. Biol. Chem. 250, 1734–1740 [PubMed] [Google Scholar]
- 37. Steinmetz C. G., Xie P., Weiner H., Hurley T. D. (1997) Structure of mitochondrial aldehyde dehydrogenase. The genetic component of ethanol aversion. Structure 5, 701–711 [DOI] [PubMed] [Google Scholar]
- 38. D'Ambrosio K., Pailot A., Talfournier F., Didierjean C., Benedetti E., Aubry A., Branlant G., Corbier C. (2006) The first crystal structure of a thioacylenzyme intermediate in the ALDH family. New coenzyme conformation and relevance to catalysis. Biochemistry 45, 2978–2986 [DOI] [PubMed] [Google Scholar]
- 39. Brauner F., Šebela M., Snégaroff J., Peč P., Meunier J. C. (2003) Pea seedling aminoaldehyde dehydrogenase. Primary structure and active site residues. Plant Physiol. Biochem. 41, 1–10 [Google Scholar]
- 40. Wang X., Weiner H. (1995) Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry 34, 237–243 [DOI] [PubMed] [Google Scholar]
- 41. Mann C. J., Weiner H. (1999) Differences in the roles of conserved glutamic acid residues in the active site of human class 3 and class 2 aldehyde dehydrogenases. Protein Sci. 8, 1922–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brouquisse R., Weigel P., Rhodes D., Yocum C. F., Hanson A. D. (1989) Evidence for a ferredoxin-dependent choline monooxygenase from spinach chloroplast stroma. Plant Physiol. 90, 322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishitani M., Arakawa K., Mizuno K., Kishitani S., Takabe T. (1993) Betaine aldehyde dehydrogenase in the Gramineae. Levels in leaves of both betaine-accumulating and non-accumulating cereal plants. Plant Cell Physiol. 34, 493–495 [Google Scholar]
- 44. Brunk D. G., Rich P. J., Rhodes D. (1989) Genotypic variation for glycinebetaine among public inbreds of maize. Plant Physiol. 91, 1122–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lerma C., Rich P. J., Ju G. C., Yang W.-J., Hanson A. D., Rhodes D. (1991) Betaine deficiency in maize. Complementation tests and metabolic basis. Plant Physiol. 95, 1113–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mäkelä P., Jokinen K., Kontturi M., Peltonen-Sainio P., Pheu E., Somersalo S. (1998) Foliar application of glycinebetaine, a novel product from sugar beet, as an approach to increase tomato yield. Ind. Crop. Prod. 7, 139–148 [Google Scholar]
- 47. Charlton A. J., Donarski J. A., Harrison M., Jones S. A., Godward J., Oehlschlager S., Arques J. L., Ambrose M., Chinoy C., Mullineaux P. M., Domoney C. (2008) Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics 4, 312–327 [Google Scholar]
- 48. Díaz-Sánchez Á. G., González-Segura L., Mújica-Jiménez C., Rudiño-Piñera E., Montiel C., Martínez-Castilla L. P., Muñoz-Clares R. A. (2012) Amino acid residues critical for the specificity for betaine aldehyde of the plant ALDH10 isoenzyme involved in the synthesis of glycine betaine. Plant Physiol. 158, 1570–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weigel P., Weretilnyk E. A., Hanson A. D. (1986) Betaine aldehyde oxidation by spinach chloroplasts. Plant Physiol. 82, 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hibino T., Meng Y. L., Kawamitsu Y., Uehara N., Matsuda N., Tanaka Y., Ishikawa H., Baba S., Takabe T., Wada K., Ishii T., Takabe T. (2001) Molecular cloning and functional characterization of two kinds of betaine-aldehyde dehydrogenase in betaine-accumulating mangrove Avicennia marina (Forsk.) Vierh. Plant Mol. Biol. 45, 353–363 [DOI] [PubMed] [Google Scholar]
- 51. Valenzuela-Soto E. M., Muñoz-Clares R. A. (1994) Purification and properties of betaine aldehyde dehydrogenase extracted from detached leaves of Amaranthus hypochondriacus L. subjected to water deficit. J. Plant Physiol. 143, 145–152 [Google Scholar]
- 52. Rippa S., Zhao Y., Merlier F., Charrier A., Perrin Y. (2012) The carnitine biosynthetic pathway in Arabidopsis thaliana shares similar features with the pathway of mammals and fungi. Plant Physiol. Biochem. 60, 109–114 [DOI] [PubMed] [Google Scholar]
- 53. Marchitti S. A., Brocker C., Stagos D., Vasiliou V. (2008) Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 4, 697–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johansson K., El-Ahmad M., Ramaswamy S., Hjelmqvist L., Jörnvall H., Eklund H. (1998) Structure of betaine aldehyde dehydrogenase at 2.1 Å resolution. Protein Sci. 7, 2106–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hjelmqvist L., Norin A., El-Ahmad M., Griffiths W., Jörnvall H. (2003) Distinct but parallel evolutionary patterns between alcohol and aldehyde dehydrogenases. Addition of fish/human betaine aldehyde dehydrogenase divergence. Cell Mol. Life Sci. 60, 2009–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tavladoraki P., Rossi M. N., Saccuti G., Perez-Amador M. A., Polticelli F., Angelini R., Federico R. (2006) Heterologous expression and biochemical characterization of a polyamine oxidase from Arabidopsis involved in polyamine back conversion. Plant Physiol. 141, 1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takahashi Y., Cong R., Sagor G. H., Niitsu M., Berberich T., Kusano T. (2010) Characterization of five polyamine oxidase isoforms in Arabidopsis thaliana. Plant Cell Rep. 29, 955–965 [DOI] [PubMed] [Google Scholar]
- 58. Raman S. B., Rathinasabapathi B. (2003) β-Alanine N-methyltransferase of Limonium latifolium. cDNA cloning and functional expression of a novel N-methyltransferase implicated in the synthesis of the osmoprotectant β-alanine betaine. Plant Physiol. 132, 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moschou P. N., Sanmartin M., Andriopoulou A. H., Rojo E., Sanchez-Serrano J. J., Roubelakis-Angelakis K. A. (2008) Bridging the gap between plant and mammalian polyamine catabolism. A novel peroxisomal polyamine oxidase responsible for a full back-conversion pathway in Arabidopsis. Plant Physiol. 147, 1845–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kamada-Nobusada T., Hayashi M., Fukazawa M., Sakakibara H., Nishimura M. (2008) A putative peroxisomal polyamine oxidase, AtPAO4, is involved in polyamine catabolism in Arabidopsis thaliana. Plant Cell Physiol. 49, 1272–1282 [DOI] [PubMed] [Google Scholar]
- 61. Paquet L., Rathinasabapathi B., Saini H., Zamir L., Gage D. A., Huang Z.-H., Hanson A. D. (1994) Accumulation of the compatible solute 3-dimethylsulfoniopropionate in sugarcane and its relatives, but not other gramineous crops. Aust. J. Plant Physiol. 21, 37–48 [Google Scholar]
- 62. Rastogi R., Davies P. J. (1990) Polyamine metabolism in ripening tomato fruit. I. Identification of metabolites of putrescine and spermidine. Plant Physiol. 94, 1449–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen S., Yang Y., Shi W., Ji Q., He F., Zhang Z., Cheng Z., Liu X., Xu M. (2008) Badh2, encoding betaine aldehyde dehydrogenase, inhibits the biosynthesis of 2-acetyl-1-pyrroline, a major component in rice fragrance. Plant Cell 20, 1850–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Niu X., Tang W., Huang W., Ren G., Wang Q., Luo D., Xiao Y., Yang S., Wang F., Lu B. R., Gao F., Lu T., Liu Y. (2008) RNAi-directed down regulation of OsBADH2 results in aroma (2-acetyl-1-pyrroline) production in rice (Oryza sativa L.). BMC Plant Biol. 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nakamura T., Nomura M., Mori H., Jagendorf A. T., Ueda A., Takabe T. (2001) An isozyme of betaine aldehyde dehydrogenase in barley. Plant Cell Physiol. 42, 1088–1092 [DOI] [PubMed] [Google Scholar]
- 66. Missihoun T. D., Schmitz J., Klug R., Kirch H. H., Bartels D. (2011) Betaine aldehyde dehydrogenase genes from Arabidopsis with different sub-cellular localization affect stress responses. Planta 233, 369–382 [DOI] [PubMed] [Google Scholar]
- 67. Charrier A., Rippa S., Yu A., Nguyen P. J., Renou J. P., Perrin Y. (2012) The effect of carnitine on Arabidopsis development and recovery in salt stress conditions. Planta 235, 123–135 [DOI] [PubMed] [Google Scholar]