

Background: Variations of agarase GH modules and their effects have been minimally elucidated.
Results: Deleting the extra fragment in the GH module or swapping the inside Tyr276 changes the degradation characteristics of the agarase rAgaG4.
Conclusion: The 276-residue sequence within the extra fragment is crucial for AgaG4 to bind and degrade neoagarooctaose.
Significance: Modification of active site residues in agarase changes the degradation pattern.
Keywords: Carbohydrate, Enzymes, Genetics, Glycobiology, Microbiology, Agarase, Flammeovirga, Gene Truncation, Glycoside Hydrolase, Active Site Residue
Abstract
Agarase hydrolyzes agarose into a series of oligosaccharides with repeating disaccharide units. The glycoside hydrolase (GH) module of agarase is known to be responsible for its catalytic activity. However, variations in the composition of the GH module and its effects on enzymatic functions have been minimally elucidated. The agaG4 gene, cloned from the genome of the agarolytic Flammeovirga strain MY04, encodes a 503-amino acid protein, AgaG4. Compared with elucidated agarases, AgaG4 contains an extra peptide (Asn246–Gly302) within its GH module. Heterologously expressed AgaG4 (recombinant AgaG4; rAgaG4) was determined to be an endo-type β-agarase. The protein degraded agarose into neoagarotetraose and neoagarohexaose at a final molar ratio of 1.5:1. Neoagarooctaose was the smallest substrate for rAgaG4, whereas neoagarotetraose was the minimal degradation product. Removing the extra fragment from the GH module led to the inability of the mutant (rAgaG4-T57) to degrade neoagarooctaose, and the final degradation products of agarose by the truncated protein were neoagarotetraose, neoagarohexaose, and neoagarooctaose at a final molar ratio of 2.7:2.8:1. The optimal temperature for agarose degradation also decreased to 40 °C for this mutant. Bioinformatic analysis suggested that tyrosine 276 within the extra fragment was a candidate active site residue for the enzymatic activity. Site-swapping experiments of Tyr276 to 19 various other amino acids demonstrated that the characteristics of this residue were crucial for the AgaG4 degradation of agarose and the cleavage pattern of substrate.
Introduction
Agarose is a complex polysaccharide composed of 3,6-anhydro-l-galactopyranose- α-1,3-d-galactose units that are joined by β1–4 bonds (1). Agarases, which are classified as glycoside hydrolases (GHs),3 catalyze the cleavage of agarose at α1–3 or β1–4 linkages, producing agaro-oligomers with repeating disaccharide units. Based on their cleaved linkage type, agarases are classified into two groups: α-agarases and β-agarases. The α-agarases (EC 3.2.1.158) degrade agarose at α1–3 linkages, producing agaro-oligosaccharides with β1-4–3,6-anhydro-l-galactopyranose as the reducing end (2), whereas the β-agarases (EC 3.2.1.81) degrade agarose at β1–4 linkages, producing neoagaro-oligosaccharides (NAOs) with α-1,3-d-galactose as the reducing end (3, 4). More than 50 agarases have been identified and characterized in different microorganisms to date (5–7). Most of these enzymes are β-agarases, whereas only two are α-agarases (from Alteromonas agarlyticus GJ1B (8) and Thalassomonas sp. JAMB-A33 (9), respectively). Agarases are modular proteins that contain an array of a catalytic GH module and one or two non-catalytic carbohydrate-binding modules (10, 11). Based on GH sequence similarity, the characterized α-agarases are classified as members of the GH96 family (12, 13). Most of the reported β-agarases belong to the GH16 family in the CAZy (carbohydrate-active enzyme) database, but some β-agarases belong to the GH50, GH86, or GH118 family (5, 14, 15). Crystal structures of the GH modules (10, 16, 17) and their catalytic mechanisms have been well elucidated (18–21). However, there have been few studies focusing on inner peptides of the GH modules and their effects on the enzymatic properties of agarases.
Flammeovirga is a bacterial genus of the newly denominated α-proteobacterial family Flammeovirgaceae. All type strains of the five species (22–25) within this genus have been described as agarolytic. In our previous studies (26), we obtained an agarolytic marine bacterial strain, Flammeovirga sp. MY04. This strain can potently degrade and utilize many different polysaccharides, particularly agarose. The degradation of agarose by the extracellular agarase system of MY04 produces neoagarotetraose (NA4) and neoagarohexaose (NA6) as the final products. In our analysis of the sequences of a fosmid clone of MY04, we found a predicted agarase gene, agaG4. Interestingly, compared with the already elucidated agarases, the predicted 503-amino acid protein of agaG4 contains an extra peptide (Asn246–Gly302) within its GH module. In this paper, we characterize this agarase and the contributions of the extra fragment on agarose degradation of the enzyme.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Growth Conditions
The strains and plasmids used in this study are listed in Table 1. Unless otherwise noted, Escherichia coli strains were cultured at 37 °C in Luria-Bertani (LB) medium supplemented with chloramphenical (12.5 μg/ml) and/or ampicillin (100 μg/ml). Flammeovirga sp. MY04 (CGMCC 2777) was cultured at 30 °C in a medium (pH 7.2) containing 3.0% (w/v) NaCl, 0.75% (w/v) KCl, 0.11% (w/v) CaCl2, 0.72% (w/v) Mg2SO4, 0.4% (w/v) tryptone, and 0.25% (w/v) yeast extract. Agar (1.5%, w/v) was used to prepare the solid medium.
TABLE 1.
Bacterial strains, plasmids, and sequencing primers used in this study
Apr, ampicillin-resistant; Cmr, chloramphenicol-resistant.

Construction of the Genomic Fosmid Library
Flammeovirga sp. MY04 cells were pretreated with lysozyme, and the genomic DNA was extracted using SDS and proteinase K treatment (27). Approximately 2 mg of the genomic DNA was mechanically sheared using a 10-μl glass syringe and separated on a preparative pulsed field gel (Bio-Rad CHEF DR®III; 0.1–10-s switch time, 6 V/cm, 0.5× TBE buffer, 120° included angle, 14 h). DNA fragments with approximate lengths ranging from 36 to 50 kb were recovered, electroeluted, and dialyzed against 0.5× TE buffer (pH 8.0). The purified DNA fragments were end-repaired with end-repaired enzyme mix, size-fractionated, and recovered from the gel. The recovered blunt-ended, 5′-phosphorylated DNA was ligated into the cloning-ready Copycontrol pCC1FOS vector, and the recombinant molecules were packaged in vitro with a MaxPlaxTM Lambda packaging kit (Epicenter Biotechnologies, Madison, WI).
Recombinant fosmids were isolated using the method of Birnboim and Doly (28). Plasmids were prepared using a TIANprep minikit (TianGen, Beijing, China). Competent E. coli cells were prepared using calcium chloride (27). Restriction enzymes, T4 DNA ligase, calf intestinal alkaline phosphatase, and DNA polymerases were purchased from Takara (Dalian, China) and used according to the manufacturer's instructions.
Screening and Cloning of Agarose-degrading Genes
Iodine staining was initially performed to identify extracellular agarase genes in the genomic fosmid library (29). Briefly, transformed EPI300 cells were initially plated on LB plates and incubated at 37 °C for 18 h. Randomly selected colonies were then transferred onto LB agar plates supplemented with inducer (1:1000 dilution, Epicenter Biotechnologies), which increases the fosmid copy number up to 50. The transferred cultures were incubated at 30 °C for 7 days, and the plates were stained using an iodine solution (2.0%, w/v). Colonies surrounded by halos were referred to as positive clones that contained agarase genes from Flammeovirga sp. MY04.
The other genetic screening strategy was also used to identify intracellular agarase genes in the fosmid genomic library. In brief, recombinant fosmids of randomly selected clones were prepared individually and sequenced using the vector primers F5FP010 and F5RP011 (Table 1) to determine the DNA sequences at the T7 end and the pCC1R end, respectively. The sequencing products were trimmed of their vector sequences, and the pertinent sequence tags were analyzed. Based on the sequence tags encoding putative agarases, gene-specific primer pairs were designed, synthesized, and applied in the PCR screening of the positive clones that targeted full-length genes. Fosmids harboring agarase genes were shotgun-sequenced.
Sequence Analysis of the Agarase Genes and Proteins
Promoter motifs of the 5′-flanking sequences upstream from the open reading frames (ORFs) or operons were identified using Primer Premier version 5.0 (PREMIER Biosoft International, Palo Alto, CA) and the Promoter 2.0 Prediction Server. The G + C content (G + C%) of the ORFs and the G + C% at the third position of the synonymous codons (GC3s) were calculated using CodonW.
In an extension of the functional annotation, similarity searches of the predicted proteins were performed using the BLAST algorithm on the National Center for Biotechnology Information (NCBI) server. Secretion signals and their types were identified using the SignalP 3.0 server and the LipoP 1.0 server. Molecular masses of polypeptides were estimated using the peptide mass tool on the ExPASy server of the Swiss Institute of Bioinformatics. Sequences were aligned using Bio-Edit version 7.0.5.3 (30). Protein modules and domains were identified in the products using the Simple Modular Architecture Research Tool (SMART), the Pfam database, and the CAZy database. A phylogenetic tree was generated using the neighbor-joining method of Nei and co-workers (31) with MEGA version 5.05.
Construction of Expression Vectors
For the expression of AgaG4, the full-length gene of AgaG4 was amplified using the primers G4BAD-f and G4BAD-r (Table 2) and high fidelity PrimeSTARTMHS DNA polymerase (Takara). The primer pairs with restriction enzyme sites (underlined) for NcoI and XbaI were designed to generate a gIII virus signal peptide for secretion at the N terminus and a His6 tag at the C terminus of the recombinant agarase (rAgaG4). The DNA amplification products were cloned into the expression vector pBAD/gIII A, and the recombinant plasmid (pBAgaG4) was transformed into E. coli TOP10 cells. Expression vectors derived from the pET-22b(+), pET-28a(+), pET-30a(+), and pCold TF plasmids were constructed for the heterologous expression of AgaG4 in E. coli BL21(DE3). The integrity of the nucleotide sequence of all newly constructed plasmids was confirmed by DNA sequencing.
TABLE 2.
Primers used in this study
Heterologous Expression and Purification of AgaG4
E. coli TOP10 cells harboring plasmid pBAgaG4 were initially cultured in LB broth. When the cell density reached an A600 of 0.8, expression of rAgaG4 was induced by the addition of 1.0 mmol/liter l-arabinose, followed by continual cultivation for an additional 6 h at 30 °C. Cells were harvested by centrifugation at 5000 × g for 15 min, washed twice with ice-cold buffer A (50 mmol/liter Tris, 150 mmol/liter NaCl, pH 8.0), resuspended in buffer A, and disrupted by sonication (60 repetitions, 5 s) in an ice-water bath. Centrifugation at 15,000 × g for 30 min allowed for crude inclusion bodies of rAgaG4 to be collected from the precipitation. Using buffer A supplemented with 1.0% (v/v) Triton X-100 and 2 mol/liter urea, the inclusion bodies were washed twice for further purification. The purified inclusion bodies were dissolved in buffer B (buffer A supplemented with 8 mol/liter urea) at 4 °C. After centrifugation, the supernatant containing the denatured proteins was loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) column (Novagen) that was pre-equilibrated with buffer B and eluted with buffer B that was supplemented with imidazole at increasing gradient concentrations (0, 10, 50, and 250 mmol/liter). Fractionated protein samples were analyzed using SDS-PAGE. To obtain active agarase, the purified 55-kDa protein was diluted and dialyzed against the universal TGE buffer (50 mmol/liter Tris, 50 mmol/liter NaCl, 0.5 mmol/liter EDTA, 5 mmol/liter DTT, 5% glycerol, pH 7.9) supplemented with 4 mol/liter urea and then 2 mol/liter urea and finally without urea. To reduce the urea concentration to lower than 1 mg/liter, the agarase solution was further dialyzed against the TGE buffer for a total of four times. Each dialysis step was performed at 4 °C for 2 h with a volume ratio of 1:50 (protein/buffer). Moreover, similar analyses were performed for the heterologous expression of AgaG4 using pET-22b(+), pET-28a(+), pET-30a(+), and pCold TF in E. coli BL21(DE3).
SDS-PAGE was performed using 13.2% (w/v) polyacrylamide gels according to Sambrook and Russell (27). Proteins were detected by staining the gel with Coomassie Brilliant Blue R-250. Protein concentrations were determined by the Folin-Lowry method using Folin Ciocalteu's phenol reagent (Sigma-Aldrich) and bovine serum albumin as a standard.
Enzyme Activity Assay
Agarase activity was assayed using the 3,5-dinitrosalicylic acid method (32). Stock solutions (0.50%, w/v) of agarose (Invitrogen) were individually prepared using deionized water. Aliquots (950 μl) were boiled and then maintained at 45 °C to prevent the polysaccharide from gelling. The solutions, referred to as melted agarose, were incubated in triplicate at 45 °C with 50 μl of appropriately diluted agarase. The mixture was incubated at 50 °C for 40 min. After adding 1 ml of 3,5-dinitrosalicylic acid, the reaction mixture was heated in boiling water (99 °C) for 10 min. The absorbance of the reducing sugar was measured at 540 nm and compared with the standard curve of d-galactose (Sigma-Aldrich). One unit of enzyme was defined as the amount of enzyme that produced 1 μmol of d-galactose/min.
Biochemical Characterization of rAgaG4
To determine the optimal conditions for rAgaG4 activity and the stability of rAgaG4, agarose solutions (0.10%, w/v) were prepared using buffers with different pH values, including 50 mmol/liter acetate buffer (pH 4.0–6.5), 50 mmol/liter HEPES buffer (pH 6.0–8.0), and 50 mmol/liter Tris-HCl buffer (pH 7.5–10). The optimal temperature for rAgaG4 was determined by monitoring its enzymatic activity at temperatures ranging from 0 to 80 °C at pH 7.5 for 40 min. The thermostability of rAgaG4 was evaluated by measuring the residual activity of the enzyme after incubation at various temperatures for 1 h. The pH dependence of rAgaG4 was assayed at 50 °C within a pH range of 4.0–10. The effect of pH on rAgaG4 stability was determined by measuring its residual activity after incubation at 4 °C and various pH values (4.0–10.0) for 1 h. The effects of metal ions and chelating agents on rAgaG4 activity were examined by determining its activity in the presence of 1 mmol/liter and 10 mmol/liter of various metal ions or chelating agents. Additionally, the substrate specificity of rAgaG4 was determined at 50 °C using 0.10% (w/v) agar, agarose, alginate, carrageenan, ι-carrageenan, chitin, chitosan, and xylan as substrates.
Analysis of the Agarose-degrading Properties of rAgaG4
The digestion pattern of agarose (0.05%, w/v) by rAgaG4 (5 units/liter) at 45 °C was traced over 72 h. Similar experiments were conducted using different final concentrations of agarose, ranging from 0.10 to 0.50% (w/v). Aliquots of the degradation products were removed for time course analysis with thin layer chromatography (TLC) using silica gel 60 plates (F254, Merck). The plates were developed using a solvent system with a ratio of 2:1:1 (v/v/v) for n-butanol/ethanol/water. The resultant oligosaccharide spots were visualized by spraying the plates with a staining solution (1% diphenylamine and aniline in acetone) and heating at 110 °C for 10 min (33). Standard NAOs ranging from 2 to 8 residues (NA2–NA8) were provided by Prof. Wengong Yu (Ocean University of China). The concentrations of the degradation products were determined using the dinitrosalicylic acid reducing sugar assay (32).
To determine the molar ratio of the individual oligosaccharides in the product, the reducing ends of ∼10 μg of the final degradation products were fluorescently labeled using 2-aminobenzamide (2-AB) and sodium cyanoborohydride reagents as described by Bigge et al. (34). Fluorescently labeled samples (20 μg/ml) were then injected into a CO-150 sampler equipped with a 20-μl sample loop and a BioSep SEC-S2000 column, and the samples were stimulated at 330 nm and monitored at 420 nm. The mobile phase for the SEC was 0.10 m NH4HCO3 with a flow rate of 0.5 ml/min. A mixture of d-galactose and standard NAOs was fluorescently labeled and used as a calibration standard. The software LCsolution version 1.25 was used for online data monitoring and analysis.
To determine the composition of the final degradation product, 100 ml of agarose (0.1%, w/v) was digested using superfluous rAgaG4 (0.10 unit/ml) at 45 °C for 72 h. The reaction mixture was heated in boiling water for 10 min, subsequently cooled to 4 °C, and centrifuged at 12,000 × g for 30 min. The supernatant was concentrated by rotary evaporation at 45 °C. The concentrated oligosaccharide products were loaded onto a pre-equilibrated Superdex Peptide 10/300G column (GE Healthcare) and eluted with 0.10 m NH4HCO3 at a flow rate of 0.5 ml/min. Oligosaccharide monomers in the fractionated samples were assayed using TLC, collected, and freeze-dried repeatedly to remove NH4HCO3 for further component identification. With MALDI-TOF MS (AXIMA-CFR plus, Shimadzu, Japan), the pure oligosaccharides of the two final degradation products were individually observed as the pseudomolecular ions [M + K]+ and [M + Na]+ via spectrometry using 2,5-dihydroxybenzoic acid as the matrix. For 13C NMR spectroscopy, 10 mg of each purified oligosaccharide was dissolved in 0.5 ml of D2O in 5-mm NMR tubes. The spectra were recorded on a JNM-ECP600 (JEOL, Japan) apparatus set at 150 MHz with acetone-d6 as the internal standard.
Analysis of the Oligosaccharide-degrading Properties of rAgaG4
To assay the smallest substrate of rAgaG4, NAOs with different length chains (NA4, NA6, NA8, and NA10) were purified using gel filtration from partially digested agarose by rAgaG4. Individual oligosaccharide solutions (0.05%, w/v) were supplemented with rAgaG4 (0.05 unit/ml), and the mixture was incubated at 45 °C for 12 h. The reaction mixture was heated in boiling water for 10 min, cooled at 4 °C, and centrifuged at 12,000 × g for 5 min. The supernatant was concentrated by rotary evaporation and labeled using 2-AB. The fluorescently labeled products were analyzed by HPLC using the BioSep SEC-S2000 column.
To determine the enzymatic degradation pattern of rAgaG4, pure oligosaccharide monomers (NA4, NA6, NA8, and NA10) were fluorescently labeled at their reducing ends using 2-AB. The labeled products were purified by HPLC and degraded with rAgaG4. The substrate and degradation products were subjected to HPLC for fluorescent analyses under similar conditions.
Functional Analyses of the Extra Peptide within the GH Module of AgaG4
To identify the role of the extra fragment (Asn246–Gly302) in agarose degradation by AgaG4, we deleted this peptide from the enzyme using a gene truncation strategy as listed in Fig. 1. PCR primers for the truncation of agaG4 and the heterologous expression of the mutant are listed in Table 2. The mutated gene product was named agaG4-T57, the recombinant protein of which (rAgaG4-T57) was overexpressed, purified, and characterized using a strategy identical to that described previously for rAgaG4.
FIGURE 1.
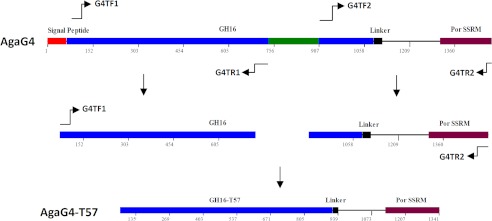
Schematic diagram of β-agarase AgaG4 and its derivatives. GH16, GH16 module; Linker, a Gly- and Ser-rich region; SSSM, secretion system sorting module. AgaG4-T57, a deviant of AgaG4 made by deleting the excess 57-amino acid peptide within the GH16 module. PCR primers used in the gene truncation of agaG4 and heterologous expression of agaG4-T57 are listed in Table 2.
Identification and Characterization of the Active Site Residue within the Extra Peptide
The protein sequence of AgaG4 was directly submitted to the SwissModel Automatic Modeling Model server to search for the homologous contribution of the three-dimensional structure online (35). The modeled structure was observed using SPD-Viewer version 3.7 (36).
The computational model showed that the Tyr276 residue was present at one entrance of the deep active channel and may affect the interaction between the enzyme and substrate. To characterize the role of Tyr276, we employed site-directed mutagenesis of the pCTFg4 plasmid by swapping the tyrosine residue into 19 other amino acid residues using rapid PCR amplification with high fidelity DNA polymerase of PrimeSTARTM MAX Premix (Takara) to form a series of gene mutation products. In addition, to characterize the function of the two conservative catalytic site residues Glu154 and Glu159, these residues were mutated into glycines. Corresponding primer pairs and gene mutation products are listed in Table 2. Amplified gene products were individually phosphorylated at the 5′-end, circulated, and transformed into E. coli BL21(DE3) cells. Isopropyl 1-thio-β-d-galactopyranoside was used to induce expression at a final concentration of 0.05 mmol/liter. After a 24-h incubation period at 16 °C, the host cells were collected by centrifugation, washed with ice-cold water, and subjected to complete sonication. In another round of centrifugation, individual supernatants were collected and used as the crude enzyme in agarose degradation tests. The enzymatic products were assayed using TLC and fluorescent HPLC techniques.
RESULTS
AgaG4 Sequence Information
To investigate the enzymes used in agarose degradation in Flammeovirga sp. MY04, we constructed a fosmid library of its genomic DNA. However, activity screening failed to identify any agarase-producing transformants. We then end-sequenced 118 random library clones to screen for agarase gene sequences. The agaG4 agarase gene was retrieved from the complete sequencing of the fosmid clone R012.
The 31,655-bp inserted DNA sequence (GenBankTM number EU414985) of the R012 clone encodes 18 ORFs, including the full-length agaG4 gene, which is adjacent to several genes predicted to encode the degradation enzymes for cellulose and mannan (supplemental Table S1 and Fig. S1). The agaG4 gene is 1512 bp and has a low GC content (40%). A BLASTp search showed that AgaG4, a 503-amino acid protein, shared the highest sequence identity (98%) to the GH16 β-agarase AgaYT from Flammeovirga yaeyamensis YT (GenBankTM number JF261095) (37), followed by the putative GH16 β-agarase MS116 from Microscilla sp. PRE1 (GenBankTM number AF339846) (63%) (38) and two GH16 β-agarases AgaD (GenBankTM number CBM41186) (44%) (16, 39) and AgaB (GenBankTM number AAF21821) (37%) (10) from Zobellia galactanivorans. Phylogenetic analysis (Fig. 2A) of β-agarases indicated that AgaG4, AgaYT, MS116, and AgaD were clustered into the same familial branch. CAZy and SMART analyses (Fig. 1A) indicated that AgaG4 contained an N-terminal signal peptide, a GH16 module, a Ser/Gly-rich linker, and a por secretion system sorting domain. Previous studies of the structures of AgaA and AgaB, two β-agarases from Z. galactanivorans Dsij, have revealed that their GH16 modules contain eight sugar binding sites and two essential catalytic site residues, which form the catalytic site (10, 18, 40). Similarly, the amino acid sequence of AgaG4 also contains these conservative functional sites residues, with the catalytic motif involving three residues (Glu154, Asp156, and Glu159) in addition to eight sugar-binding residues (Trp74, Trp145, Trp166, His172, Phe178, Phe183, Trp194, and Trp332). Online analyses using Signal P3.0 and Liop P1.0 showed that the signal peptide of AgaG4 was composed of 20 amino acids at the N terminus and belonged to the class of type I signal peptides, whereas the presence of the por secretion system sorting domain at the C terminus suggested that the protein was associated with the cell surface. Multiple sequence alignments (Fig. 2) revealed that, compared with the previously studied GH16 β-agarases, the GH16 module (Gln21–Lys363) of AgaG4 contained an extra function-unknown fragment (Asn246–Phe302), which, partially conservative, is also present in AgaYT (Asn246–Gly302) from F. yaeyamensis YT, MS116 (Lys262–Ser315) from Microscilla sp. PRE1, and AgaD (Asp262–Asn313) from Z. galactanivorans.
FIGURE 2.

Phylogenetic analysis (A) and sequence alignment (B) of AgaG4 based on the protein sequences. The phylogenetic analysis was performed using the neighbor-joining method in MEGA version 5.1. The numbers on branches indicate the bootstrap confidence values from 1000 replicates. The bar is equal to the distance corresponding to one amino acid substitution per 10 amino acids. The multiple-sequence alignment of the amino acid sequences of AgaG4 with other GH16 β-agarases shows the presence of the extra fragments in a red frame and the position of Tyr276 with a black arrow.
Heterologous Expression of agaG4 in E. coli
The full-length sequence of the agaG4 gene was amplified directly from the genomic DNA of Flammeovirga sp. MY04 and cloned into the pBAD/gIII A vector, following a PBAD promoter. In this AgaG4 expression vector, a gIII secretion peptide was added at the N terminus, and a His6 tag was added at the C terminus. SDS-PAGE analysis indicated that the rAgaG4 protein had the correct molecular mass (i.e. 55 kDa). However, the recombinant proteins formed inclusion bodies and rarely formed soluble products. Expression of the agaG4 gene in E. coli BL21(DE3) cells using pET-22b(+), pET-28a(+), or pET-30a(+) vectors was attempted, but all attempts failed to produce soluble protein. Although the pCold TF plasmid produced soluble products in E. coli BL21(DE3) cells, the yield was less than 1% of the total protein (data not shown). The inclusion bodies, which contained rAgaG4, were extracted from cultures of pBAgaG4-containing E. coli TOP10 cells using sonication and centrifugation, followed by step-washing with buffer A supplemented with 1.0% (w/v) Triton X-100 and 2 m urea, respectively. The rAgaG4 protein in the inclusion bodies was denatured and dissolved using urea, purified by Ni-NTA affinity chromatography, and refolded via dialysis. SDS-PAGE analysis (supplemental Fig. S2) showed that the denatured rAgaG4 protein was eluted from the Ni-NTA column by gradient concentrations of imidazole from 50 to 250 mmol/liter, yielding products with >97% purity and ∼70% recovery. The purified rAgaG4 was refolded by dialyzing against TGE buffer at an initial concentration of 150 μg/ml.
Enzymatic Characteristics of rAgaG4 and Its Degradation of Agarose
The refolded and purified rAgaG4 was able to hydrolyze agar and agarose but not alginate, carrageenan, ι-carrageenan, chitin, chitosan, or xylan. The optimal temperature for the degradation of agarose was 50 °C. The activity of rAgaG4 was stable at lower temperatures and retained more than 90% of its activity after incubation at various temperatures ranging up to 50 °C. The optimal pH was 7.5. The agarase activity of rAgaG4 was stable at pH 6–9 and retained more than 80% of its activity after 1 h of incubation. The agarase activity of rAgaG4 was completely inhibited by Ag+, Hg2+, and Pb2+ and slightly inhibited by Co2+, Fe3+, Ni2+, Zn2+, EDTA, SDS, and cetyltrimethylammonium bromide. In contrast, Na+, Li+, Ca2+, and Mn2+ slightly increased the activity. Notably, 10 mmol/liter DTT increased the enzymatic activity up to 839%, suggesting that breaking disulfide linkages may increase the enzymatic activity of rAgaG4. Under the optimal conditions of 50 °C and pH 7.5, the specific activity of the rAgaG4 agarase was 144 units/mg.
When incubated with agarose as a substrate, rAgaG4 initially produced high molecular mass oligosaccharides, which were progressively converted into smaller oligomers, a typical digestion pattern of random endolytic agarases (Fig. 3A). The final products contained two oligosaccharides, which had Rf values identical to NA4 and NA6, respectively. The two final oligosaccharide products were purified using a Superdex peptide 10/300 GL column and identified by MALDI-TOF MS, which provided molecular masses (630 and 936 Da) that were consistent with agarose-derived tetra- and hexasaccharides, respectively. Additionally, these two oligosaccharides showed resonances at ∼97 and 93 ppm in the 13C NMR spectrum (supplemental Fig. S3), which are characteristic of the β- or α-anomeric forms of galactose residues at the reducing ends of neoagaro-oligosaccharides (41).
FIGURE 3.

TLC detection of the agarose degradation products by recombinant agarases rAgaG4 (A) and rAgaG4-T47 (B). M, standard markers of neoagaro-oligosaccharides ranging from NA2 to NA8.
The dinitrosalicylic acid reducing sugar assay revealed that rAgaG4 produced ∼0.48–0.52 mmol/liter reducing oligosaccharides as final products when 0.05% (w/v) agarose was used as a substrate. To identify their composition, the final products of agarose degradation by rAgaG4 were labeled using superfluous 2-AB and analyzed using a fluorescent HPLC system equipped with a BioSep Sec 2000 column. Fluorescently labeled NA4 and NA6 accounted for more than 99% of the total agarose degradation products with a molar ratio of ∼1.5:1 (Fig. 4A). When the concentration of agarose was increased to 0.25% (w/v) or when the enzyme rAgaG4 was brought to a final concentration of 0.10 unit/ml, the products and their ratio were similar. The molar ratio of NA4 to NA6 indicated that individual oligosaccharides accounted for half the weight of the final products. These results indicate that AgaG4 is an endo-β-agarase with NA4 and NA6 as the final agarose degradation products.
FIGURE 4.
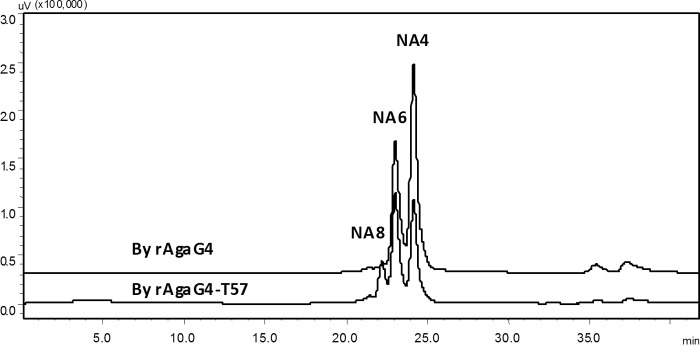
Florescent HPLC analyses of the 2-AB-labeled agarose degradation products by recombinant agarases of rAgaG4 (A) and rAgaG4-T57 (B).
Properties of rAgaG4 in the Degradation of Neoagaro-oligosaccharides
We next prepared agarose oligomers from agarose that was partially digested by rAgaG4 using a Superdex peptide 10/300 GL column, which yielded NA4, NA6, NA8, and NA10 oligomers (for the corresponding determinations, see supplemental Fig. S4), for the investigation of the oligosaccharide degradation properties. After the enzymatic reactions, the final products were labeled using superfluous 2-AB and analyzed with HPLC. The results showed that rAgaG4 did not digest NA4 or NA6 but divided NA8 into two NA4 molecules (Fig. 5, a-A) and NA10 into NA4 and NA6 with equal molar proportions (Fig. 5, a-B). To determine the degradation pattern, the purified neoagaro-oligosaccharides were individually labeled using superfluous 2-AB and then enzymatically digested. Fluorescent HPLC analysis indicated that rAgaG4 did not degrade 2-AB-labeled NA4 or NA6 but digested 2-AB-labeled NA8 into equal amounts of unlabeled NA4 and fluorescently labeled NA4 (Fig. 5, b-A). When 2-AB-labeled NA10 was used as the substrate, degradation by rAgaG4 produced unlabeled NA4 and fluorescently labeled NA6 in equimolar concentrations (Fig. 5, b-B). Thus, NA8 is the smallest substrate of rAgaG4, and the smallest final product is NA4, which was cleaved from the non-reducing ends of the agarose oligomers or polymers.
FIGURE 5.

Fluorescent HPLC analyses of oligosaccharide degradation products by rAgaG4 and rAgaG4-T57. a, NA8 (A) and NA10 (B) as the substrates; b, 2-AB-labeled NA8 (A) and NA10 (B) as the substrates.
Deletion of the Extra Fragment from AgaG4 and Characteristics of the Mutant Protein
To investigate the possible functions of the extra peptide in agarose degradation, we created a mutant gene by deleting the extra fragment (Asn246–Phe302) from the GH module of AgaG4 (Fig. 1). The mutated gene was heterologously expressed in E. coli, refolded, and purified using procedures similar to those described for rAgaG4. The expressed mutant protein, which contained a His6 tag at its C terminus, was named rAgaG4-T57. Similar to the degradation patterns of rAgaG4, rAgaG4-T57 only degraded agarose rather than other polysaccharides. The optimal pH value for the enzymatic activity remained at 7.5, but the optimal temperature for rAgaG4-T57 shifted to 40 °C. Moreover, rAgaG4-T57 lost its agarase activity at temperatures above 60 °C. The presence of 10 mmol/liter DTT increased the enzymatic activity of rAgaG4-T57 up to 863%. Interestingly, the maximum specific degradation activity of rAgaG4-T57 on agarose was 77.5 ± 0.3 units/mg at 40 °C, which was ∼7-fold higher than that of rAgaG4 (11.2 ± 0.2 units/mg).
Also similar to rAgaG4, no NA2 was produced in the degradation of agarose by rAgaG4-T57 (Fig. 3B). Notably, there were three final products from the degradation of agarose by rAgaG4-T57: NA4, NA6, and NA8, with a molar ratio of 2.7:2.8:1. To confirm these results, we performed the catalytic reaction under various conditions, including doubling the amount of enzyme and changing the temperature to 40 or 50 °C, but the products and composition ratios were identical. HPLC analyses (Fig. 4B) indicated that, similar to rAgaG4, the rAgaG4-T57 protein could not degrade NA4 or NA6 but degraded NA10 into NA6 and NA4, the latter of which was produced from the non-reducing end (Fig. 5, b-B). However, the mutant protein was unable to degrade NA8, which could be degraded into two molecules of NA4 by rAgaG4.
Function of the Tyr276 Residue in the Extra Fragment of AgaG4 in Agarose Degradation
Allouch et al. (10) reported the structures of AgaA and AgaB in Z. galactanivorans Dsij and determined that the Glu147 and Glu152 sites in AgaA and the Glu184 and Glu189 sites in AgaB were responsible for the cleavage of glycosidic bonds. Multiple alignments showed similar conserved sites (Glu154 and Glu159) in AgaG4. We created site-directed mutations at Glu154 and Glu159, changing the amino acids at these two sites to glycine. The mutant was expressed using the pCold TF expression system, and the soluble recombinant mutant rTFAgaG4-EEMGG was assayed for its degradation ability. Although rTFAgaG4 degraded agarose into NA4 and NA6, the mutant rTFAgaG4-EEMGG was no longer able to degrade agarose, detected using TLC and fluorescent HPLC techniques (Fig. 6, A and B). The results suggest that AgaG4 share a similar agarose degradation mechanism with AgaA and AgaB.
FIGURE 6.

Final degradation products of agarose by crude enzymes of rTFAgaG4 and its mutant proteins, using the TLC (A) and fluorescent HPLC (B) techniques. One-letter amino acid codes are shown.
To screen the essential sites for the functions of the extra fragment, we analyzed the computational structure of AgaG4 by homology modeling using the online SWISS-MODEL program, with the crystal structure of the agarase AgaD (Protein Data Bank entry 4ASM chain B) from Z. galactanivorans Dsij as a model. The extra fragment extended the substrate binding cleft of AgaG4 and resulted in a deeper and steeper active cleft, which is similar to that of the partially conservative fragment in AgaD (39). The Tyr276 residue was present at one entrance of the deep active channel, which suggested that the residue may limit the interaction between the enzyme and substrate. Accordingly, we created a series of mutations at the Tyr276 site by swapping the tyrosine residue with 19 different amino acid residues using site-directed mutagenesis and heterologously expressed the mutants in E. coli BL21(DE3) cells using the pCold TF expression system. The mutated proteins were partially purified for enzymatic assays.
When the Tyr276 residue was changed to phenylalanine (Y276F) or methionine (Y276M), the agarose degradation products of the mutants and the molar ratios were similar to those produced by rAgaG4 (i.e. NA4 and NA6 as the final products; Fig. 4) and rTFAgaG4 (Fig. 6, A and B). However, the mutants containing a residue swapped with 13 other amino acid residues produced NA4, NA6, and NA8 as the final degradation products (Fig. 6, A and B), which was similar to the result with the truncated rAgaG4-T57 (Fig. 4). It is noted that when the Tyr276 residue was changed to glycine (Y276G) or proline (Y276P), the mutated proteins markedly lost their ability to degrade agarose (TLC analysis present in Fig. 6A). Further fluorescent HPLC detection determined that the oligosaccharide productions from the agarose degradation by these two mutants were both decreased to less than 1% of the original production by rTFAgaG4 (Fig. 6B). Swapping the tyrosine to lysine (Y276K) or arginine (Y276R) also led to a significant decrease of the agarose degradation ability (less than 25% of the original, calculated from the fluorescent HPLC results). These four mutants produced NA4, NA6, NA8, NA10, and higher oligomers (Fig. 6B), seemingly a mixture of incomplete degradation products. The above results suggest that the Tyr276 residue has a crucial function in the agarolytic activity of AgaG4, especially for the digestion of neoagarooctaose. The benzene ring of tyrosine and phenylalanine or methyl sulfide of methionine at this site is decisive for the degradation of neoagarooctaose.
DISCUSSION
The GH module of agarase is well known to be responsible for its agarolytic activities, producing a series of oligosaccharides with repeating disaccharide units (6, 7). Recent site-directed mutagenesis studies of two GH16 β-agarases, AgaA and AgaB from Z. galactanivorans, have demonstrated that the residues adjacent to some of the active site residues have significant effects on the properties of the enzyme, such as the enzymatic activity, optimal catalytic temperature, and thermostability (20, 21). DNA shuffling of AgaB, a GH118 β-agarase of Pseudoalteromonas sp. CY24, also indicated that hydrophobic interactions are essential for increasing its thermostability (19). The studies present in this paper provide further insights into the influences of sequence variations in the GH module on the enzymatic characteristics of the enzyme.
Among the studied β-agarases, AgaYT of F. yaeyamensis strain YT (37), AgaD of Z. galactanivorans DSij (39), the predicted protein MS116 of Microscilla sp. PRE1 (38), and AgaG4 of Flammeovirga sp. MY04 are clustered into their own branch of the phylogenetic tree of β-agarases, mainly due to the presence of a particularly conservative extra peptide in the GH module of these proteins. The final agarose degradation products of rAgaG4 were chemically characterized as NA4 and NA6 but no NA2, with a molar ratio of ∼1.5:1. Similar to AgaG4, AgaD also produced NA4 and NA6 as the final products of agarose degradation (39). Deleting the extra fragment from the GH module of AgaG4 (rAgaG4-T57) not only changed its enzymatic characteristics, such as enzymatic activity, optimal catalytic temperature, and the range of the optimal catalytic temperature, but also the composition of the final agarose degradation products. The truncated protein was unable to degrade NA8 but retained the ability to degrade NA10. The results showed that the extra fragment within the GH16 module of AgaG4 plays multiple roles in the degradation of agarose, particularly in the digestion of the intermediate product, NA8.
Hehemann et al. (39) found that the AgaD extra peptide formed a loop structure. The extra peptide loop extended, together with two other loop insertions within the GH16 module, into the substrate-binding cleft, forming a larger size than those of AgaA and AgaB, which do not harbor the extra peptide. The Tyr296 residue within the peptide, along with the aromatic sugar-binding residues in the catalytic groove of AgaD, constructed a hydrophobic binding site platform, and it was considered the key site essential for binding to oligosaccharides at their reducing ends (39). The tyrosine residue is conserved in AgaD (Tyr296), AgaYT (Tyr276), MS116 (Tyr290), and AgaG4 (Tyr276). The computational model also showed that Tyr276 was present at one entrance of the deep active channel of AgaG4 and may affect the interaction between the enzyme and substrate. We swapped the residue with 19 different amino acid residues. The results showed that the Tyr276 residue within the extra fragment is essential for the degradation of neoagarooctaose. When this site residue harbored a benzene ring (the original tyrosine or swapped phenylalanine) or a methyl sulfide-based structure (methionine), the enzyme retained its rAgaG4 activity (i.e. completely degrading agarose into NA4 and NA6). If the Tyr276 residue was mutated to other amino acids, the mutant proteins retained their ability to produce NA4 and NA6 in agarose degradation but with NA8 accumulation at different degrees. Previous studies reported that the sugar binding sites involved in the hydrophobic stacking interactions with glycoside rings usually contain aromatic structures at their residual side chains, such as the benzene rings of tyrosine and phenylalanine, the benzazole ring of tryptophan, and the imidazole ring of histidine (10). Our swapping experiments indicated that both the benzene ring and the methyl sulfide at position 276 were decisive for recognizing and binding to neoagarooctaose.
In conclusion, AgaG4 is a GH16 β-agarase that degrades agarose into NA4 and NA6 as the final products. The GH module of AgaG4 contains a 57-amino acid extra peptide that is not only essential for its ability to degrade NA8 but also affects the enzymatic characteristics of the enzyme. The Tyr276 site residue within this particular fragment is decisive for binding and degrading NA8. A benzene ring of tyrosine or phenylalanine or a methyl sulfide of methionine at the 276 site is suggested to be essential for degrading agarose completely into NA4 and NA6. Therefore, modification of active site residues of an agarase may markedly change its degradation pattern.
This work was financially supported by National Natural Science Foundation of China Grants 30825001, 31130004, and 30870001; Independent Innovation Foundation of Shandong University Grant 2012GN014; and State Key Laboratory of Microbial Technology Grant M2010–12.

This article contains supplemental Table S1 and Figs. S1–S4.
- GH
- glycoside hydrolase
- NAO
- neoagaro-oligosaccharide
- rAgaG4
- recombinant AgaG4
- Ni-NTA
- nickel-nitrilotriacetic acid
- TLC
- thin layer chromatography
- 2-AB
- 2-aminobenzamide.
REFERENCES
- 1. Rees D. A. (1969) Structure, conformation, and mechanism in the formation of polysaccharide gels and networks. Adv. Carbohydr. Chem. Biochem. 24, 267–332 [DOI] [PubMed] [Google Scholar]
- 2. Rochas C., Potin P., Kloareg B. (1994) NMR spectroscopic investigation of agarose oligomers produced by an α-agarase. Carbohydr. Res. 253, 69–77 [DOI] [PubMed] [Google Scholar]
- 3. Morrice L. M., McLean M. W., Long W. F., Williamson F. B. (1983) β-Agarases I and II from Pseudomonas atlantica. Substrate and specificities. Eur. J. Biochem. 137, 149–154 [DOI] [PubMed] [Google Scholar]
- 4. Morrice L. M., McLean M. W., Williamson F. B., Long W. F. (1983) β-Agarases I and II from Pseudomonas atlantica. Purifications and some properties. Eur. J. Biochem. 135, 553–558 [DOI] [PubMed] [Google Scholar]
- 5. Michel G., Nyval-Collen P., Barbeyron T., Czjzek M., Helbert W. (2006) Bioconversion of red seaweed galactans. A focus on bacterial agarases and carrageenases. Appl. Microbiol. Biotechnol. 71, 23–33 [DOI] [PubMed] [Google Scholar]
- 6. Fu X. T., Kim S. M. (2010) Agarases. Review of major sources, categories, purification method, enzyme characteristics and applications. Mar. Drugs. 8, 200–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chi W. J., Chang Y. K., Hong S. K. (2012) Agar degradation by microorganisms and agar-degrading enzymes. Appl. Microbiol. Biotechnol. 94, 917–930 [DOI] [PubMed] [Google Scholar]
- 8. Potin P., Richard C., Rochas C., Kloareg B. (1993) Purification and characterization of the α-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur. J. Biochem. 214, 599–607 [DOI] [PubMed] [Google Scholar]
- 9. Ohta Y., Hatada Y., Miyazaki M., Nogi Y., Ito S., Horikoshi K. (2005) Purification and characterization of a novel α-agarase from a Thalassomonas sp. Curr. Microbiol. 50, 212–216 [DOI] [PubMed] [Google Scholar]
- 10. Allouch J., Jam M., Helbert W., Barbeyron T., Kloareg B., Henrissat B., Czjzek M. (2003) The three-dimensional structures of two β-agarases. J. Biol. Chem. 278, 47171–47180 [DOI] [PubMed] [Google Scholar]
- 11. Michel G., Barbeyron T., Kloareg B., Czjzek M. (2009) The family 6 carbohydrate-binding modules have coevolved with their appended catalytic modules toward similar substrate specifity. Glycobiology. 19, 615–623 [DOI] [PubMed] [Google Scholar]
- 12. Henrissat B., Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 [DOI] [PubMed] [Google Scholar]
- 13. Flament D., Barbeyron T., Jam M., Potin P., Czjzek M., Kloareg B., Michel G. (2007) α-agarases define a new family of glycoside hydrolases distinct from β-agarase families. Appl. Environ. Microbiol. 73, 4691–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma C., Lu X., Shi C., Li J., Gu Y., Ma Y., Chu Y., Han F., Gong Q., Yu W. (2007) Molecular cloning and characterization of a novel β-agarase, AgaB, from marine Pseudoalteromonas sp. CY24. J. Biol. Chem. 282, 3747–3754 [DOI] [PubMed] [Google Scholar]
- 15. Dong J., Hashikawa S., Konishi T., Tamaru Y., Araki T. (2006) Cloning of the novel gene encoding β-agarase C from a marine bacterium, Vibrio sp. strain PO-303, and characterization of the gene product. Appl. Environ. Microbiol. 72, 6399–6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hehemann J. H., Michel G., Barbeyron T., Czjzek M. (2010) Expression, purification and preliminary x-ray diffraction analysis of the catalytic module of a β-agarase from the flavobacterium Zobellia galactanivorans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ren A., Xia Z. X., Yu W., Zhou J. (2010) Expression, crystallization and preliminary x-ray analysis of an anomeric inverting agarase from Pseudoalteromonas sp. CY24. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1635–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jam M., Flament D., Allouch J., Potin P., Thion L., Kloareg B., Czjzek M., Helbert W., Michel G., Barbeyron T. (2005) The endo-β-agarase AgaA and AgaB from the marine bacterium Zobellia galactanivorans. Two paralogue enzymes with different molecular organizations and catalytic behaviours. Biochem. J. 385, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi C., Lu X., Ma C., Ma Y., Fu X., Yu W. (2008) Enhancing the thermostability of a novel β-agarase AgaB through directed evolution. Appl. Biochem. Biotechnol. 151, 51–59 [DOI] [PubMed] [Google Scholar]
- 20. Jang M. K., Lee S. W., Lee D. G., Kim N. Y., Yu K. H., Jang H. J., Kim S., Kim A., Lee S. H. (2010) Enhancement of the thermostability of a recombinant β-agarase, AgaB, from Zobellia galactanivorans by random mutagenesis. Biotechnol. Lett. 32, 943–949 [DOI] [PubMed] [Google Scholar]
- 21. Lee S., Lee D. G., Jang M. K., Jeon M. J., Jang H. J., Lee S. H. (2011) Improvement in the catalytic activity of β-agarase AgaA from Zobellia galactanivorans by site-directed mutagenesis. J. Microbiol. Biotechnol. 21, 1116–1122 [DOI] [PubMed] [Google Scholar]
- 22. Nakagawa Y., Hamana K., Sakane T., Yamasato K. (1997) Reclassification of Cytophaga aprica (Lewin 1969) Reichenbach 1989 in Flammeovirga gen. nov. as Flammeovirga aprica comb. nov., and of Cytophaga diffluens (ex Stanier 1940; emend. Lewin 1969) Reichenbach 1989 in Persicobacter gen. nov. as Persicobacter diffluens comb. nov. Int. J. Syst. Evol. Microbiol. 47, 220–223 [Google Scholar]
- 23. Takahashi M., Suzuki K., Nakagawa Y. (2006) Emendation of the genus Flammeovirga and Flammeovirga aprica with the proposal of Flammeovirga arenaria nom. rev., comb. nov., and Flammeovirga yaeyamensis sp. nov. Int. J. Syst. Evol. Microbiol. 56, 2095–2100 [DOI] [PubMed] [Google Scholar]
- 24. Hosoya S., Yokota A. (2007) Flammeovirga kamogawensis sp. nov., isolated from coastal seawater in Japan. Int. J. Syst. Evol. Microbiol. 57, 1327–1330 [DOI] [PubMed] [Google Scholar]
- 25. Xu H., Fu Y., Yang N., Ding Z., Lai Q., Zeng R. (2012) Flammeovirga pacifica sp. nov., isolated from deep-sea sediment. Int. J. Syst. Evol. Microbiol. 62, 937–941 [DOI] [PubMed] [Google Scholar]
- 26. Han W. J., Gu J. Y., Yan Q. J., Li J. G., Wu Z. H., Gu Q. Q., Li Y. Z. (2012) A polysaccharide-degrading marine bacterium Flammeovirga sp. MY04 and its extracellular agarase system. J. Ocean Univ. China 11, 375–382 [Google Scholar]
- 27. Sambrook J., Russel D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed, pp. A8.40-A8.47, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 28. Birnboim H. C., Doly J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki H., Sawai Y., Suzuki T., Kawai K. (2002) Purification and characterization of an extracellular β-agarase from Bacillus sp. MK03. J. Biosci. Bioeng. 93, 456–463 [DOI] [PubMed] [Google Scholar]
- 30. Hall T. A. (1999) BioEdit. A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 [Google Scholar]
- 31. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5. Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller G. L. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 [Google Scholar]
- 33. Wang S. Y., Hu W., Lin X. Y., Wu Z. H., Li Y. Z. (2012) A novel cold-active xylanase from the cellulolytic myxobacterium Sorangium cellulosum So9733-1. Gene cloning, expression, and enzymatic characterization. Appl. Microbiol. Biotechnol. 93, 1503–1512 [DOI] [PubMed] [Google Scholar]
- 34. Bigge J. C., Patel T. P., Bruce J. A., Goulding P. N., Charles S. M., Parekh R. B. (1995) Nonselective and efficient fluorescent labelling of glycans using 2-amino benzamide and anthranilic acid. Anal. Biochem. 230, 229–238 [DOI] [PubMed] [Google Scholar]
- 35. Nielsen M., Lundegaard C., Lund O., Petersen T. N. (2010) CPHmodels-3.0. Remote homology modelling using structure guided sequence profiles. Nucleic Acids Res. 38, W576–W581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer. An environment for comparative protein modelling. Electrophoresis. 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 37. Yang J. I., Chen L. C., Shih Y. Y., Hsieh C., Chen C. Y., Chen W. M., Chen C. C. (2011) Cloning and characterization of β-agarase AgaYT from Flammeovirga yaeyamensis strain YT. J. Biosci. Bioeng. 112, 225–232 [DOI] [PubMed] [Google Scholar]
- 38. Zhong Z., Toukdarian A., Helinski D., Knauf V., Sykes S., Wilkinson J. E., O'Bryne C., Shea T., DeLoughery C., Caspi R. (2001) Sequence analysis of a 101-kilobase plasmid required for agar degradation by a Microscilla isolate. Appl. Environ. Microbiol. 67, 5771–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hehemann J. H., Correc G., Thomas F., Bernard T., Barbeyron T., Jam M., Helbert W., Michel G., Czjzek M. (2012) Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J. Biol. Chem. 287, 30571–30584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hehemann J. H., Correc G., Barbeyron T., Helbert W., Czjzek M., Michel G. (2010) Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912 [DOI] [PubMed] [Google Scholar]
- 41. Rochas C., Lahaye M., Yaphe W., Phan Viet M. T. (1986) 13C-NMR spectroscopic investigation of agarose oligomers. Carbohydr. Res. 148, 199–207 [DOI] [PubMed] [Google Scholar]