

Background: The myosin superfamily has many classes that have evolved to carry out different functions.
Results: Mouse myosin-18A binds actin weakly in an ATP-independent manner and has very low enzymatic activity.
Conclusion: Not all myosins exhibit motor activity.
Significance: This work demonstrates that myosins may have functions unrelated to their ability to hydrolyze ATP.
Keywords: Actin, ATPases, Cell Motility, Enzyme Kinetics, Myosin, Actin-binding Protein, Optical Tweezers
Abstract
The Mus musculus myosin-18A gene is expressed as two alternatively spliced isoforms, α and β, with reported roles in Golgi localization, in maintenance of cytoskeleton, and as receptors for immunological surfactant proteins. Both myosin-18A isoforms feature a myosin motor domain, a single predicted IQ motif, and a long coiled-coil reminiscent of myosin-2. The myosin-18Aα isoform, additionally, has an N-terminal PDZ domain. Recombinant heavy meromyosin- and subfragment-1 (S1)-like constructs for both myosin-18Aα and -18β species were purified from the baculovirus/Sf9 cell expression system. These constructs bound both essential and regulatory light chains, indicating an additional noncanonical light chain binding site in the neck. Myosin-18Aα-S1 and -18Aβ-S1 molecules bound actin weakly with Kd values of 4.9 and 54 μm, respectively. The actin binding data could be modeled by assuming an equilibrium between two myosin conformations, a competent and an incompetent form to bind actin. Actin binding was unchanged by presence of nucleotide. Both myosin-18A isoforms bound N-methylanthraniloyl-nucleotides, but the rate of ATP hydrolysis was very slow (<0.002 s−1) and not significantly enhanced by actin. Phosphorylation of the regulatory light chain had no effect on ATP hydrolysis, and neither did the addition of tropomyosin or of GOLPH3, a myosin-18A binding partner. Electron microscopy of myosin-18A-S1 showed that the lever is strongly angled with respect to the long axis of the motor domain, suggesting a pre-power stroke conformation regardless of the presence of ATP. These data lead us to conclude that myosin-18A does not operate as a traditional molecular motor in cells.
Introduction
The myosin superfamily has been ordered into 35 classes based on amino acid sequence homology of motor domains (1). Generally, myosins have a motor domain that catalyzes ATP hydrolysis and binds F-actin. Within the superfamily, the enzymatic and mechanical properties of each class of motors vary widely, depending on the role each specific motor plays within the cell (2).
The founding member of class-18 of the myosin superfamily was MysPDZ, later named myosin-18A, which contains a PDZ domain upstream of the motor (3). PDZ domains are common components of scaffolding proteins, signaling cascades, and molecular complexes within the cell (4). The gene products of the mouse myosin-18A gene (Myo18A) encode two reported splice variants; myosin-18Aα contains a lysine- and glutamic acid-rich (KE-rich) sequence at the extreme N terminus, followed by a short amino acid sequence before the PDZ domain (3, 5). Myosin-18Aβ lacks the KE-rich sequence and the PDZ domain and, instead, has a short leading sequence upstream of the motor (Fig. 1) (5). Both isoforms have a predicted single canonical IQ motif and a much less conserved one followed by a long sequence predicted to form a 100-nm coiled-coil tail, analogous to the 155-nm tail of myosin-2. A second mouse class-18 gene, termed Myo18B, lacks the N-terminal extension characteristic of myosin-18A but instead has a long N-terminal extension with unknown function or domain structure (6, 7).
FIGURE 1.

Domain organization of M. musculus myosin-18A. The domain structure of myosin-18A isoforms was analyzed using SMART (Simple Modular Architecture Research Tool) at EMBL (myosin-18Aα, GenBankTM accession number AB026497; myosin-18Aβ RIKEN clone, GenBankTM accession number AK171342), and the schematic diagrams of the two isoforms were created (top two structures). Using this information, HMM-like constructs with coiled-coil regions truncated at Leu1463 for myosin-18Aα and Leu1128 for myosin-18Aβ were cloned for baculoviral expression. To create single-headed S1-like constructs, the α isoform was truncated at Leu1242, and the 18Aβ isoform was truncated at Leu907. C-terminal FLAG epitopes were added to each construct as an affinity purification aid. Note that these schematics are not drawn to scale.
Class-18 myosins have been implicated in many physiological events in mammalian cells and tissues, including stromal cell differentiation (3) and tumor suppression (7–9). In cells, myosin-18A isoforms have been reported to be involved in maintenance of trans-Golgi structure (10), in organization of the actin cytoskeleton in the lamellipodia and membrane ruffles (11), and as receptors for surfactant proteins in the immunological response (12). Biochemical analysis of murine myosin-18Aα suggested an ATP-insensitive actin binding site in the KE-rich region or the region between the KE-rich and PDZ domains, but myosin-18Aβ did not bind to actin even in the absence of ATP in vitro and did not strongly co-localize with actin in vivo (13, 14). A previous report analyzing the biochemical properties of Drosophila melanogaster myosin-18 isoforms also reported ATP-insensitive actin binding and suggested that myosin-18 may act as a dynamic actin tether (15).
In this study, we present a biochemical analysis of recombinant Mus musculus myosin-18Aα and -18Aβ single-headed (subfragment-1; S1)4 and double-headed (heavy meromyosin; HMM) constructs purified from the baculovirus/Sf9 (S. frugiperda) cell system. Our data show that myosin-18A isoforms bound both regulatory (RLC) and essential light chains (ELC). Both isoforms weakly bound nucleotide and only hydrolyzed ATP very slowly. They bound weakly to actin in an ATP-insensitive manner. We speculate on the structural reasons behind these behaviors.
EXPERIMENTAL PROCEDURES
Cloning of M. musculus Myosin-18A
Full-length Myo18A heavy chain sequences were amplified in 5′- and 3′-halves of the sequence using primer pairs designed as follows: 5′ segment of 18Aα(1–4438) upstream primer (AATTACTCAGATCTATGTTTAACCTCATG), 5′ segment of 18Aβ(1–3382) upstream primer (AATTACTCAGATCTATGCTGGCCAAGACGGAA), 5′ segment of shared downstream primer (GCTCACTGTCGAACCTCCTC); 3′ segment (18Aα(2924–6155); 18Aβ(1867–5100)) of shared upstream primer (CTGCAGGACTCCCAGAAAAA), 3′ segment of shared downstream primer (TAATACTAGTGCACTGGTCTCTGTCA). Each PCR product was generated using a template vector encoding for either myosin-18Aα (GenBankTM accession number AB026497) or myosin-18Aβ (RIKEN clone, GenBankTM accession number AK171342) provided by Dr. Melanie Barzik and Dr. John Hammer III and amplified with Platinum HiFi Supermix (Invitrogen) and standard PCR techniques (PTC-200 Thermocycler, MJ Research). Each PCR product was ligated into the pCR4-TOPO vector (Invitrogen). 5′ and 3′ segments were then combined by an initial restriction double digestion of the 3′ segments with BglII and BamHI (New England Biolabs) to insert into pFastBac1 (Invitrogen) containing a C-terminal FLAG tag (DYKDDDDK) between the NotI and XbaI sites (pFastBac1-NX), followed by a double digestion of the 5′ segments with BamHI and SpeI for upstream in-frame insertion. For subsequent cloning of HMM and S1 truncations, the full-length clones were used as templates in PCRs using the aforementioned 5′ segment upstream primers for myosin-18Aα and myosin-18Aβ using the following downstream primers: HMM (18Aα(1–4438); 18Aβ(1–3382)) downstream primer (ATTATACTAGTTCACTGTCGAACCTCCTC); S1 (18Aα(1–3725); 18Aβ(1–2720)) downstream primer (ATTATACTAGTAGGGGCCGCACGGTGGTGAA). Truncations were cloned into pFastBac1-NX between the BglII and SpeI restriction sites.
Baculoviral Expression of S1 and HMM Constructs
All myosin-18A constructs were recombinantly expressed in the baculovirus/Sf9 cell system along with a baculovirus encoding both chicken RLC and bovine ELC (16). Infected cells were harvested by sedimentation after 48 h. Cell pellets were quick frozen in liquid nitrogen and were either processed immediately or stored at −80 °C for later processing. Purification of proteins using the C-terminal FLAG tag was performed as described (17). Proteins were first purified via a FLAG affinity column, followed by separation on a 1-ml Q-Sepharose slurry pre-equilibrated with 0.1 m NaCl, 10 mm MOPS (pH 7.2), 0.1 mm EGTA, 3 mm NaN3, 1 mm DTT, and 0.1 mm PMSF. Fractions were eluted with buffer containing 0.5 m NaCl, 10 mm MOPS (pH 7.2), 0.1 mm EGTA, 3 mm NaN3, 1 mm DTT, and 0.1 mm PMSF, followed by dialysis in this buffer, substituting NaCl with either 0.1 or 0.5 m KCl. Expected masses for each heavy chain construct based on amino acid sequence were 164 kDa for myosin-18Aα-HMM, 138 kDa for myosin-18Aα-S1, 127 kDa for myosin-18Aβ-HMM, and 101 kDa for myosin-18Aβ-S1. The identity of the purified proteins was confirmed by LC-MS/MS.
Preparation of Other Proteins
Skeletal muscle actin was purified from rabbit skeletal muscle (18). Skeletal muscle HMM (SkHMM) was prepared from rabbit skeletal muscle (19). A clone for rat calmodulin (generous gift of Dr. Eva Forgacs, Eastern Virginia Medical School) was expressed in Escherichia coli. A baculovirus driving the expression of FLAG-tagged rabbit smooth muscle myosin light chain kinase (MLCK) was provided by Dr. Zenon Grabarek (Boston Biomedical Research Institute). The protein was purified from Sf9 cells using FLAG affinity chromatography (20). Nonmuscle myosin-2A-S1 (a generous gift of Dr. Attila Nagy) was expressed in the baculovirus/Sf9 system with co-expressed chicken RLC and bovine ELC and purified by FLAG affinity chromatography.
Negative Staining Electron Microscopy
Samples were diluted to 50–100 nm myosin with a buffer containing 10 mm MOPS (pH 7.0), 2 mm MgCl2, 0.1 mm EGTA, and 50 mm KCl (+1 mm ATP where indicated). For actin decoration experiments, 100 nm myosin-18Aα-S1 (or 100 nm nonmuscle myosin-2A-S1 as a control) was mixed with equimolar F-actin in the absence of nucleotide in the buffer detailed above. A 5-μl drop of sample was applied to a carbon-coated copper grid (pretreated with UV light) and stained with 1% uranyl acetate. Micrographs were recorded at ×60,000 on a JEOL 1200EX II microscope. Data were recorded on an AMT XR-60 CCD camera. Catalase crystals were used as a size calibration standard. Image processing was carried out using SPIDER software as described (36). Initial data sets contained 1321 (myosin-18Aα-S1, no nucleotide), 2185 (myosin-18Aβ-S1, no nucleotide), and 1001 (myosin 18Aα-S1, presence of 1 mm ATP) particles. In each case, misaligned particles were removed following an initial round of alignment and classification. The number of particles in each final classification was 1108 (myosin 18Aα-S1, no nucleotide), 1341 (myosin-18Aβ-S1, no nucleotide), and 817 (myosin 18Aα-S1, presence of 1 mm ATP).
In Silico Modeling
A homology model of murine myosin-18Aβ motor domain (amino acids 1–864) in the pre-power stroke state was generated with the program Modeler 9v8 (21), using the crystal structure of Dictyostelium discoideum myosin-2 motor domain (2XEL) as a template. M. musculus myosin-18Aβ and D. discoideum myosin-2 motor domains share 45.2% similarity and 25.7% identity at the amino acid level.
Phosphorylation of RLC
In applications requiring analysis of myosin with phosphorylated RLC, purified myosins with bound light chains in dialysis buffer containing 0.1 m KCl were supplemented with 0.2 mm CaCl2, 0.5 μm calmodulin, and 0.9 μm MLCK. Phosphorylation of RLC on myosin-18A-HMM proteins was confirmed with a gel shift assay using Phos-tag acrylamide reagent (Wako Chemicals, Richmond, VA). The Phos-tag reagent was incorporated at 25 μm into a 15% SDS-PAGE resolving gel along with 50 μm MnCl2. Resolving gels were poured with a Bio-Rad Mini-Protean gel apparatus followed by a 4% stacking gel, and gels were run at a constant 30 mA/gel and then stained with Coomassie Blue.
Characterization of Nucleotide Interaction
Actin-activated MgATPase activity of myosin-18A-S1 was assayed with an NADH-coupled assay at 25 °C using 1 μm S1 and up to 60 μm actin (22, 23), in a solution comprising 50 mm KCl, 10 mm MOPS (pH 7.2), 2 mm MgCl2, 0.15 mm EGTA, 2 mm ATP, 40 units/ml lactate dehydrogenase, 200 units/ml pyruvate kinase, 1 mm phosphoenol pyruvate, and 200 μm NADH. Changes in A340 were monitored using a Varian Cary 50 spectrophotometer. Alterations of this basic steady-state ATPase assay included increasing the final concentrations of myosin-18A-S1 to 2.5 μm for myosin-18Aα and 4.4 μm for myosin-18Aβ, increasing salt concentration from 50 to 150 mm KCl, phosphorylating the bound RLC by adding MLCK with calmodulin and 0.4 mm CaCl2, analyzing 0.2 μm myosin-18-S1 in the presence of 20 μm F-actin and 6.7 μm smooth muscle tropomyosin over a 60-min time course, and using myosin-18A-HMM constructs at 0.2 μm in the presence of 0.4 μm GOLPH3 protein (Proteintech Group, Chicago, IL).
Binding of N-methylanthraniloyl derivatives of ATP (mantATP) and ADP (mantADP) to myosin-18A-S1 constructs was assayed using a stopped-flow apparatus (HiTech, TgK Scientific, Bradford-on-Avon, UK) by excitation at 297 nm using a 400-nm long pass filter. The conditions were 100 mm KCl, 25 mm HEPES (pH 7.0 at 25 °C), and 5 mm MgCl2, with assays performed at 20 °C using 0.25 μm postmixing myosin-18A-S1 and postmixing concentrations of N-methylanthraniloyl-nucleotide analogs ranging up to 20 μm for mantATP and to 12.5 μm for mantADP. The total fluorescence changes at high ADP concentrations were in the range of 6–8%. ADP dissociation from myosin-18A-S1 constructs was measured by premixing 5 μm mantADP + 40 μm unlabeled ADP with 0.5 μm myosin-18A-S1 and chasing with 2 mm unlabeled ATP. As with nucleotide binding experiments, all transients for ADP release were fit to single exponentials prior to compilation.
Filter binding assays used nitrocellulose membrane pre-equilibrated with buffer containing 0.25 m KCl, 10 mm MOPS (pH 7.2), 4 mm MgCl2, 0.1 mm EGTA, and 1 mm DTT. The membrane was placed under vacuum and was blotted in duplicate with 1 μm myosin-18A-S1 or SkHMM that had been preincubated with 20 μm [α-32P]ATP (1.5 × 1015 cpm/mol) in the buffer mentioned above, at 22 °C for 60 s. The membrane was washed with 2 ml of equilibration buffer under vacuum and dried. For quantification, dried membranes were exposed to Fujifilm BAS-MS phosphor imager screens for 1 h and scanned on a Fuji FLA-5000 series Image Analyzer (Fuji Medical Systems, Stamford, CT). Quantitation was done using Image Gauge software (version 3.0, Fuji Medical Systems). The amount of radionucleotide bound to the membrane in a no-protein control was subtracted from the amount bound to the motor blots, and remaining portions were normalized to the value associated with the SkHMM-positive control.
Radiometric K+-EDTA ATPase assays were performed at 25 °C as described (24), in a solution containing 0.5 m KCl, 20 mm MOPS (pH 7.0), 2 mm EDTA, and 0.5 mm [γ-32P]ATP (PerkinElmer Life Sciences).
Characterization of Actin Binding
Binding of myosin-18A-S1 to F-actin was assayed by co-sedimentation using 1 μm S1 mixed with different concentrations of phalloidin-stabilized F-actin in either the absence of ATP or the presence of 1 mm ATP or 1 mm ADP, respectively. Variations of the assay included the phosphorylation of the RLC by MLCK. Incubations were for 10 min at 22 °C in buffer containing 0.1 m KCl, 20 mm MOPS (pH 7.0), 5 mm MgCl2, 0.05 mm EGTA, 1 mm NaN3, and 1 mm DTT. Following incubation, the solution was sedimented for 10 min at 100,000 × g in a Beckman TLA-100 rotor at 4 °C. The supernatant was removed, and the pellet was resuspended in 1× SDS sample buffer to an equivalent volume. Supernatants and pellets were fractionated on a 4–20% Tris-glycine SDS-polyacrylamide gel, stained with Coomassie Blue, and analyzed by densitometry (Li-Cor Biosciences, Odyssey version 3.0, Lincoln, NE). Data points were fitted to a hyperbolic equation, correcting for the amount of myosin that pellets in the absence of actin, typically around 1% (SigmaPlot 11.0, Systat Software, Inc., San Jose, CA). For repeat co-sedimentation assays, the supernatant from the first sedimentation at 40 μm actin was brought to the same actin concentration again, incubated as before, and then resedimented under the same assay conditions. For myosin-18Aβ-S1, the same co-sedimentation procedure was repeated sequentially a total of four times.
Simulation of Actin Binding Data
Because the reaction of myosin-18Aα and myosin-18β and actin does not behave as a simple one-step equilibrium binding reaction in the co-sedimentation studies, we had to use a two-step reaction mechanism (Scheme 1) to accurately describe the binding of actin. In this scheme, it is assumed that there is a competent form of myosin (M*) that can bind to actin (A) and an isomer of myosin-18A (M), which was incompetent to bind actin. In the absence of actin, there is an equilibrium between M and M*, but with the addition of actin, this equilibrium is shifted to the right.
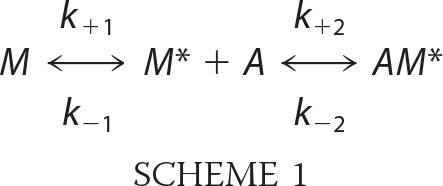 |
Differential equations describing the reactions for each reactant and product were written for each over small time increments (0.001 s) describing this reaction and were solved numerically over the experimental time frame using Euler's theorem for AM*/MT (where T is the total concentration of myosin-18A (M + M* + AM) using a program written in LabVIEW 2010 (National Instruments, Austin, TX). The differential equations for Scheme 1 and the LabVIEW program used to solve them with respect to time are available on request. The rate constants for the forward (positive subscripts) and backward rate constants (negative subscripts) were selected so that the computed fraction of myosin-18Aα-S1 or myosin-18Aβ-S1 bound to actin corresponded to the observed values obtained in the co-sedimentation experiments. To solve the differential equations with respect to time, it was assumed that prior to the 10-min incubation and centrifugation of S1 with actin, the M and M* forms were at equilibrium. The duration that the actin reacted with the S1 was taken as the sum of the 10-min incubation period and the first 5 min in the 10-min centrifugation (assuming that by 5 min into the centrifugation, the actin had pelleted). The duration of the incubation and time for separation of the bound and unbound myosin-18A fragments is insufficient for Scheme 1 to come to equilibrium (with respect to the rates of the first and second reactions in Scheme 1) in the time frame of the experiments. However, if the reactants are allowed to equilibriate for more than 2 h, the system does come to an equilibrium in which the concentration of the bound myosin-18A is given by Equation 1 (see supplemental material for the derivation of Equation 1),
where Ki = k+i/k−i.
Actin Gliding Assays
Motility assays were performed in buffer containing 50 mm KCl, 20 mm MOPS (pH 7.4), 5 mm MgCl2, 0.1 mm EGTA, 1 mm ATP, 25 μg/ml glucose oxidase, 45 μg/ml catalase, 2.5 mg/ml glucose, and 50 mm DTT (25). All experiments were performed at 30 °C. SkHMM and myosin-18A-HMM fragments were mixed at various molar ratios, keeping the total myosin concentration added to the flow cell at 0.2 mg/ml. Visualization of filaments and quantification of motility were performed as described (26).
Optical Trapping
Three bead assays (27) were performed using a similar dual-beam optical trapping apparatus as reported previously (28). An assay chamber (volume ∼30–40 μl), was constructed using two coverslips, one of which was sparsely decorated with 2.1-μm diameter glass microspheres (Bangs Laboratories, Fishers, IN), suspended in 0.1% (v/v) nitrocellulose, and assembled using Scotch double-sided adhesive tape. These glass microspheres were used as “pedestals” for the myosin-18A in the three-bead assay. Experiments were performed using the same motility buffer as used in the actin gliding assay.
Myosin-18A-HMM constructs were diluted to a concentration of ∼10–30 pm and allowed to bind nonspecifically inside the chamber. Approximately four chamber volumes of motility buffer with 1 mg/ml bovine serum albumin (BSA) were used to wash the chamber to reduce nonspecific binding of polystyrene beads and actin filaments. Actin/polystyrene bead “dumbells” were made using 0.2 nm rhodamine-phalloidin (Invitrogen)-labeled, 10% biotinylated actin filaments, linked to NeutrAvidin- coated 1-μm biotin-labeled polystyrene beads (Invitrogen), which was also conjugated with TRITC-BSA. Optical trapping experiments were performed in a motility buffer supplemented with 2 mm creatine phosphate, 0.1 mg/ml creatine phosphokinase, and 1 mm ATP. Under fluorescence imaging, a single actin filament was attached to two 1-μm beads, via manipulation of the optical traps. These beads/actin dumbbells (length ∼4–6 μm) were pulled taut using the optical traps and positioned above pedestals in the chamber to record transient actomyosin-18A interactions. One of ∼15–20 pedestals exhibited actomyosin interactions, providing statistical evidence that most probably only a single myosin-18A-HMM molecule was capable of interacting with the actin filament. Experiments were performed using optical trap stiffness of ∼0.015–0.025 piconewton/nm. Similarly, as reported previously (29), data were collected at 20 kHz while sine waves (200 Hz) of amplitudes ∼200 nm, peak-to-peak, were applied to one of the optical traps. The decrease in the S.D. of the noise level of this sine wave was used to distinguish periods with or without myosin-18A attachments. Analysis was performed using custom software written in LabVIEW 8.5 (National Instruments, Corp., Austin, TX), and histograms were plotted using Origin 8.5 (OriginLab Corp., Northampton, MA). Detachment rate and power stroke histograms were compiled from data collected from seven pedestals each for both myosin-18A-HMMs (15).
RESULTS
Protein Production and Light Chain Phosphorylation
We engineered truncated clones of both mouse myosin-18A isoforms, α and β, corresponding to S1- and HMM-like fragments that were produced as C-terminally FLAG-tagged constructs in the Sf9/baculovirus system (Figs. 1 and 2A). Sequence analysis of myosin-18A has generally detected a single canonical IQ motif for light chain binding, located between the motor domain and the coiled-coil tail (1, 30, 31) (Fig. 2C). However, inspection of the sequence immediately downstream of this IQ motif revealed a WPWW motif that is known to be the site of the sharp bend in the heavy chain of the myosin-2 RLC binding site (32). An alignment of this region of myosin-18 sequences across subclasses and phyla and comparison with myosin-2 (Fig. 2B) indeed show a striking similarity between myosin-18 and myosin-2 in several functionally important respects. First, the spacing between the start of IQ1 and the start of IQ2 is identical in both myosins (26 residues). Second, the WPWW motif is in the same place. Third, the proline that marks the start of the predicted coiled-coil in myosin-2 is also present in myosin-18, is the same distance from the WPWW motif, and (like in myosin-2) occupies the d position in the coiled-coil heptad repeat. Finally, there is high sequence conservation between the IQ regions of the two classes. Therefore, we co-expressed both ELC and RLC with the myosin-18A heavy chain. Myosin-18A-S1 constructs, truncated at the start of the predicted coiled-coil (Leu1242 for the α isoform or Leu907 for the β isoform), and HMM constructs, truncated after ∼32 heptad repeats of coiled-coil (Leu1463 for the α isoform or Leu1128 for the β isoform), were successfully co-purified with both light chains (Fig. 2A). The RLC bound to myosin-18A could be phosphorylated by MLCK (Fig. 2B). Negative staining electron microscopy demonstrated that the S1 was single-headed and that the HMM was double-headed, as expected (Fig. 3, A and B).
FIGURE 2.

Overproduction of M. musculus myosin-18A. A, purification of myosin-18A-S1 and -HMM constructs. Lanes are marked as either molecular weight markers (M) or purified fractions of each motor construct. The dashed line indicates that the image is a composite of two separate gel scans. Note that previous studies of nonmuscle myosin 2 and smooth muscle isoforms have revealed that the ELC binds Coomassie dye poorly and always appears lighter in intensity than does the RLC (16, 48). B, confirmation of phosphorylation of the bound RLC using a gel shift Phos-tag assay. Each of the myosin fragments described at the top was either untreated (−) or treated (+) with MLCK to phosphorylate the RLC as described under “Experimental Procedures.” C, alignments of the neck region of representative myosin-18 and myosin-2 heavy chain sequences. Genome sources are M. musculus (Mm), Homo sapiens (Hs), Xenopus tropicalis (Xt), and D. melanogaster (Dm). Myosin-2 H. sapiens genes are MYH9 (nm myo-2A), MYH7 (card myo-2), and MYH4 (sk myo-2). Blue highlighting shows (from left to right) the consensus first IQ motif; the second, more divergent motif that forms the binding site for RLC in the aligned motors; the bend region; and the Pro that marks the end of the neck. The heptad register for the beginning of the coiled-coil motif is shown at the bottom.
FIGURE 3.

Electron microscopic images of myosin-18A fragments. A, field of negatively stained myosin-18Aα HMM molecules. Scale bar, 50 nm. B, field of negatively stained myosin-18Aα-S1 molecules. Scale bar, 50 nm. C, myosin-18Aα-S1 mixed with equimolar actin in the absence of nucleotide demonstrates the weakness of actin binding. D, nonmuscle myosin-2A-S1 mixed with actin under the same conditions shows the classic arrowhead decoration.
Myosin-18A Has a Slow Basal Rate of ATP Hydrolysis That Is Not Activated by F-actin
To measure the steady-state ATPase of myosin-18A, we tested S1 constructs using an ATP-regenerating, NADH-coupled, actin-activated ATPase assay under a variety of conditions ranging up to 30 μm actin. The basal and actin-activated ATPase traces of myosin-18Aα-S1 and myosin-18Aβ-S1 showed that neither isoform hydrolyzed ATP at an appreciable rate, with upper limits on the basal activity of 0.0015 and 0.0014 s−1 for the α and β isoforms, respectively, and rates of 0.002 and 0.0006 s−1 for the myosin-18Aα and -18Aβ isoforms in the presence of actin, respectively (Fig. 4). Similar data were obtained with myosin-18A HMM isoforms. Phosphorylation of the RLC did not activate the ATPase activity of these molecules either. The measurement of the ATPase activity in the presence of actin was greatly complicated by the slow hydrolysis of ATP by actin preparations. The ATPase was not significantly enhanced by raising the actin concentration to 60 μm, increasing the salt concentration from 50 to 150 mm KCl, phosphorylation of the bound RLC, the addition of smooth muscle tropomyosin, or the addition of the reported myosin-18 binding partner GOLPH3 (data not shown). The ATPase activity of myosin-18A-S1 isoforms was also tested with the radiometric basal K+-EDTA ATPase assay with no detectable release of Pi above the rate seen in a no-protein control (data not shown). Together, these data suggest that murine myosin-18A fragments hydrolyze ATP only very slowly under any of the experimental conditions tested.
FIGURE 4.

The MgATPase activity of myosin-18A isoforms is low and is poorly activated by actin. Time course of ATP hydrolysis measured used an NADH-coupled assay where the decline in absorbance at 340 nm reflects the course of ATP hydrolysis. Open triangle, 30 μm F-actin alone; open diamond, myosin-18Aα-S1 alone; closed diamond, 30 μm actin plus myosin-18Aα-S1; open square, myosin-18Aβ-S1 alone; closed square, myosin-18Aβ-S1 plus 30 μm F-actin. The data were arithmetically normalized to a starting absorbance value of 1.0. Experiments shown were conducted at 25 °C in a buffer containing 50 mm KCl, 10 mm MOPS (pH 7.2), 2 mm MgCl2, 0.15 mm EGTA, 2 mm ATP, 40 units/ml lactate dehydrogenase, 200 units/ml pyruvate kinase, 1 mm phosphoenolpyruvate, and 200 μm NADH. The concentration of myosin-18A motors used in these traces was 1 μm. AU, absorbance units.
Myosin-18A Isoforms Bind Adenosine Nucleotides Weakly
We tested the ability of myosin-18A isoforms to bind adenosine nucleotides using several methods. In a filter binding assay, myosin-18A-S1 isoforms bound [α-32P]ATP above base-line binding of the radionucleotide to the membrane but only at 10–20% of the levels obtained with the same concentration of rabbit SkHMM (Fig. 5A). To confirm this binding and further analyze the kinetics of nucleotide binding to myosin-18A-S1 isoforms, the fluorescent nucleotide analogs mantATP and mantADP were employed in a series of transient state kinetic experiments in a stopped-flow apparatus using FRET from intrinsic tryptophan residues (Fig. 5, B–D). Although myosin-18A lacks the conserved tryptophans corresponding to Trp113, Trp131, and Trp512 in chicken skeletal muscle myosin-2 that are associated with efficient FRET emission in other myosins, there are other tryptophan residues within the motor domain that contribute to this signal. Based on a homology modeling approach, eight of these tryptophan residues are positioned within a 3-nm radius from the nucleotide (Trp22, Trp201, Trp332, Trp461, Trp542, Trp598, Trp606, Trp715) and could contribute to the observed FRET signal. mantADP binding to myosin-18A splice variants was assayed from the fluorescence increase upon mixing myosin and mantADP under pseudo-first order conditions in a stopped-flow spectrophotometer. The fluorescence transients were well fitted by single exponentials (Fig. 5B). The second order plot of the observed rate constants versus [mantADP] defines a line with a slope of k+D = 0.17 ± 0.02 μm−1 s−1 and k+D = 0.10 ± 0.02 μm−1 s−1 for myosin-18Aα and -18Aβ, respectively. The corresponding y intercepts reflect the ADP dissociation rates, which were k−D = 9.58 ± 0.15 s−1 and k−D = 10.87 ± 0.16 s−1, respectively, for myosin-18Aα-S1 and -18Aβ-S1. The mantADP release rates were independently determined from the fluorescence decrease upon chasing myosin·mantADP with excess ATP (Fig. 5C and Table 1). Single-exponential fits indicated the respective dissociation rates of k−D = 10.01 ± 0.23 s−1 and k−D = 10.30 ± 0.30 s−1 for myosin-18Aα-S1 and -18Aβ-S1, in excellent agreement with the values determined from the ordinate intercept of the secondary plots shown above (Fig. 5B). Dissociation equilibrium constants of 56.4 and 108.7 μm were calculated from the corresponding release and binding rate constants (k−D/k+D) for myosin-18Aα-S1 and -18Aβ-S1 splice variants.
FIGURE 5.

Nucleotide interactions of myosin-18A isoforms. A, binding of [α-32P]ATP to rabbit SkHMM, myosin-18Aα-S1, and myosin-18Aβ-S1 in a filter-based assay. The top spot is a no-protein control. See “Experimental Procedures” for methods. The right-hand column demonstrates the relative extent of binding compared with values obtained for SkHMM. B, mantADP binding to myosin-18Aα-S1 (closed circles) and myosin-18Aβ-S1 (open circles) as a function of mantADP concentration. The slopes define the apparent second order rate constants for mantADP binding to myosin-18Aβ-S1 and myosin-18Aβ-S1 of k+D = 0.17 ± 0.02 μm−1 s−1 and k+D = 0.10 ± 0.02 μm−1 s−1. The corresponding y intercepts reflect the ADP dissociation rates (k−D = 9.58 ± 0.15 s−1 and k−D = 10.87 ± 0.16 s−1 for myosin-18Aα-S1 and -18Aβ-S1. The inset shows a representative trace when a final concentration of 12.5 μm mantADP is mixed with 0.25 μm myosin-18Aα-S1. The single exponential fit to the data gave a rate of 11.5 s−1. C, representative fluorescence decay observed after mixing myosin-18A-S1·mantADP with excess ATP. Release rate constants of k−D = 10.01 ± 0.23 s−1 and k−D = 10.30 ± 0.30 s−1 were obtained for myosin-18Aα (inset) and myosin-18Aβ from single-exponential fits to the data. D, mantATP binding to myosin-18Aα-S1 (closed circles) and myosin-18Aβ-S1 (open circles) as a function of mantATP concentration. Second order rate constants for mantATP binding (KT) were calculated from the slopes to be 0.42 ± 0.03 μm−1 s−1 and 0.12 ± 0.01 μm−1 s−1 for myosin-18Aα-S1 and myosin-18Aβ-S1, respectively. ATP dissociation rate constants of k−T = 7.29 ± 0.26 s−1 for myosin-18Aα and k−T = 10.16 ± 0.09 s−1 for myosin-18Aβ-S1 were deduced from the ordinate intercepts. The inset shows a representative trace when a final concentration of 10 μm mantATP is mixed with 0.25 μm myosin-18Aβ-S1. The single exponential fit to the data gave a rate of 11.4 s−1.
TABLE 1.
Kinetic properties of myosin-18A isoforms
KA and KTA were determined by hyperbolic fits to the binding data.
| Parameter | Signal or calculation | M. musculus MY018Aα | M. musculus MY018Aβ |
|---|---|---|---|
| ATP binding | |||
| KT (μm−1 s−1) | mantATP | 0.42 ± 0.03 | 0.12 ± 0.01 |
| k−T (s−1) | mantATPa | 7.29 ± 0.26 | 10.16 ± 0.09 |
| ADP interaction | |||
| k+D (μm−1 s−1) | mantADP | 0.17 ± 0.02 | 0.10 ± 0.02 |
| k−D (s−1) | mantADPb | 9.58 ± 0.15 | 10.87 ± 0.16 |
| mantADPc | 10.01 ± 0.23 | 10.3 ± 0.3 | |
| KD (μm) | k−D/k+D | 56.35 ± 6.69 | 108.70 ± 21.79 |
| Actin interaction | |||
| KA (μm) | Co-sedimentation | 5.9 ± 1.7 | 47.9 ± 9.2 |
| KTA (μm) | Co-sedimentationd | 4.9 ± 1.6 | 54.0 ± 29 |
a Ordinate of the kobs versus [mantATP] plot.
b Ordinate of the kobs versus [mantADP] plot.
c ATP chasing experiment.
d In the presence of 1 mm ATP.
The second order rate constant for mantATP binding (KT) was determined to be 0.42 ± 0.03 and 0.12 ± 0.01 μm−1 s−1 for myosin-18Aα-S1 and myosin-18Aβ-S1, respectively, by fitting plots of the observed rates of fluorescence increase as a function of [mantATP] to straight lines (Fig. 5D). Distinct from most other kinetically characterized conventional and unconventional myosins, the ordinate intercepts were different from zero, and these yield ATP dissociation rate constants of k−T = 7.29 ± 0.26 s−1 for myosin-18Aα-S1 and k−T = 10.16 ± 0.09 s−1 for myosin-18Aβ-S1, respectively, suggesting weak ATP interactions. The results show that ATP binds somewhat faster to myosin-18Aα-S1 than it does to myosin-18Aβ-S1, whereas ADP bound to each isoform with more similar rates.
Myosin-18A Binds Actin with Low Affinity, Insensitive to ATP
The binding of myosin-18A-S1 isoforms to F-actin was analyzed through co-sedimentation assays in the absence of ATP (Fig. 6, A and B). Dissociation constants (Kd) determined by fitting the binding curves to a hyperbolic function were determined to be 5.9 ± 1.7 μm for myosin-18Aα-S1 and 47.9 ± 9.2 μm for myosin-18Aβ-S1. The addition of 1 mm ATP to these co-sedimentations (Fig. 6, A and B) did not appreciably change the dissociation constants, with Kd = 4.9 ± 1.6 and 54 ± 29 μm for myosin-18Aα-S1 and -18Aβ-S1, respectively (Fig. 6, A and B). The binding of S1 fragments of both isoforms determined by a hyperbolic fit to the data plateaued at values below 100% at high actin concentrations. Myosin-18Aα-S1 saturated at a maximum bound proportion of 77 ± 5% in the absence of nucleotide and 69 ± 6% in the presence of 1 mm ATP. In the absence of nucleotide, myosin-18Aβ-S1 reached a maximum bound of 41 ± 4% (and in the presence of 1 mm ATP, 44 ± 12%). Phosphorylation of myosin-18Aβ-S1 by MLCK did not significantly affect the fractional bound population (Table 2). Similarly, the presence of ADP did not significantly affect either parameter (Table 2).
FIGURE 6.

Binding of myosin-18A isoforms to actin. A, binding of myosin-18Aα-S1 to actin at a final concentration of 1 μm motor in the presence (open circles) or absence (closed circles) of 1 mm ATP. Conditions were as follows: 0.1 m KCl, 20 mm MOPS (pH 7.0), 5 mm MgCl2, 0.05 mm EGTA, 1 mm NaN3, and 1 mm DTT, actin concentrations as indicated and ATP present or not. Kd values were obtained by fitting the data to a rectangular hyperbolic function, AM*/MT = (AM*/MT)max × A/(Kd + A). Values were Kd = 5.9 ± 1.7 μm in the absence of ATP (red curve) and Kd = 4.9 ± 1.6 μm in the presence of 1 mm ATP (blue curve) for myosin-18Aα-S1. For these experiments, myosin-18Aα-S1 binding saturated at 77 ± 5% bound in the absence of nucleotide and at 69 ± 6% in the presence of ATP. B, binding of myosin-18Aβ-S1 to actin. In the absence of ATP (closed circles), Kd = 47.9 ± 9.2 μm (red curve), and in the presence of 1 mm ATP (open circles), Kd = 41 ± 4 μm (blue curve). For myosin-18Aβ, saturation of binding occurred at 44 ± 12% in the absence of ATP, and in the presence of 1 mm ATP, saturation occurred at 24.5 ± 7.1%. In both A and B, the black lines shown are the simulation of the binding data using the model described in Scheme 1. Rate constants used for the simulations were as follows: k1 = 0.001 s−1, k−1 = 0.00012−1 s, k2 = 2.5 × 106 m−1 s−1, k−2 = 10 s−1 for the interaction of myosin-18Aα-S1 with actin and k1 = 0.0005 s−1, k−1 = 0.0045 s−1, k2 = 7.5 × 105 m−1 s−1, k−2 = 10 s−1 for the interaction of myosin-18Aβ-S1 with actin. C, sequential actin rebinding experiments. Lanes 1 and 2, supernatant (S) and pellet (P) of myosin-18Aβ-S1 sedimented in the absence of actin. Lanes 3 and 4, supernatant and pellet of myosin-18A-S1 sedimented in the presence of 40 μm actin. Lanes 5 and 6, the supernatant from the first sedimentation was mixed with 40 μm actin and sedimented to give supernatant and pellet fractions. Lanes 7 and 8, the supernatant from the second sedimentation was mixed with 40 μm actin and sedimented to give supernatant and pellet fractions. Lanes 9 and 10, the supernatant from the third sedimentation was mixed with 40 μm actin and sedimented to give supernatant and pellet fractions. The percentage of myosin-18Aβ-S1 found in the pellet is given below the pairs of lanes for each experiment. Error bars, S.D.
TABLE 2.
Effect of nucleotide and RLC phosphorylation on binding of myosin-18Aβ to actin
Data show the extent of binding of myosin-18Aβ-S1 to 10 μm actin for cases where the myosin was first phosphorylated with MLCK or where the binding was conducted in the presence of ADP or ATP or in the absence of nucleotide (APO). The amount of binding predicted by the simulation is marked as “Simulation” (see below).
| Assay | Fraction of myosin-18Aβ-S1 bound |
|---|---|
| Apo | 0.048 ± 0.011 |
| ATP | 0.032 ± 0.006 |
| ADP | 0.061 ± 0.027 |
| MLCK | 0.074 ± 0.0002 |
| Simulation | 0.074 ± 0.0431 |
There are at least two possible mechanisms to account for the apparent substoichiometric binding. It is possible that a portion of the protein was permanently incapable of binding to actin. In this case, if a second sedimentation is carried out with the supernatant fractions of the first sedimentation (performed at high actin concentrations), then little or no additional binding should occur. The second possibility is the myosin-18A is in an equilibrium between a state that is incompetent to bind to actin and a state that can bind actin. In this latter case, resedimentation of the supernatant fractions with added actin should result in an additional amount of actin binding, the magnitude of which will be dependent on the rates of the myosin equilibrium between the two states, incompetent and competent to bind to actin. To test this hypothesis, the unbound fraction of myosin-18Aβ-S1 after an initial sedimentation at 40 μm actin was resedimented in the presence of 40 μm actin, and the supernatant fraction was analyzed for the percentage of myosin bound. In both the first sedimentation and in several subsequent resedimentations, ∼20% of the myosin was bound (Fig. 6C). When the supernatant of the first spin at 40 μm actin was done with myosin-18Aα, about 60% of the myosin-18Aα pelleted in both spins, replicating the result obtained with myosin-18Aβ. This behavior is consistent with the second model outlined above. This peculiar behavior was successfully simulated using a model in which there is a slow equilibrium between incompetent and competent actin binding states of myosin and is shown by the black lines in Fig. 6, A and B. See Scheme 1 for the reaction model used and the legend of Fig. 6, A and B, for the rate constants used. The value for k−2 was taken as 10 s−1, because in optical trapping studies described below (Fig. 7), there were frequent interactions between actin and myosin-18A, whose duration averaged 50–100 ms (see below). We also found that the fits of the observed AM/MT versus actin seen in Figs. 6A and 4B were the same as the value of k−2, varying between 0.01 and 10 s−1, as long as the K2 was held constant at 2.5 × 105 m−1 for myosin-18Aα-S1 and 7 × 104 m−1 for myosin-18Aβ-S1. This result further supports the hypothesis that there is an isomerization from an incompetent actin-binding myosin form to a competent myosin form that can bind actin. The model predicts that at long incubation time (1 h or more) and at high actin concentration, the fraction of myosin-18A isoforms bound to actin should approach unity, and we find in preliminary data with myosin-18A HMM isoforms that this does occur. Note that the apparent Kd values determined in the absence of nucleotide are 50–1000 times greater than those for other myosins (33–35).
FIGURE 7.

Optical trapping analysis of myosin-18A interactions. Actomyosin attachment lifetime data of myosin-18Aα-HMM (A) and myosin-18Aβ-HMM interactions (B) collected in the optical trap were fitted to a single exponential curve. Myosin-18Aα-HMM (A) yielded a detachment rate of 16.7 ± 0.4 s−1, and myosin-18Aβ-HMM (B) yielded a detachment rate of 19.7 ± 0. 3s−1. Insets in A and B show the time axis extended to 1 s instead of 0.3 s. C, actomyosin-18Aα-HMM displacement data from the optical trap were fit to a Gaussian distribution centered at 0.5 ± 0.4 nm. D, actomyosin-18Aβ-HMM displacement data from the optical trap were fit to a Gaussian distribution centered at 0.9 ± 0.3 nm. No significant displacements (i.e. power strokes) were observed for either construct. Data were collected at 20 °C in a buffer containing 25 mm KCl, 25 mm imidazole (pH 7.4), 4 mm MgCl2, 1 mm EGTA, 2 mm creatine phosphate, 50 mm DTT, 1 mm ATP, 0.1 mg/ml creatine phosphokinase, 3 mg/ml glucose, 0.1 mg/ml glucose oxidase, and 0.02 mg/ml catalase. The number of actomyosin-18Aα-HMM interactions was 1804, and the number of actomyosin-18Aβ-HMM interactions was 697.
We were unable to measure the rate or extent of binding of myosin-18A-S1 isoforms to actin using pyrene-actin quenching or light scattering. This is probably related to two problems. First, the sedimentation results presented above indicate that the affinity of both myosin-18A S1 isoforms to actin is weak. Second, given the weak affinity and lack of effect of ATP, it is possible that myosin-18A-S1 resides in a weakly bound conformation that would not quench pyrene fluorescence upon binding. Attempts to observe the binding of myosin-18Aα-S1 to actin in the absence of ATP were consistent with this. Fig. 3D shows an actin filament decorated with nonmuscle myosin-2A-S1 in the absence of ATP as a control. The familiar chevron structures showing regular decoration of myosin with actin are clearly visible. In contrast, mixing actin with a similar concentration of myosin-18Aα-S1 shows little or no binding of the myosin to actin, consistent with the weak binding constant for actin observed for this myosin (Fig. 3C).
Myosin-18A Shows No Power Stroke on F-actin in the Optical Trap
Interactions of both myosin-18A-HMM isoforms with actin filaments were studied using the optical trapping apparatus via the three-bead assay. Three-bead assays were performed using an induced oscillation of 200 Hz on one of the traps to monitor increases in system stiffness of the dumbbell during actomyosin attachment. Both isoforms exhibited intermittent interactions with actin filaments, as detected by the decrease in the Brownian noise of the bead. Raw data collected using a position-sensitive quadrant photodiode were analyzed to determine detachment rates and attachment positions to determine power strokes from each actomyosin-18A interaction (Figs. 7, A–D). Detachment rates were determined from a single-exponential fit to the respective histograms of lifetimes of actomyosin-18A interactions. The detachment rates for both myosin-18Aα-HMM and myosin-18Aβ-HMM constructs showed approximately similar detachment rates, of 16.7 ± 0.4 s−1 myosin-18A-α and 19.7 ± 0.3 s−1 for the myosin-18Aβ-HMM (Fig. 7, A and B). Unlike a mechanically active myosin, whereby a “shift” in the peak of the Gaussian distribution by the size of its power stroke would be detected, the myosin-18A power stroke histograms for both constructs during attachment (Fig. 7, C and D) were centered at ∼0 nm (0.5 ± 0.4 nm for myosin-18Aα-HMM and 0.9 ± 0.3 nm for the myosin-18Aβ-HMM), indicating that mammalian myosin-18A does not produce a power stroke even in the presence of 1 mm ATP. These results suggest that the myosin-18A does not function as a typical molecular motor and does not perform work against an actin filament.
Myosin-18A Inhibits Translocation of F-actin by Myosin-2
To test this, we used the gliding actin in vitro motility assay, in which myosin bound to a surface interacts with actin. The myosin-18A-HMM isoforms did not move actin filaments in this assay. Myosin-18Aα-HMM tethered actin filaments to the surface but did not move them. At the same concentration of myosin bound to the surface, myosin-18Aβ-HMM only weakly tethered actin to the surface regardless of whether ATP was present or not. To further explore the actin binding potential of myosin-18A isoforms, we tested the ability of these motors to impede the locomotor activity of an actively cycling myosin, specifically SkHMM, in an in vitro motility assay (Fig. 8). Mixing increasing ratios of myosin-18A-HMM motors with SkHMM while keeping the total myosin protein concentration added to the flow cell constant at 0.2 mg/ml slowed the rate of F-actin filament translocation by the SkHMM. The actin filaments in the SkHMM/myosin-18Aα-HMM mixing experiments ceased translocating when the total myosin content was ∼60% myosin-18Aα-HMM, whereas the translocation of the filaments in the presence of myosin-18Aβ-HMM did not fully cease motility until 100% of the myosin content was myosin-18A. The decline in translocation rate with added myosin-18A-HMM fragments was not due to merely lowering the concentration of SkHMM in the assay because control experiments showed that 0.02 mg/ml SkHMM alone added to the flow cell translocated actin at the same rate as the 0.2 mg/ml sample. Combined, these data suggest that on a population scale, mammalian myosin-18A acts to induce a frictional drag against an actively cycling myosin, and the different actin affinities seen between the myosin-18Aα and -β isoforms in co-sedimentation assays can be recapitulated through the actin tethering seen in the in vitro motility assays.
FIGURE 8.

Myosin-18A attenuates the motility of actively cycling SkHMM in vitro. Keeping the final myosin motor concentration in the assay at 0.2 mg/ml, increasing concentrations of murine myosin-18Aα-HMM (closed circles) or myosin-18Aβ-HMM (open circles) were premixed with SkHMM prior to binding to the coverslip surface. The rate of actin filament sliding was measured by centroid tracking from the CellTrak program (Motion Analysis). The translocation speed of SkHMM alone, which varied between preparations from 4.5 to 7.2 μm s−1, was used for normalization. Assays were performed at 30 °C in buffer containing 50 mm KCl, 20 mm MOPS (pH 7.4), 5 mm MgCl2, 0.1 mm EGTA, 1 mm ATP, 25 μg/ml glucose oxidase, 45 μg/ml catalase, 2.5 mg/ml glucose, and 50 mm DTT. Error bars, S.D.
Shape of Myosin-18 Heads by Electron Microscopy
Taken together, the F-actin binding data, the optical trapping data, the in vitro motility data and the lack of rapid ATP hydrolysis suggest that myosin-18A isoforms assume a conformation that binds only weakly to actin. The conformational state of myosin-18A-S1 isoforms in the absence of nucleotide was examined by negative staining electron microscopy. The conformation observed for both myosin-18A isoform was one in which the lever arm was strongly angled with respect to the long axis of the motor domain (Fig. 9A). A similar conformation was seen for myosin 2B-S1 in the presence of nucleotide (Fig. 9C, right) but not in nucleotide-free conditions (Fig. 9C, left), as has been demonstrated previously for other myosins (36, 37). In these other myosins, in contrast to myosin-18A, the lever arm is in line with respect to the long axis of the motor domain in the absence of nucleotide. In accord with the lack of effect of ATP on the actin binding, we found that there was no effect of ATP on the shape of myosin-18A isoforms (Fig. 9, compare A with B).
FIGURE 9.

The gross conformation of myosin-18A is not affected by nucleotide. Shown are class averages of myosin-18Aα S1 (1108 molecules) (A) and myosin-18Aβ-S1 (1341 molecules) (B) in the absence of nucleotide. C, myosin-18Aα S1 in the presence of ATP (n = 817). C, surface representations showing the three major conformations of Argopectan irradians myosin 2 S1 determined by x-ray crystallography. Crystal structures are oriented to show the most similarity to class averages. Red, pre-power stroke state (Protein Data Bank entry 1QVI) (69); blue, near rigor state (Protein Data Bank entry 1SR6) (70); green, internally uncoupled state (Protein Data Bank entry 1KK8) (71). Images were prepared using UCSF Chimera. Shown are class averages of myosin 2B-S1 in the absence (786 molecules) (E) and presence of ATP (933 molecules) (F). The ATP concentration was 1 mm when present.
DISCUSSION
There is considerable enzymatic diversity among the members of the myosin superfamily, with actin-activated ATPase rates and F-actin translocation rates varying by 3 orders of magnitude (33). This represents adaptations made in the various myosins to perform their particular cellular role (38). Class-18 myosins represent the extreme edge of this diversity. Previous work demonstrated that D. melanogaster myosin-18 bound ATP poorly and showed no evidence for an ATPase activity and that it bound actin in an ATP-insensitive manner (15). Earlier biochemical studies using myosin-18A fragments expressed in mammalian cells suggested that it may lack the enzymatic activity that is usually associated with myosins and may function more like an actin-binding protein than as a motor protein, although enzymatic measurements were not directly performed (13, 14). By examining the nucleotide and actin binding properties of soluble single-headed and double-headed fragments of mammalian myosin-18A isoforms, we now quantify these behaviors.
On a gross structural level, there are many similarities between class-18 myosins and the conventional class-2 myosins. We have shown that myosin-18A, like myosin-2, has two IQ motifs separated by 26 amino acids following the motor domain and that these bind an ELC and an RLC. A previous study provided evidence for RLC association with myosin-18A using immunoprecipitation (11). Both classes have a WPWW motif near the end of the lever arm heavy chain sequence that introduces a sharp bend into the lever arm and a conserved proline located 8 amino acids from this motif, which marks the start of the coiled-coil motif. The myosin-18A-bound RLC is a substrate for MLCK. A long coiled-coil-forming domain follows the IQ motifs in both classes to produce two-headed myosins. These similarities argue strongly for a common evolutionary origin for class-18 and class-2 myosins. However, the similarity fades when examining the structure and function of the motor domains of the two classes. In fact, it has been concluded that myosin-18 shares a phylogeny with myosin-6 and myosin-1 based on motor domain sequence (1). The biochemical data presented in this paper suggest that the myosin-18A motor domain is markedly different from that of class-1, -2, or -6 myosins. All class-1, -2, or -6 myosins examined to date bind and hydrolyze ATP, and the rate of ATP hydrolysis is markedly activated by F-actin (38). Furthermore, the binding of these myosins to F-actin is dramatically weaker when the myosins have ATP or both of the hydrolysis products, ADP and Pi, bound to the active site compared with their binding affinity for actin when no nucleotide or only ADP is bound. Our data show that M. musculus myosin-18A-S1 exhibits ATP-insensitive actin binding with affinities more similar to those of the weakly bound myosin states described above and that it does not hydrolyze ATP at a significant rate.
Mammalian myosin-18Aα-S1 in the absence of nucleotide binds actin with an affinity (Kd ∼5.0 μm) similar to that previously reported for the isoforms of D. melanogaster myosin-18 (15). Mammalian myosin-18Aβ, however, exhibits a lower actin affinity around 50 μm. The higher affinity of the myosin-18Aα isoform is probably related to the actin binding sequence located in the extended N-terminal region between the PDZ domain and the KE-rich region of that isoform (14). The differences in actin affinity of the myosin-18Aα and myosin-18Aβ isoforms coupled with the similar attachment lifetimes suggest that the β isoform may bind to actin ∼10 times more slowly than does the α isoform. Unfortunately, we cannot verify this directly because on rates cannot be determined in the optical trapping experiments, and we see no quenching of pyrene-actin fluorescence in stopped-flow experiments. The weak affinity was not affected by phosphorylation of bound RLC, the addition of smooth muscle tropomyosin, or the presence of the putative myosin-18A binding partner, GOLPH3 (10), into the assay. Most significantly, the actin binding affinity was not affected by the presence of ATP. The actin affinities of the myosin-18A isoforms in the absence of ATP are 3–5 orders of magnitude weaker than those of myosin-2 family members under the same conditions (2). Even with weak F-actin binding potential, both isoforms of myosin-18A hindered the translocation of actin by an actively cycling SkHMM in an in vitro motility assay, suggesting that myosin-18A motors induce a significant frictional drag on a population scale despite their weak affinity for F-actin. Similar behavior is observed in the in vitro motility assay when inactive, unphosphorylated smooth muscle myosin-2 is mixed with active, phosphorylated smooth muscle myosin-2 (39) or when actin-binding proteins are mixed with actively cycling myosins (40). This is consistent with the optical trapping results showing transient interactions with actin in a nucleotide-independent manner that do not result in a net displacement of the actin filament. Interestingly, unphosphorylated nonmuscle myosin 2B HMM also gave a 0-nm displacement when interacting with actin in the optical trap (41). The weak F-actin affinity of myosin-18A could still be physiologically significant because this myosin is localized in the lamellipodia of some cells (11, 42) where the actin concentration is in the range of 500 μm (43).
Only very low rates of ATP hydrolysis were measured with myosin-18A-HMM and -S1 fragments. We found no conditions where this rate was activated. This included the addition of F-actin, the addition of F-actin plus smooth muscle tropomyosin, phosphorylation of the RLC, the addition of calcium, or the addition of GOLPH3. There was no detectable K+-ATPase activity measured at high salt. We considered the possibility that we have isolated a regulated, “off,” state of myosin-18A, similar to the state of smooth or nonmuscle myosin-2 isoforms with unphosphorylated RLCs (44–46). However, several lines of evidence argue against this. First, phosphorylation of the RLCs by MLCK did not activate myosin-18A. Second, the regulated state of myosin-2 family members involves an asymmetric interaction between the two heads of a single molecule, and single-headed fragments of these myosins are constitutively active in the presence of F-actin (46–48). Myosin-18A-S1 isoforms remained as inactive as the HMM isoform. Third, the myosin exhibited no detectable K+-ATPase activity, which in class-2 myosins is not affected by phosphorylation of the RLC (49). Fourth, ATP bound weakly and reversibly to myosin-18A isoforms. The reversible nature of the ATP binding is in striking contrast to that seen for most other myosin superfamily members (38).
The unusual properties of myosin-18 can be rationalized by comparison of the amino acid sequence and a structural homology model of myosin-18A (Fig. 10A) with the sequence and crystal structures of class-2 myosins. This demonstrated several deviations from the consensus sequence of the myosin superfamily, which might impact ATP binding and hydrolysis and the coupling of ATP binding to actin affinity. There are significant deviations in the amino acid sequence of the three highly conserved loops in the nucleotide binding pocket of myosins (P-loop, switch-1, and switch-2) (Figs. 11 and 12A). Of these differences, the most significant might be the presence of a glutamine residue (Asn457) at the end of switch-2 instead of the consensus glutamic acid (Figs. 11 and 12A). (Note that all amino acid numbers refer to the sequence of the myosin-18Aβ isoform unless otherwise noted.) In other myosins, this glutamic acid (Glu459 in D. discoideum myosin-2) forms a salt bridge with an arginine (Arg238 in D. discoideum myosin-2) at the end of switch-2 to help close the pocket around the nucleotide in preparation for catalysis (50). Mutation of either of these charged amino acids to an alanine in class-2 myosins dramatically reduces the ATPase activity of these myosins (51, 52). It is likely that Asn457 in myosin-18A would not be as effective at forming this salt bridge, and this evolutionary substitution alone would probably be sufficient to explain the low ATPase activity.
FIGURE 10.

Homology model of myosin-18A structure. A, structural overview of the myosin-18A motor domain based on a homology modeling approach. The nucleotide is shown in brown. B, selected view of the superposition of myosin-18A and Dictyostelium myosin-2 activation loops. The myosin-18A activation loop (blue) is longer than that of Dictyostelium myosin-2 (green). C, amino acid alignment of the activation loop of myosin-18A compared with other myosins. Genome sources are M. musculus (Mm), D. discoideum (Dd), Gallus gallus (Gg), and Sus scrofus (Ss). sm myo-2, smooth muscle myosin-2.
FIGURE 11.

Comparison of active site residues of myosin-18Aβ with those of Dictyostelium myosin-2. Amino acid residues in black are from Dictyostelium myosin-2, whereas those in gray are from myosin-18Aβ. This figure was adapted from Sellers (33).
FIGURE 12.

Sequence/structural alignment showing consensus sequence motifs and myosin-18A-specific insertions within myosin motor domains. A, alignment of P-loop, switch-1, and switch-2 regions. Overall, the alignment indicates a high degree of conservation in this triad of nucleotide-interacting loops (P-loop, switch-1, and switch-2) across the myosin superfamily and highlights the exceptional role/deviation of myosin-18A, which accounts for its unique kinetic properties. B, alignment of a region of myosins in which myosin-18A contains a unique myosin-18A-specific extension (myosin-18A loop) within the myosin motor domain, which does not share any homology to other myosins identified so far. In both A and B, blue indicates the position of the typically invariantly conserved glycine residue, which is an arginine residue in myosin-18A; orange indicates myosin-18A-specific amino acid sequences. Genome sources are M. musculus (Mm), D. discoideum (Dd), G. gallus (Gg), A. irradians (Ai), Placopecten magellanicus (Pm), and S. scrofa (Ss). sm myo-2, smooth muscle myosin 2; st myo-2, striated muscle myosin 2.
Myosin-18A has a proline in the middle of the switch-2 consensus sequence (X position of the DIXGFE motif, marked by an arrow at the top of the alignment) not found in other myosin classes (Fig. 12A). This modification might possibly form a kink in the loop to alter the position of other critical residues. Switch-2 in myosin-18A is followed by a small loop (myosin-18Aα Trp796–Arg801; myosin-18Aβ Trp461–Arg466 shown in orange in Fig. 12A), which is predicted to protrude into the nucleotide binding site and may sterically reduce nucleotide accessibility (15). In myosins and kinesins, conformational changes in the γ-phosphate sensor switch-1 upon ATP binding are coupled to changes in the track affinity of the motor. The myosin-18A switch-1 sequence greatly differs from the consensus sequence, which might alter the movement of this loop and impair γ-phosphate sensing (Fig. 12). In particular, the highly conserved SSR sequence of this region is most typically ATR in myosin 18 isoforms. A serine to alanine substitution for the first of these serines is known to reduce motor cycling in other myosins (53, 54) (Figs. 11 and 12). A serine to threonine mutation at this residue is associated with a perturbed actin activation of the steady-state ATPase activity and blocked motile properties (53). The second serine is also important for myosin activity. A serine to threonine mutation in Dictyostelium myosin knocks out most of the actin-activated MgATPase activity and all of the in vitro motility activity (54). Both of the switch-1 serines have been implicated in the ATPase activity of the microtubule-dependent motor kinesin, either by site-directed mutagenesis or from analysis of crystal structures (55, 56).
A long myosin-18A-specific insert (denoted as Myo-18A loop in Fig. 10A and shown in orange in the alignment in Fig. 12B) is located between the fifth strand of the central β sheet and the SH1 and SH2 helices and might sterically hinder the accessibility of nucleotide to the nucleotide binding site and impair transducer function. There is a conserved glycine (Gly680 in the D. discoideum myosin-2 sequence) within the myosin superfamily that lies between the SH1 and SH2 helices and is thought to be in the vicinity of the rotation point of the lever arm. Mutation of this residue to amino acids other than glycine typically alters the nucleotide binding, hydrolysis, and release kinetics and dramatically lowers the in vitro motility (57). Recently, D. discoideum myosin-2 motor domain constructs bearing mutations to this residue were crystallized in different nucleotide states (58). The mutation affected the interactions of the SH1 and SH2 helices with the relay helix. In addition, the switch-1/switch-2 salt bridge was not formed. This glycine residue is very well conserved among most myosin classes but is an arginine in myosin-18A (33) (Fig. 12B).
The inability of F-actin to activate the steady-state ATPase activity of myosin-18A might be due to the alterations in the actin binding sites on myosin. Loop-2, an important surface loop that is involved in forming the weak binding interactions with actin, is longer in myosin-18A than in D. discoideum myosin-2. There are also alterations in the recently described activation loop (59), which contains a conserved arginine residue (Arg520 in D. discoideum myosin-2) shown to directly interact with the negatively charged N-terminal region of actin. Mutation of this residue dramatically reduces the actin-activated ATPase of D. discoideum myosin-2 while not affecting its motility or ADP release. It also appears to reduce the force-generating capacity of the myosin by reducing the duty ratio (59). This loop is longer in myosin-18A than in myosin-2 and lacks the conserved arginine (Fig. 11, B and C).
The structure of the molecules observed in the electron microscope is consistent with the observed weak binding to actin. Despite the absence of ATP, the appearance of myosin-18A was inconsistent with the nucleotide-free (lever-down) state seen in other myosins (36, 60). The addition of ATP did not result in a marked conformational change in the electron microscopic images. For D. discoideum myosin-2, it was shown that the M·ADP·Pi (pre-power stroke, lever-up) state binds F-actin very weakly with a Kd of 57 μm (61). This value is in good agreement with the values obtained for myosin-18Aβ-S1 (Table 1). A motor mechanically trapped in the pre-power stroke conformation indicates chemo-mechanical uncoupling of the reciprocal relationship of this myosin from the usual allosteric triggers, actin and ATP. The low coupling efficiency between the nucleotide binding site and the actin binding region does not trigger a conformational change toward the lever-down state (post-power stroke state) in myosin-18A. Based on our structural model, we speculate that the low nucleotide and F-actin affinities in combination with a perturbed transducer function account for the compromised chemo-mechanical coupling of myosin-18A. This perturbed coupling was also reflected in the nucleotide-insensitive F-actin interaction of myosin-18A-S1 splice variants observed in the co-sedimentation assays.
The unusual actin binding properties, in which a population of the myosins does not bind to actin even at saturating actin concentrations, required a kinetic model that assumes an equilibrium between a myosin-18A state that does not bind actin and a state that does bind. However, the model predicts that at much longer incubation times and at higher actin concentrations, all of the myosin-18A protein should bind to actin, and preliminary experiments support this. Similar actin-binding behavior was previously observed with D. melanogaster myosin-18 (15) and Limulus polyphemus (horseshoe crab) myosin-3 (now termed myosin-21) (62). Interestingly, we were also unable to demonstrate nucleotide binding to either of these myosins. This raises the question of whether myosin-18B will have similar properties.
These in vitro biochemical data suggest that myosin-18A molecules do not act as actively translocating motors, which would negate the possibility of them exerting an actively pulling force against Golgi membranes, as has been proposed (10). Instead, our data support the possibility of mammalian myosin-18A providing a tethering activity within the cell, perhaps anchoring other cellular components to the actin cytoskeleton and resisting the strain induced by actively cycling myosin-2 motors within the cell body, or serving as a scaffold for other proteins. In this regard, several studies have identified myosin-18A-associated proteins, such as GOLPH3 (10), myotonic dystrophy kinase-related Cdc4-binding kinase (MRCK) (11) (a kinase that phosphorylates the RLC of myosin-18A), and the PAK2/βPIX/GITI complex, which are involved in cytoskeletal dynamics and cell motility (42).
Two recent papers made point mutations in myosin-18Aα (G520S/K521A; equivalent to G172S/K173A in myosin-18Aβ) designed to eliminate its “ATPase” activity without carrying out any in vitro work to ascertain the enzymatic status of the protein. These mutations were based on comparable mutations previously shown to abolish ATPase activity in smooth muscle myosin-2 in vitro (53). Both mutations resulted in observable phenotypes in cells. In one case, abnormal Golgi structures were observed following expression of the mutation (10), and in another, MRCK was mislocalized (11). This raises the question of how one “inactivates” an already inactive protein. In this regard, it is interesting to note a recent study by Suijkerbuijk et al. (63) of BUBR1, a pseudokinase involved in a mitotic checkpoint. BUBR1 shares significant similarity to known kinases in terms of both sequence and protein domain architecture, but many residues known to be critical for enzymatic activity have been replaced during evolution, rendering it inactive as a kinase. However, a triad of critically conserved consensus residues remains unperturbed in this inactive kinase. Mutation of one of these residues results in phenotypes in cells (10, 11), but the paper by Suijkerbuijk et al. (63) shows that this is probably due to a reduced conformation stability of the protein and not to elimination of kinase activity (because such activity never existed), as suggested by the authors of the papers. Similarly with mammalian myosin-18A, many of the amino acids important for ATP hydrolysis in the myosin consensus sequences have been replaced with ones that would not be able to carry out the same function, but the two residues mutated in the aforementioned papers still adhere to the consensus sequence for the P-loop. Perhaps the phenotypes observed in these studies are due to reduced protein stability.
An alternative mechanism, not yet tested by our experiments or any other published results, would be the possibility of stretch activation of this motor. This mechanism would be particularly interesting in light of its potential role in the maintenance of the cisternal structure of trans-Golgi, where stretch induced on a bound motor may increase its affinity for the actin filament. Future studies will seek to determine whether tension against the motor may induce some level of activation.
The demonstrated binding of RLC to myosin-18A should be noted by investigators intent on studying the localization and dynamics of nonmuscle myosin-2 isoforms in cells by overexpressing GFP-tagged RLC (64–67). Such studies may be revealing the location of other myosins in addition to the nonmuscle myosin-2 being investigated.
In conclusion, a recent review suggested that there were four kinetic behaviors that classified most myosins as fast movers, slow/efficient force holders, strain sensors, or gated, processive movers (38). These classifications are based on the duty ratio, the equilibrium constant for the step that opens the nucleotide binding pocket, the load dependence of ADP release, and the thermodynamic and kinetic coupling between actin and ADP binding to myosin. Because myosin-18A does not appear to have an actin-activated ATPase cycle, it must define a fifth class of myosins, which may also include members of the invertebrate class-3 (recently renamed as class-21) myosins (1, 62). Interestingly, there are parallels in the kinesin superfamily. Vik1 is a kinesin family member that lacks nucleotide binding activity but does possess strong microtubule binding (68). The crystal structure of this molecule reveals a typical kinesin motor domain fold, but it is missing the characteristic sequences typically found in the conserved loop L1, switch-1, and switch-2 kinesin motifs.
Acknowledgments
We thank Fang Zhang for technical assistance with myosin-18A experiments and protein purification, Melanie Barzik and John A. Hammer III (NHLBI, National Institutes of Health) for providing a full-length clone of mouse myosin-18Aα and -β as starting points for cloning in this project, and Kathryn Callahan (NIDDK, National Institutes of Health) for assistance with filter binding assays. We thank the NHLBI Proteomics Core Facility, which provided excellent service. We thank the Electron Microscopy Core Facility of the NHLBI for support and the use of facilities.

This article contains supplemental Equations 1–8.
- S1
- subfragment-1
- ELC
- essential light chain
- HMM
- heavy meromyosin
- SkHMM
- skeletal muscle HMM
- mantATP
- N-methylanthraniloyl derivative of ATP
- mantADP
- N-methylanthraniloyl derivative of ADP
- RLC
- regulatory light chain
- MLCK
- myosin light chain kinase
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Odronitz F., Kollmar M. (2007) Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 8, R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sellers J. R. (2000) Myosins. A diverse superfamily. Biochim. Biophys. Acta 1496, 3–22 [DOI] [PubMed] [Google Scholar]
- 3. Furusawa T., Ikawa S., Yanai N., Obinata M. (2000) Isolation of a novel PDZ-containing myosin from hematopoietic supportive bone marrow stromal cell lines. Biochem. Biophys. Res. Commun. 270, 67–75 [DOI] [PubMed] [Google Scholar]
- 4. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain. Molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 [DOI] [PubMed] [Google Scholar]
- 5. Mori K., Furusawa T., Okubo T., Inoue T., Ikawa S., Yanai N., Mori K. J., Obinata M. (2003) Genome structure and differential expression of two isoforms of a novel PDZ-containing myosin (MysPDZ) (Myo18A). J. Biochem. 133, 405–413 [DOI] [PubMed] [Google Scholar]
- 6. Ajima R., Kajiya K., Inoue T., Tani M., Shiraishi-Yamaguchi Y., Maeda M., Segawa T., Furuichi T., Sutoh K., Yokota J. (2007) HOMER2 binds MYO18B and enhances its activity to suppress anchorage independent growth. Biochem. Biophys. Res. Commun. 356, 851–856 [DOI] [PubMed] [Google Scholar]
- 7. Nishioka M., Kohno T., Tani M., Yanaihara N., Tomizawa Y., Otsuka A., Sasaki S., Kobayashi K., Niki T., Maeshima A., Sekido Y., Minna J. D., Sone S., Yokota J. (2002) MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc. Natl. Acad. Sci. U.S.A. 99, 12269–12274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakano T., Tani M., Nishioka M., Kohno T., Otsuka A., Ohwada S., Yokota J. (2005) Genetic and epigenetic alterations of the candidate tumor-suppressor gene MYO18B, on chromosome arm 22q, in colorectal cancer. Genes Chromosomes Cancer 43, 162–171 [DOI] [PubMed] [Google Scholar]
- 9. Yanaihara N., Nishioka M., Kohno T., Otsuka A., Okamoto A., Ochiai K., Tanaka T., Yokota J. (2004) Reduced expression of MYO18B, a candidate tumor-suppressor gene on chromosome arm 22q, in ovarian cancer. Int. J. Cancer 112, 150–154 [DOI] [PubMed] [Google Scholar]
- 10. Dippold H. C., Ng M. M., Farber-Katz S. E., Lee S. K., Kerr M. L., Peterman M. C., Sim R., Wiharto P. A., Galbraith K. A., Madhavarapu S., Fuchs G. J., Meerloo T., Farquhar M. G., Zhou H., Field S. J. (2009) GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan I., Yong J., Dong J. M., Lim L., Leung T. (2008) A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136 [DOI] [PubMed] [Google Scholar]
- 12. Yang C. H., Szeliga J., Jordan J., Faske S., Sever-Chroneos Z., Dorsett B., Christian R. E., Settlage R. E., Shabanowitz J., Hunt D. F., Whitsett J. A., Chroneos Z. C. (2005) Identification of the surfactant protein A receptor 210 as the unconventional myosin 18A. J. Biol. Chem. 280, 34447–34457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mori K., Matsuda K., Furusawa T., Kawata M., Inoue T., Obinata M. (2005) Subcellular localization and dynamics of MysPDZ (Myo18A) in live mammalian cells. Biochem. Biophys. Res. Commun. 326, 491–498 [DOI] [PubMed] [Google Scholar]
- 14. Isogawa Y., Kon T., Inoue T., Ohkura R., Yamakawa H., Ohara O., Sutoh K. (2005) The N-terminal domain of MYO18A has an ATP-insensitive actin-binding site. Biochemistry 44, 6190–6196 [DOI] [PubMed] [Google Scholar]
- 15. Guzik-Lendrum S., Nagy A., Takagi Y., Houdusse A., Sellers J. R. (2011) Drosophila melanogaster myosin-18 represents a highly divergent motor with actin tethering properties. J. Biol. Chem. 286, 21755–21766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pato M. D., Sellers J. R., Preston Y. A., Harvey E. V., Adelstein R. S. (1996) Baculovirus expression of chicken nonmuscle heavy meromyosin II-B. Characterization of alternatively spliced isoforms. J. Biol. Chem. 271, 2689–2695 [DOI] [PubMed] [Google Scholar]
- 17. Wang F., Harvey E. V., Conti M. A., Wei D., Sellers J. R. (2000) A conserved negatively charged amino acid modulates function in human nonmuscle myosin IIA. Biochemistry 39, 5555–5560 [DOI] [PubMed] [Google Scholar]
- 18. Spudich J. A., Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 19. Margossian S. S., Lowey S. (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85, 55–71 [DOI] [PubMed] [Google Scholar]
- 20. Wang F., Chen L., Arcucci O., Harvey E. V., Bowers B., Xu Y., Hammer J. A., 3rd, Sellers J. R. (2000) Effect of ADP and ionic strength on the kinetic and motile properties of recombinant mouse myosin V. J. Biol. Chem. 275, 4329–4335 [DOI] [PubMed] [Google Scholar]
- 21. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics, Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trentham D. R., Bardsley R. G., Eccleston J. F., Weeds A. G. (1972) Elementary processes of the magnesium ion-dependent adenosine triphosphatase activity of heavy meromyosin. A transient kinetic approach to the study of kinases and adenosine triphosphatases and a colorimetric inorganic phosphate assay in situ. Biochem. J. 126, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang F., Kovacs M., Hu A., Limouze J., Harvey E. V., Sellers J. R. (2003) Kinetic mechanism of non-muscle myosin IIB. Functional adaptations for tension generation and maintenance. J. Biol. Chem. 278, 27439–27448 [DOI] [PubMed] [Google Scholar]
- 24. Pollard T. D., Korn E. D. (1973) Acanthamoeba myosin. I. Isolation from Acanthamoeba castellanii of an enzyme similar to muscle myosin. J. Biol. Chem. 248, 4682–4690 [PubMed] [Google Scholar]
- 25. Sellers J. R., Cuda G., Wang F., Homsher E. (1993) Myosin-specific adaptations of motility assays. Meth. Cell Biol. 39, 23–49 [DOI] [PubMed] [Google Scholar]
- 26. Homsher E., Wang F., Sellers J. R. (1992) Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am. J. Physiol. 262, C714–C723 [DOI] [PubMed] [Google Scholar]
- 27. Finer J. T., Simmons R. M., Spudich J. A. (1994) Single myosin molecule mechanics. Piconewton forces and nanometre steps. Nature 368, 113–119 [DOI] [PubMed] [Google Scholar]
- 28. Takagi Y., Homsher E. E., Goldman Y. E., Shuman H. (2006) Force generation in single conventional actomyosin complexes under high dynamic load. Biophys. J. 90, 1295–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baboolal T. G., Sakamoto T., Forgacs E., White H. D., Jackson S. M., Takagi Y., Farrow R. E., Molloy J. E., Knight P. J., Sellers J. R., Peckham M. (2009) The SAH domain extends the functional length of the myosin lever. Proc. Natl. Acad. Sci. U.S.A. 106, 22193–22198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foth B. J., Goedecke M. C., Soldati D. (2006) New insights into myosin evolution and classification. Proc. Natl. Acad. Sci. U.S.A. 103, 3681–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamashita R. A., Sellers J. R., Anderson J. B. (2000) Identification and analysis of the myosin superfamily in Drosophila. A database approach. J. Muscle Res. Cell Motil. 21, 491–505 [DOI] [PubMed] [Google Scholar]
- 32. Brown J. H., Kumar V. S., O'Neall-Hennessey E., Reshetnikova L., Robinson H., Nguyen-McCarty M., Szent-Györgyi A. G., Cohen C. (2011) Visualizing key hinges and a potential major source of compliance in the lever arm of myosin. Proc. Natl. Acad. Sci. U.S.A. 108, 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sellers J. R. (1999) Myosins, 2nd Ed., Oxford University Press, Oxford [Google Scholar]
- 34. De La Cruz E. M., Wells A. L., Rosenfeld S. S., Ostap E. M., Sweeney H. L. (1999) The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. U.S.A. 96, 13726–13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kovács M., Wang F., Sellers J. R. (2005) Mechanism of action of myosin X, a membrane-associated molecular motor. J. Biol. Chem. 280, 15071–15083 [DOI] [PubMed] [Google Scholar]
- 36. Burgess S., Walker M., Wang F., Sellers J. R., White H. D., Knight P. J., Trinick J. (2002) The prepower stroke conformation of myosin V. J. Cell Biol. 159, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Y., Baboolal T. G., Siththanandan V., Chen M., Walker M. L., Knight P. J., Peckham M., Sellers J. R. (2009) A FERM domain autoregulates Drosophila myosin 7a activity. Proc. Natl. Acad. Sci. U.S.A. 106, 4189–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bloemink M. J., Geeves M. A. (2011) Shaking the myosin family tree. Biochemical kinetics defines four types of myosin motor. Semin. Cell Dev. Biol. 22, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cuda G., Pate E., Cooke R., Sellers J. R. (1997) In vitro actin filament sliding velocities produced by mixtures of different types of myosin. Biophys. J. 72, 1767–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janson L. W., Sellers J. R., Taylor D. L. (1992) Actin-binding proteins regulate the work performed by myosin II motors on single actin filaments. Cell Motil. Cytoskeleton 22, 274–280 [DOI] [PubMed] [Google Scholar]
- 41. Nagy A., Takagi Y., Billington N., Sun S. A., Hong D. K., Homsher E., Wang A., Sellers J. R. (2013) Kinetic characterization of nonmuscle myosin IIb at the single molecule level. J. Biol. Chem. 288, 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hsu R. M., Tsai M. H., Hsieh Y. J., Lyu P. C., Yu J. S. (2010) Identification of MYO18A as a novel interacting partner of the PAK2/βPIX/GIT1 complex and its potential function in modulating epithelial cell migration. Mol. Biol. Cell 21, 287–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koestler S. A., Rottner K., Lai F., Block J., Vinzenz M., Small J. V. (2009) F- and G-actin concentrations in lamellipodia of moving cells. PLoS One 4, e4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sellers J. R. (1985) Mechanism of the phosphorylation-dependent regulation of smooth muscle heavy meromyosin. J. Biol. Chem. 260, 15815–15819 [PubMed] [Google Scholar]
- 45. Trybus K. M. (1989) Filamentous smooth muscle myosin is regulated by phosphorylation. J. Cell Biol. 109, 2887–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cremo C. R., Wang F., Facemyer K., Sellers J. R. (2001) Phosphorylation-dependent regulation is absent in a nonmuscle heavy meromyosin construct with one complete head and one head lacking the motor domain. J. Biol. Chem. 276, 41465–41472 [DOI] [PubMed] [Google Scholar]
- 47. Wendt T., Taylor D., Messier T., Trybus K. M., Taylor K. A. (1999) Visualization of head-head interactions in the inhibited state of smooth muscle myosin. J. Cell Biol. 147, 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sweeney H. L., Chen L. Q., Trybus K. M. (2000) Regulation of asymmetric smooth muscle myosin II molecules. J. Biol. Chem. 275, 41273–41277 [DOI] [PubMed] [Google Scholar]
- 49. Adelstein R. S., Conti M. A. (1975) Phosphorylation of platelet myosin increases actin-activated myosin ATPase activity. Nature 256, 597–598 [DOI] [PubMed] [Google Scholar]
- 50. Reubold T. F., Eschenburg S., Becker A., Kull F. J., Manstein D. J. (2003) A structural model for actin-induced nucleotide release in myosin. Nat. Struct. Biol. 10, 826–830 [DOI] [PubMed] [Google Scholar]
- 51. Furch M., Geeves M. A., Manstein D. J. (1998) Modulation of actin affinity and actomyosin adenosine triphosphatase by charge changes in the myosin motor domain. Biochemistry 37, 6317–6326 [DOI] [PubMed] [Google Scholar]
- 52. Onishi H., Morales M. F., Kojima S., Katoh K., Fujiwara K. (1997) Functional transitions in myosin. Role of highly conserved Gly and Glu residues in the active site. Biochemistry 36, 3767–3772 [DOI] [PubMed] [Google Scholar]
- 53. Li X. D., Rhodes T. E., Ikebe R., Kambara T., White H. D., Ikebe M. (1998) Effects of mutations in the γ-phosphate binding site of myosin on its motor function. J. Biol. Chem. 273, 27404–27411 [DOI] [PubMed] [Google Scholar]
- 54. Shimada T., Sasaki N., Ohkura R., Sutoh K. (1997) Alanine scanning mutagenesis of the switch I region in the ATPase site of Dictyostelium discoideum myosin II. Biochemistry 36, 14037–14043 [DOI] [PubMed] [Google Scholar]
- 55. Cochran J. C., Sindelar C. V., Mulko N. K., Collins K. A., Kong S. E., Hawley R. S., Kull F. J. (2009) ATPase cycle of the nonmotile kinesin NOD allows microtubule end tracking and drives chromosome movement. Cell 136, 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parke C. L., Wojcik E. J., Kim S., Worthylake D. K. (2010) ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J. Biol. Chem. 285, 5859–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patterson B., Ruppel K. M., Wu Y., Spudich J. A. (1997) Cold-sensitive mutants G680V and G691C of Dictyostelium myosin II confer dramatically different biochemical defects. J. Biol. Chem. 272, 27612–27617 [DOI] [PubMed] [Google Scholar]
- 58. Preller M., Bauer S., Adamek N., Fujita-Becker S., Fedorov R., Geeves M. A., Manstein D. J. (2011) Structural basis for the allosteric interference of myosin function by reactive thiol region mutations G680A and G680V. J. Biol. Chem. 286, 35051–35060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Várkuti B. H., Yang Z., Kintses B., Erdélyi P., Bárdos-Nagy I., Kovács A. L., Hári P., Kellermayer M., Vellai T., Málnási-Csizmadia A. (2012) A novel actin binding site of myosin required for effective muscle contraction. Nat. Struct. Mol. Biol. 19, 299–306 [DOI] [PubMed] [Google Scholar]
- 60. Sweeney H. L., Houdusse A. (2010) Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys. 39, 539–557 [DOI] [PubMed] [Google Scholar]
- 61. Gyimesi M., Kintses B., Bodor A., Perczel A., Fischer S., Bagshaw C. R., Málnási-Csizmadia A. (2008) The mechanism of the reverse recovery step, phosphate release, and actin activation of Dictyostelium myosin II. J. Biol. Chem. 283, 8153–8163 [DOI] [PubMed] [Google Scholar]
- 62. Tóth J., Kovács M., Wang F., Nyitray L., Sellers J. R. (2005) Myosin V from Drosophila reveals diversity of motor mechanisms within the myosin V family. J. Biol. Chem. 280, 30594–30603 [DOI] [PubMed] [Google Scholar]
- 63. Suijkerbuijk S. J., van Dam T. J., Karagöz G. E., von Castelmur E., Hubner N. C., Duarte A. M., Vleugel M., Perrakis A., Rüdiger S. G., Snel B., Kops G. J. (2012) The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev. Cell 22, 1321–1329 [DOI] [PubMed] [Google Scholar]
- 64. Komatsu S., Yano T., Shibata M., Tuft R. A., Ikebe M. (2000) Effects of the regulatory light chain phosphorylation of myosin II on mitosis and cytokinesis of mammalian cells. J. Biol. Chem. 275, 34512–34520 [DOI] [PubMed] [Google Scholar]
- 65. Peterson L. J., Rajfur Z., Maddox A. S., Freel C. D., Chen Y., Edlund M., Otey C., Burridge K. (2004) Simultaneous stretching and contraction of stress fibers in vivo. Mol. Biol. Cell 15, 3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Uchimura T., Fumoto K., Yamamoto Y., Ueda K., Hosoya H. (2002) Spatial localization of mono- and diphosphorylated myosin II regulatory light chain at the leading edge of motile HeLa cells. Cell Struct. Funct. 27, 479–486 [DOI] [PubMed] [Google Scholar]
- 67. Beach J. R., Licate L. S., Crish J. F., Egelhoff T. T. (2011) Analysis of the role of Ser1/Ser2/Thr9 phosphorylation on myosin II assembly and function in live cells. BMC. Cell Biol. 12, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Allingham J. S., Sproul L. R., Rayment I., Gilbert S. P. (2007) Vik1 modulates microtubule-Kar3 interactions through a motor domain that lacks an active site. Cell 128, 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gourinath S., Himmel D. M., Brown J. H., Reshetnikova L., Szent-Györgyi A. G., Cohen C. (2003) Crystal structure of scallop Myosin s1 in the pre-power stroke state to 2.6 Å resolution. Flexibility and function in the head. Structure. 11, 1621–1627 [DOI] [PubMed] [Google Scholar]
- 70. Risal D., Gourinath S., Himmel D. M., Szent-Györgyi A. G., Cohen C. (2004) Myosin subfragment 1 structures reveal a partially bound nucleotide and a complex salt bridge that helps couple nucleotide and actin binding. Proc. Natl. Acad. Sci. U.S.A. 101, 8930–8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Himmel D. M., Gourinath S., Reshetnikova L., Shen Y., Szent-Györgyi A. G., Cohen C. (2002) Crystallographic findings on the internally uncoupled and near-rigor states of myosin. Further insights into the mechanics of the motor. Proc. Natl. Acad. Sci. U.S.A. 99, 12645–12650 [DOI] [PMC free article] [PubMed] [Google Scholar]