

Abstract
Mutations in the genes encoding leucine-rich repeat kinase 2 (LRRK2) and α-synuclein are associated with both autosomal dominant and idiopathic forms of Parkinson’s disease (PD). α-Synuclein is the main protein in Lewy bodies, hallmark inclusions present in both sporadic and familial PD. We show that in PD brain tissue, the levels of LRRK2 are positively related to the increase in α-synuclein phosphorylation and aggregation in affected brain regions (amygdala and anterior cingulate cortex), but not in the unaffected visual cortex. In disease-affected regions, we show co-localization of these two proteins in neurons and Lewy body inclusions. Further, in vitro experiments show a molecular interaction between α-synuclein and LRRK2 under endogenous and over-expression conditions. In a cell culture model of α-synuclein inclusion formation, LRRK2 co-localizes with the α-synuclein inclusions, and knocking down LRRK2 increases the number of smaller inclusions. In addition to providing strong evidence for an interaction between LRRK2 and α-synuclein, our results shed light on the complex relationship between these two proteins in the brains of patients with PD and the underlying molecular mechanisms of the disease.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-012-0984-y) contains supplementary material, which is available to authorized users.
Keywords: LRKK2, α-Synuclein, Parkinson’s disease, Lewy bodies, Interaction
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder resulting from the loss of dopamine neurons in the substantia nigra and the abnormal deposition of cytoplasmic inclusions known as Lewy bodies and Lewy neurites in widespread regions of the brain [1]. The aetiology of PD is multifactorial, with a growing number of genetic abnormalities identified [2]. The first PD causative gene was α-synuclein (SNCA), which encodes for the presynaptic protein α-synuclein [3]. Rare patients have missense mutations (A53T, A30P, and E46K) or multiplications of SNCA [2], but all PD patients accumulate phosphorylated α-synuclein in the form Lewy pathologies [4, 5]. Leucine-rich repeat kinase 2 (LRRK2) has been identified as the second and more common gene responsible for autosomal-dominant PD [6–9]. The function of the large LRRK2 protein remains unclear, although its serine–threonine/tyrosine kinase function is considered most important for PD aetiology due to the occurrence of the most common LRRK2 mutation (G2019S) in this domain [2]. As phosphorylation of α-synuclein is central to PD and the most common autosomal-dominant mutation occurs in a kinase, there has been intense debate about whether α-synuclein physically interacts with LRRK2 and whether it might be one of its substrates [10]. However, to date, only one report has shown that α-synuclein interacts with, and is phosphorylated by, LRRK2 and only under pathological and non-physiological oxidative stress conditions [11].
Co-immunoprecipitation is the gold standard for assessing direct protein interactions but relies on antibody specificity, a previous problem for LRRK2 antibodies that has been recently solved with the aid of resources from the Michael J. Fox Foundation (MJFF). It is now possible to revisit the question of a LRRK2 and α-synuclein interaction using these new and well-characterized LRRK2 antibodies. The aim of the present study was to establish whether LRRK2 and α-synuclein interact in human brain samples and to investigate the significance of the interaction in cell models. We report a molecular interaction between LRRK2 and α-synuclein under endogenous and over-expression conditions. We show in affected PD brain regions that the amount of LRKK2 protein is increased in association with increasing levels of phosphorylated α-synuclein. At the neuronal level, we confirm co-localization of LRRK2 and α-synuclein in Lewy bodies in PD patients and show co-localization in a cell model of α-synuclein inclusion formation. In addition, knockdown of LRRK2 in this cell model increases the number but reduces the size of α-synuclein inclusions. Altogether, our data provide strong evidence for an interaction between LRRK2 and α-synuclein in PD and opens novel avenues for the investigation of the interplay between different PD genes and their exploitation as targets for therapeutic intervention.
Materials and methods
Human and mouse brain samples
Human brain tissue was obtained from the Sydney Brain Bank and the NSW Tissue Resource Centre as part of the Australian Brain Bank Network funded by the National Health and Medical Research Council of Australia (NHMRC) with appropriate institutional ethics approvals. Frozen brain tissue samples and formalin-fixed paraffin-embedded tissue sections from different brain regions considered to be progressively affected by α-synuclein deposition in PD [12] were received from ten sporadic PD cases and ten matched controls (see Supplementary Information). The regions were the amygdala (affected pre-clinically in PD), the midbrain and anterior cingulate cortex (affected when symptoms are apparent) and the visual cortex (remains free of α-synuclein pathology even at end-stage disease).
Crude soluble human brain proteins were extracted from the frozen tissue as previously described [13]. Briefly, tissue was homogenized with a pre-chilled dounce homogenizer using ice-cold tris-buffered saline (TBS, pH 7.4) lyses buffer (LB) containing protease and phosphatase inhibitor cocktails (Roche, Dee Why, Australia and Thermo Fisher Scientific, Waltham, MA, USA). The TBS-soluble supernatant fraction was collected after centrifugation at 16,000×g for 25 min at 4 °C, and the pellets were solubilised in LB containing 5 % SDS (SDS-soluble fraction). Protein concentration was measured using a Nanodrop1000 (Thermo scientific) for all samples. Ethics approval for the human tissue studies was from the University of New South Wales Human Research Ethics Committee.
Frozen mouse brain samples from LRRK2 knockout C57BL/6J adult mice and age-matched controls were kindly provided by Dr. Mark Cookson and Dr. Iakov Rudenko (NIH (NIA), Bethesda, MD, USA). Mouse brain tissue was lysed in RIPA buffer (25 mM Tris–HCl pH 7.6; 150 mM NaCl; 0.1 % SDS; 1 % NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche diagnostics, Mannheim, Germany) using a mechanic homogenizer. Lysates were incubated in a rotor for 1 h at 4 °C and then sonicated. Following centrifuge separation (at 10,000×g for 10 min at 4 °C), the supernatants were kept and total protein concentration quantified using BCA assay (Thermo Fisher Scientific, Rockford, IL, USA).
Immunoprecipitation and western blot analyses
Immunoprecipitation experiments were performed using 1 mg (cells) or 6 mg (brain) of total protein. Lysates were pre-cleared by incubation with 20 μl of protein G beads (Invitrogen, Barcelona, Spain) for 30 min at 4 °C in rotation. Supernatants were recovered and incubated with 2 μg of the corresponding immunoprecipitation antibody: anti-α-synuclein (C-20, Santa Cruz Biotechnologies, Santa Cruz, CA, USA), anti-Myc (Cell Signaling, Danvers, MA, USA) or anti-LRRK2 (c41-2 MJFF), followed by overnight rotation at 4 °C. The next day, 40 μl of protein G beads were added for 3 h in a rotator at 4 °C. Beads were washed 5× with immunoprecipitation buffer, then re-suspended in 20 μl of protein sample buffer (50 mM Tris–HCl pH 6.8; 2 % SDS; 10 % glycerol; 1 % β-mercaptoethanol; 0.02 % bromophenol blue) and boiled at 95 °C for 5 min. Supernatants were resolved on 12 % SDS-PAGE gels. Proteins were transferred overnight to nitrocellulose membranes and blocked in 5 % non-fat dry milk in TBS-Tween for 1 h. The membranes were incubated overnight at 4 °C with the primary antibodies using the following dilutions: anti-α-synuclein (syn-1, BD Biosciences, San Jose, CA, USA, 1:1,000), anti-Myc (Cell Signaling, Danvers, MA, USA, 1:4,000) and anti-LRRK2 (c41-2 MJFF, 1:1,000). Immunoblots were washed with TBS-Tween and incubated for 1 h at room temperature with the corresponding HRP-labelled secondary antibody (GE Healthcare, Bucks, UK, 1:10,000). Immunoreactivity was visualised by chemiluminescence using an ECL detection system (Millipore, Billerica, MA, USA) and subsequent exposure to autoradiographic film.
Standard Western blotting was used to assess the relative amounts of LRRK2 [14] and α-synuclein [15] in 25 μg of the TBS-soluble protein from human brain samples compared with β-actin (loading control). The following primary antibodies were used: LRRK2 (c41-2, MJFF, 1:1,000), anti-α-synuclein (syn-1, BD Biosciences, San Jose, CA, USA, 1:2,000), anti-S129 phosphorylated α-synuclein (Elan Pharmaceuticals Inc. [16], 1:10,000 or WAKO, Richmond, VA, USA, 1:10,000) and anti-mouse β-actin (Sapphire Biosciences, Waterloo, Australia, 1:10,000). LRRK2 protein was separated on pre-cast NuPAGE 3–8 % gradient Tris–acetate gels (Invitrogen, Carlsbad, CA, USA) with constant voltage of 150 V for 50 min, and transferred onto polyvinylidene difluoride membranes (BioRad Laboratories, Hercules, CA, USA) at 30 V constant for 2 hours. α-Synuclein and S129 phosphorylated α-synuclein was separated on 12 % SDS-PAGE gels with constant voltage of 100 V for 90 min, and transferred onto 0.22-μm nitrocellulose membranes (BioRad Laboratories, Hercules, CA, USA) at 75 V constant for 45 min. To normalise the data between the different gels, the same control sample was loaded on all gels and probed for β-actin in addition to the proteins of interest. Immunoreactivity was visualised by chemiluminescence using an ECL detection system (GE Healthcare Biosciences, Pittsburgh, PA, USA) and the intensity of each band quantified using ImageJ software (http://rsbweb.nih.gov/ij/) with the relative expression normalised to the β-actin of the internal standard. Multivariate linear regression analysis (SPSS IBM, New York, NY, USA) was used to identify any differences in protein levels between groups and regions, and linear regression modelling (SPSS IBM, New York, NY, USA) was used to determine if LRRK2 and α-synuclein protein levels were related to each other and could predict group status. Age and post-mortem interval were co-factored into all analyses. The mean difference and standard error are given for all values.
Routine cell culture, plasmids and transfections
Both wild-type (WT) and G2019S forms of LRRK2 plasmids (pCMV-Tag3B-2xMyc-LRRK2, a kind gift from Dr. Mark Cookson, NIH (NIA), Bethesda, MD, USA) and WT pSI-α-synuclein plasmids (a kind gift from Dr. Bradley Hyman, Massachusetts General Hospital, USA) were used to over-express proteins for co-immunoprecipitation experiments.
Human embryonic kidney 293 cells (HEK-293) were cultured in DMEM media (Invitrogen, Barcelona, Spain) supplemented with 10 % foetal bovine serum and 1 % penicillin–streptomycin in 5 % CO2 at 37 °C. One day before transfection, 1.5 × 106 cells were seeded in 10-cm plates. Cells were transiently transfected using a total of 6 μg of plasmid DNA using FuGENE®6 (Roche diagnostics, Mannheim, Germany). Forty-eight hours later, cells were washed with PBS, harvested in immunoprecipitation buffer supplemented with protease and phosphatase inhibitors and sonicated. Lysates were centrifuged at 10,000×g for 10 min at 4 °C. Pellets were discarded and the total protein concentration of the supernatants quantified using BCA assay (Pierce).
α-Synuclein aggregation model in H4 cells
A gene construct encoding for tagged version of α-synuclein (SynT, a kind gift from Dr. Bradley Hyman, Massachusetts General Hospital, USA) was co-transfected with the synphilin-1 into H4 cells to recreate Lewy body-like inclusions, as previously described [17]. Briefly, H4 cells were cultured in OPTIMEM media (Gibco, Invitrogen, Barcelona, Spain) supplemented with 10 % of foetal bovine serum and 1 % of penicillin-streptomycin in an atmosphere of 37 °C and 5 % CO2. Twenty-four hours before the transfection, 2.0 × 105 cells were seeded in a 35-cm dish (Ibidi, Munich, Germany). Cells were transfected with 2 μg of each synphilin-1 and SynT plasmids using FuGENE®6 (Roche diagnostics, Mannheim, Germany). After transfection, cells were maintained for 48 h prior to further manipulations. H4 cells with reduced LRRK2 expression were created using lentiviral particles encoding LRRK2 shRNAs or a control scramble shRNA sequence (see Supplementary Information) and the model for α-synuclein inclusions (described above) recreated in H4-LRRK2 knockdown cell lines.
Confocal microscopy in cells and tissue sections
LRRK2 and α-synuclein localization was performed in human tissue sections of the midbrain and anterior cingulate cortex and in transfected H4 cells with and without shRNA knockdown of LRRK2 expression. In the human tissue sections, both routine peroxidase immunohistochemistry and double immunofluorescence were performed. In H4 cells, single and double immunofluorescence were performed. Details of experiments showing antibody specificity are provided in Supplementary Information.
Adjacent human sections were pre-treated with 99 % formic acid for 3 min and citrate buffer (pH 6.0) for 3 min, then incubated sequentially with anti-LRRK2 (MJFF c41-2, 1:200 and L955 Abgent, 1:500), anti-α-synuclein (BD Biosciences, San Jose, CA, USA, 1:200) and anti-S129 phosphorylated α-synuclein (Elan Pharmaceuticals Inc. [16], 1:10,000) antibodies, biotinylated secondary antibodies (anti-mouse IgG for α-synuclein and anti-rabbit IgG for LRRK2; Vector, Burlingame, CA, USA), and then the avidin–biotin complex (Vectastain Elite ABC Kit, Vector, Burlingame, CA, USA) prior to visualisation with DAB substrate (Sigma, St. Louis, MO, USA) in 0.1 % H2O2. Sections were counterstained with cresyl violet.
LRRK2 and α-synuclein were co-localized in human sections and the H4 cell α-synuclein aggregation model using double immunofluorescence. Briefly, cells were washed, fixed with 4 % PFA, permeabilized with 0.5 % Triton, blocked with 1.5 % normal goat serum, then incubated in anti-α-synuclein (BD Biosciences, San Jose, CA, USA, 1:1,000) and anti-LRRK2-2 (MJFF c41-2, 1:50) antibodies, while human sections were pre-treated as above and incubated with anti-α-synuclein (BD Biosciences, San Jose, CA, USA, 1:200) and anti-LRRK2 (L955 Abgent, 1:500) antibodies or with anti-S129 phosphorylated α-synuclein (Elan, 1:10,000) and anti-LRRK2 (L955 Abgent, 1:500) antibodies. Then, a cocktail of secondary antibodies was used: for α-synuclein and S129 phosphorylated α-synuclein anti-mouse IgG conjugated with Alexa Fluor 488 (Molecular probes, Eugene, OR, USA, 1:500) and for LRRK2 anti-rabbit IgG conjugated with Alexa Fluor 568 (Molecular Probes, 1:250). Fluorescent images were captured either using a Nikon Microscope ECLIPSE 90i confocal microscope (for human tissue sections) or using a Leica Microsystems confocal microscope (for H4 cells).
The proportion of neurons in the human brain sections that co-localized LRRK2 and α-synuclein was quantified in each section (total number of LRRK2 positive neurons/the total number of α-synuclein positive neurons) and double labelling of Lewy bodies assessed (average number sampled/section varied from 1 to 63, depending on the region assessed). Pearson correlation coefficients were used to determine whether there was any relationship between the numbers of Lewy bodies containing α-synuclein and those also containing LRRK2 in the PD cases examined. Quantification of the aggregation pattern of α-synuclein inclusions in H4 cells was performed. Briefly, for each condition (control and LRRK2-KD), a total of 40–60 cells containing α-synuclein inclusions were analysed, and a total of three independent experiments were performed. Cells were classified into two groups: cells with <5 inclusions and cells with ≥5 inclusions, and the results were expressed as a percentage of the total number of cells with inclusions. The average size of inclusions per cell was also quantified using the ImageJ software (http://rsbweb.nih.gov/ij/).
Results
Co-immunoprecipitation of LRRK2 and α-synuclein
In order to investigate the interaction between LRRK2 and α-synuclein, we used mouse brain samples from WT and LRRK2 knockout animals. The immunoprecipitation of α-synuclein from mouse brain lysates pulled down LRRK2 in WT samples, but not in knockout samples (Fig. 1a). We also verified the interaction between LRRK2 and α-synuclein when the immunoprecipitations were performed in human brain lysates (data not shown).
Fig. 1.
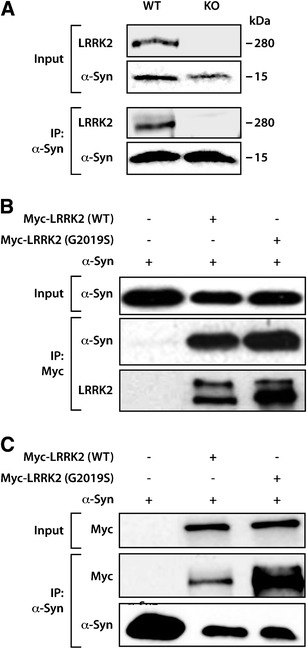
Co-immunoprecipitation of LRRK2 and α-synuclein. a Western blots showing the immunoprecipitation of endogenous α-synuclein in lysates from WT and LRRK2 knockout mouse brains. The co-immunoprecipitation with endogenous LRRK2 occurs in WT but not in the LRRK2 knockout brain sample. b, c Over-expression of Myc-LRRK2 (WT or G2019S) together with α-synuclein in HEK-293 cells showed the co-immunoprecipitation of LRRK2 (WT or G2019S) with α-synuclein using anti-Myc as the capture antibody and anti-α-synuclein and anti-LRRK2 antibodies for Western blotting (b) or using anti-α-synuclein as the capture antibody and anti-α-synuclein and anti-Myc antibodies for Western blotting (c)
In order to investigate whether LRRK2 mutations alter the interaction with α-synuclein, we over-expressed WT or G2019S mutant LRRK2 together with α-synuclein in HEK-293 cells. Immunoprecipitation of LRRK2 from cells over-expressing Myc-LRRK2 (WT or G2019S mutant) together with α-synuclein pulled down α-synuclein (Fig. 1b). Consistently, when α-synuclein was immunoprecipitated, the LRRK2 proteins (WT and G2019S) were also co-immunoprecipitated (Fig. 1c). We did not find significant alterations in the pattern of co-immunoprecipitation between WT and G2019S mutant, indicating that the interaction between the two proteins is not disturbed by this mutation.
Co-localization of LRRK2 and α-synuclein in PD brain and cell model
PD brain samples were examined to determine whether LRRK2 and α-synuclein or phosphorylated α-synuclein were co-localized. We found that LRRK2 increases along with α-synuclein in neurons prior to Lewy body formation (Fig. 2a) as well as depositing in some but not all of the hallmark inclusions (Fig. 2b–d). LRRK2 was also observed in phosphorylated α-synuclein-immunoreactive inclusions, often centralized to a radiating pattern of phosphorylated α-synuclein fibrils (Fig. 2f–h). Quantitation of the numbers of α-synuclein inclusions immunopositive for LRRK2 in ten PD cases (Table 1) indicates that 60 % of cingulate Lewy bodies and 43 % of nigral Lewy bodies contained both proteins (Fig. 2b, c). The specificity of the co-localization can be taken as genuine, as no 280-kDa LRRK2 band was detected on Western blot and no immunoreactivity in tissue sections in peptide pre-absorption experiments (see Supplementary Figure). There was no correlation between the number of α-synuclein-positive Lewy bodies and those also containing LRRK2 across the cases examined.
Fig. 2.
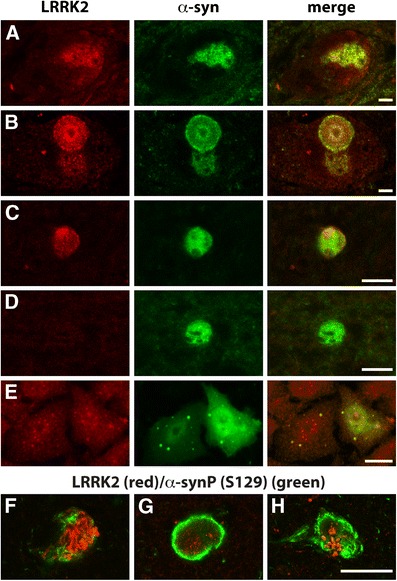
Co-localization of LRRK2 and α-synuclein in PD brain and cell models. In PD brains (a–d, f–g), merged images clearly outline single neurons in the substantia nigra (a, b) and Lewy bodies (b–d, f–g) using double-labelling immunofluorescence. There is an increase of LRRK2 and α-synuclein immunoreactivity in brainstem neurons without Lewy body formation (a), with LRRK2 co-localizing with α-synuclein in Lewy bodies (donut inclusion in b) in these neurons. The co-localisation of LRRK2 and α-synuclein was also observed in cortical Lewy bodies (c). Cortical Lewy bodies without LRRK2 immunoreactivity were also observed (d). S129 phosphorylated α-synuclein antibody also confirmed co-localisation of LRRK2 with phosphorylated α-synuclein, with LRRK2 often centralized to a radiating pattern of phosphorylated α-synuclein fibrils (f–h). In the H4 cell model, double-labelling immunofluorescence for α-synuclein inclusion formation shows that endogenous LRRK2 co-localizes with α-synuclein inclusions (e). Scales in all panels are equivalent to 10 μm
We also interrogated an in vitro model that reproduces the formation of α-synuclein inclusions in H4 cells [17]. Using this model, we observed co-localization of endogenous LRRK2 with the α-synuclein-positive inclusions (Fig. 2e).
Knocking down LRRK2 expression reduces α-synuclein aggregation
To further investigate the effect of LRRK2 on α-synuclein aggregation, LRRK2 expression was knocked down using shRNAs in the H4 cell model. Knocking down LRRK2 expression did not produce significant changes in endogenous α-synuclein or phosphorylated α-synuclein levels (Fig. 3a). Transiently transfecting these LRRK2-deficient cells with SynT and synphilin-1 expression plasmids showed that LRRK2 silencing significantly increased the number and decreased the size of α-synuclein inclusions resulting in a greater number of cells bearing smaller α-synuclein inclusions (Fig. 3b–d).
Fig. 3.
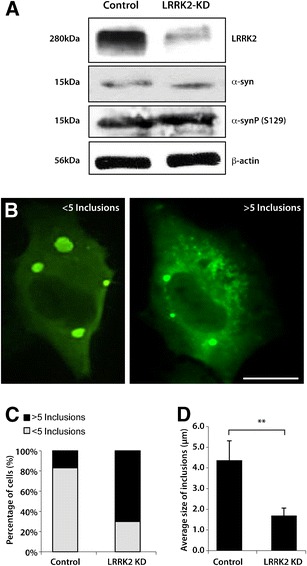
Knockdown of LRRK2 expression alters the size and number of α-synuclein inclusions. a Western blots showing that H4 cells infected with LRRK2-shRNA have the expected knockdown of LRRK2 protein (LRRK2-KD) compared with the scramble shRNA control, but have no significant change on the level of endogenous α-synuclein or phosphorylated α-synuclein at S129. b The model for α-synuclein inclusions was reproduced in a LRRK2 knockdown cell line and in parental control cells. Cells were classified into two groups according to the number of α-synuclein-immunoreactive inclusions observed: cells with five or more inclusions and cells with less than five inclusions. Scale bar = 10 μm. c Data from three independent experiments shows a greater proportion of cells containing five or more inclusions in the LRRK2 knockdown cells compared with controls. d LRRK2 silencing (LRRK2-KD) promotes a significant reduction in the average size of the inclusions, resulting in a more punctate aggregation pattern in the cells. Student’s test (n = 3; **p < 0.01). Error bars = SEM
Correlations between the levels of α-synuclein and LRRK2 in PD
To explore the relationship between protein levels of LRRK2 and α-synuclein in PD, 20 cases (controls and Braak PD stages IV and V, Supplementary Table) were analysed. Multivariate analysis factoring in age and post-mortem delay showed that the levels of total and phosphorylated α-synuclein were significantly increased over control levels only in PD brain regions with Lewy bodies (p < 0.001). In the cases examined, all stage IV cases had high Lewy body densities in the amygdala (Fig. 4a), while significant densities of cingulate Lewy bodies were observed in all stage V cases (Fig. 4b). No Lewy bodies were seen in the visual cortex of any case, although very small phosphorylated deposits were observed in stage V cases (Fig. 4c). In PD, there was a substantial 220 ± 20 % increase over controls in α-synuclein protein levels in the amygdala and a less substantial 48 ± 6 % increase in the cingulate cortex, with no change in the visual cortex (Fig. 4d, e). This pattern of regional increase in α-synuclein levels was even more striking when assessing phosphorylated α-synuclein protein levels (p = 0.01), as very low levels of phosphorylated α-synuclein were observed across all regions in controls compared to PD (Fig. 4e). There was a very large 60 ± 18-fold increase in phosphorylated α-synuclein protein levels in the PD amygdala, a 32 ± 4-fold increase in the PD cingulate cortex and an 8 ± 3-fold change in the PD visual cortex relative to controls (Fig. 4d, e).
Fig. 4.
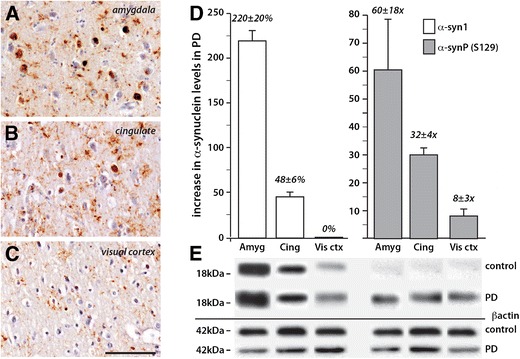
Increased levels of total and S129 phosphorylated α-synuclein in PD brain. a–c Peroxidase immunohistochemistry of brain sections from the same PD case showing the regional density of Lewy pathology as revealed by immunohistochemistry using phosphorylated α-synuclein antibody and counterstained with cresyl violet. Scale inc = 100 μm and is equivalent for a and b. Severe pathology is observed in the amygdala (a) with moderate pathology in the anterior cingulate cortex (b). Neuronal inclusions are not observed in the visual cortex (c). d, e Quantitation (d) of Western blots (e) in the same three brain regions in the PD cases (represented as an increase over control levels) confirmed the regional changes noted histologically in PD and showed considerably more phosphorylated α-synuclein compared with total α-synuclein in each regions (note the percentage at left versus fold change at right in d). Error bars = SEM
The expression of LRRK2 was analysed using the same methods in the same brain extracts (Fig. 5). Multivariate analysis co-varying for age or post-mortem delay showed that the levels of LRRK2 were increased in PD compared with controls in regions containing Lewy bodies (p < 0.04), with no difference between the LRRK2 levels in these Lewy body-containing regions (p = 0.6). Within these regions, there was a small but significant 23 ± 6 % increase over controls in full-length LRRK2 levels (Fig. 5a, b).
Fig. 5.
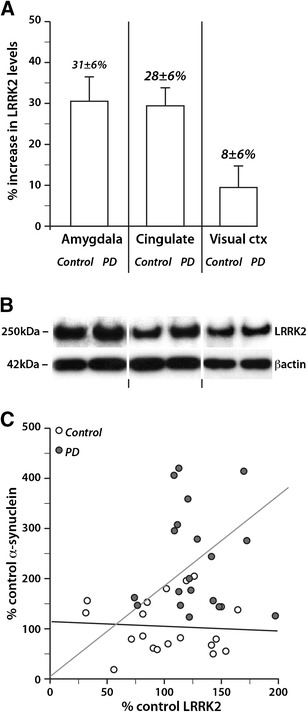
LRRK2 levels correlated with α-synuclein levels in PD brain. Quantitation (a) of LRRK2 Western blots (b) in the same brain regions in the PD cases (represented as an increase over control levels) and correlations with α-synuclein levels (c). The protein levels of LRRK2 were increased in the disease-affected areas (amygdala and cingulate) compared to the non-affected area (visual cortex) (a). Error bars = SEM. Multivariate analysis revealed a significant correlation between the increasing levels of α-synuclein and LRRK2 only in PD but not controls (c)
To determine the relationship between LRRK2 and α-synuclein levels and PD, linear regression modelling was used assessing the protein levels obtained in the amygdala and cingulate cortex. This analysis revealed that increasing levels of LRRK2 and total and phosphorylated α-synuclein correlated with each other in PD but not controls (Fig. 5c, p < 0.001, β coefficients = 0.27, 0.33 and 0.37, respectively).
Discussion
Mutations in LRRK2 and α-synuclein proteins are known to be responsible for autosomal dominant forms of PD [2]. Due to the growing interest in the potential interaction of these proteins in the pathogenesis of PD [10, 18], we investigated such an interaction using a variety of techniques. Co-immunoprecipitation showed that endogenous LRRK2 and α-synuclein interact in cells, mouse and human brain tissue. We also confirmed this interaction in over-expression studies in HEK-293 cells. In this model, we found that the G2019S mutation did not alter the ability of LRRK2 to interact with α-synuclein. Nevertheless, we cannot exclude that the interaction with G2019S is not potentiated due to the over-expression of the protein in HEK cells. The G2019S mutation is located in the kinase domain of LRRK2, and shows an enhanced kinase activity compared to WT LRRK2 [19]. Our data indicate that the kinase domain and therefore the phosphorylation capacity of LRRK2 do not play a large role in its interaction with α-synuclein. This is consistent with recent evidence showing that the levels of rather than mutations in LRRK2 are related to the deposition of neuropathology [20]. Overall, these results unequivocally demonstrate, for the first time, a definite interaction between endogenous LRRK2 and α-synuclein, a finding that had only been detected under pathological and oxidative stress conditions [11].
In human PD brains, we show co-localization of LRRK2 and α-synuclein as well as S129 phosphorylated α-synuclein in Lewy bodies and also co-localization in neurons that have not formed Lewy bodies in Lewy body-producing regions. We have also replicated this co-localization of LRRK2 and α-synuclein in an established cell model for α-synuclein inclusion formation. Of interest, our quantitation in the PD cases showed that LRRK2 co-localized in more cortical compared with brainstem Lewy bodies. According to Braak PD staging [12], cortical Lewy bodies develop later in PD, further suggesting an early association between LRRK2 and α-synuclein in Lewy body formation. In these neurons, S129 phosphorylated α-synuclein fibrils often appeared to radiate from more centralized LRRK2 within Lewy bodies. Overall, our results suggest that the interaction between LRRK2 and α-synuclein or S129 phosphorylated α-synuclein is enhanced prior to and during the formation of α-synuclein aggregation and fibrilization. These data are also consistent with other studies in brain tissue showing the co-localization of LRRK2 in α-synuclein-immunoreactive Lewy bodies [21–23], although questions regarding the specificity of the different LRRK2 antibodies used have been raised [24, 25]. While variance in the numbers of Lewy bodies co-localizing LRRK2 may be due to the age of the Lewy bodies assessed (see above) and account for some of the differences described in the literature, we are certain of the specificity of the antibodies used in the current study, as a number of specificity experiments confirmed that the protein we localized to early forming Lewy bodies was LRRK2 (see Supplementary Figure).
To test this association further, we knocked down LRRK2 in a cell model of α-synuclein inclusion formation and found that reduced LRRK2 expression altered α-synuclein inclusions, resulting in an increased number of smaller inclusions per cell. In this model, α-synuclein is co-expressed and co-aggregates with synphilin-1, as observed in Lewy bodies [26, 27]. Synphilin-1 recruits and binds α-synuclein leading to inclusion formation [26], and synphilin-1 and 14-3-3 proteins accumulate with mutant α-synuclein in A53T transgenic mice [28]. While there is little indication that synphilin-1 closely associates with LRRK2, 14-3-3 proteins are not only known to interact with and stabilise phosphorylated LRRK2 [29] but also have a preference for binding S129 phosphorylated α-synuclein [30] and accumulate in Lewy bodies [31]. 14-3-3 proteins and α-synuclein have opposing effects on regulating the activity of many enzymes [31], and such regulation may occur within a complex associated with LRRK2. The microtubule binding protein tau phosphorylation complex has been shown to require both 14-3-3 and α-synuclein [32, 33], and it is of interest that over-expression of tau has a similar effect on the formation of α-synuclein inclusions in this cellular model [34] to the knockdown of LRRK2 (present study), as well as sequestering phosphorylated tau into the inclusions [34]. Either reducing the amount of unbound α-synuclein by enhanced recruitment into the tau phosphorylation complex and/or increased tau partnering of 14-3-3 to shift its binding from and decrease the phosphorylation and activity of LRRK2, would seem to produce similar effects on the formation of α-synuclein inclusions in this cellular model of inclusion formation. In mouse models, LRRK2 over-expression enhances the progression of α-synuclein-mediated neuropathological changes, and LRRK2 deletion delays the progression of pathology [20]. All of these data are consistent with an interaction between LRRK2 and α-synuclein in patients with PD.
This is the first study showing correlations between the relative protein levels of LRRK2 and phosphorylated and total α-synuclein in PD human brain tissue extracts, but not in controls. A small increase in the levels of LRRK2 in the brain tissue from PD patients directly correlated with much larger regional increases in α-synuclein levels, and more strikingly with a widespread α-synuclein S129 phosphorylation. In cell models, an increase in LRRK2 expression significantly increases α-synuclein mRNA [35], and elevated α-synuclein mRNA levels are co-regulated with increased LRRK2 transcription [36]. The positive feedback in turn activates the ERK signalling pathway leading to phosphorylation of α-synuclein [35]. It is of interest that in long-duration PD cases, the gene expression levels of both LRRK2 [37] and α-synuclein [38] are decreased in multiple brain regions forming Lewy bodies. This is possibly as a self-protective mechanism to the high levels of these proteins that accumulate within neurons in these regions, and suggests that deficits in protein degradation mechanisms play a significant role in the progression of pathology overtime.
Beyond the endogenous interaction of LRRK2 and α-synuclein that we have shown in this study, we are still not able to determine whether the nature of LRRK2 interaction with α-synuclein is a direct protein–protein binding or an indirect binding within a protein complex. As it has been extensively suggested, LRRK2 interacts with other proteins also implicated in PD to form protein complexes [10, 18, 20, 29, 37, 39]. While a fine analysis of the molecular determinants of the interaction between LRRK2 and α-synuclein is still required, our study has unequivocally established that there is an interaction between LRRK2 and α-synuclein, and that this interaction appears to be enhanced in patients with PD and in cell models of α-synuclein inclusion formation. Importantly, we also provide evidence showing that the levels of LRRK2 impact on α-synuclein pathology, consistent with studies in animal models of PD [20]. Ultimately, our work paves the way for the understanding of the molecular interplay between two central players in PD.
Electronic supplementary material
(PDF 2212 kb)
Acknowledgements
Human brain tissue samples were received from the Australian Brain Bank Network, which is supported by the NHMRC, specifically from the Sydney Brain Bank (also supported by Neuroscience Research Australia and the University of New South Wales) and the NSW Tissue Resource Centre (also supported by the University of Sydney, the Schizophrenia Research Institute and NIH (NIAAA) R24AA012725). The authors would like to thank Dr. Mark Cookson and Dr. Iakov Rudenko for their kind gift of LRRK2 knockout mouse tissue. This work was also supported by the NHMRC (510186, GH and YH), an EMBO Installation Grant and a Marie Curie International Reintegration Grant (Neurofold, TFO). The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-20013) under grant agreement No. 241791 European Project on Mendelian Forms of PD (MEFOPA). PSG is supported by a fellowship from the Fundação para a Ciência e a Tecnologia (SFRH/BD/61495/2009). YH is supported by a UNSW Goldstar Award. GMH has a NHMRC Senior Principal Research Fellowship 630434. S129 phosphorylated α-synuclein antibodies were kindly provided by Dr. John Anderson from Elan Pharmaceuticals, South San Francisco, CA, USA. LRRK2 antibodies were kindly provided by the Michael J. Fox Foundation. LRRK2 plasmids were a kind gift from Dr. Mark Cookson (NIH (NIA), Bethesda, MD, USA), and α-synuclein plasmids were a kind gift from Dr. Bradley Hyman (Massachusetts General Hospital, USA). We would also like to thank Heidi Cartwright for the preparation of the figures, and Dr. Federico Herrera, Dr. Rita Oliveira, Dr. John Anderson and Prof. Brett Garner for critical reading of the manuscript.
Conflicts of interest
The authors have no additional disclosures or conflicts of interest to declare.
Footnotes
Patrícia Silva Guerreiroa and Yue Huang are co-first authors.
Contributor Information
Tiago Fleming Outeiro, Email: touteiro@gmail.com.
Glenda Margaret Halliday, Phone: +61-2-93991704, FAX: +61-2-93991105, Email: g.halliday@neura.edu.au.
References
- 1.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 2.Gasser T. Mendelian forms of Parkinson’s disease. Biochim Biophys Acta. 2009;1792:587–596. doi: 10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 4.Wakabayashi K, Takahashi H. Pathology of familial Parkinson’s disease. Brain Nerve. 2007;59:851–864. [PubMed] [Google Scholar]
- 5.Cavallarin N, Vicario M, Negro A. The role of phosphorylation in synucleinopathies: focus on Parkinson’s disease. CNS Neurol Disord Drug Targets. 2010;9:471–481. doi: 10.2174/187152710791556140. [DOI] [PubMed] [Google Scholar]
- 6.Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, et al. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet. 2004;74:11–19. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Halliday GM, Vandebona H, Mellick GD, Mastaglia F, Stevens J, Kwok J, Garlepp M, Silburn PA, Horne MK, et al. Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson’s disease. Mov Disord. 2007;22:982–989. doi: 10.1002/mds.21477. [DOI] [PubMed] [Google Scholar]
- 9.Lesage S, Lohmann E, Tison F, Durif F, Durr A, Brice A. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J Med Genet. 2008;45:43–46. doi: 10.1136/jmg.2007.051854. [DOI] [PubMed] [Google Scholar]
- 10.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qing H, Wong W, McGeer EG, McGeer PL. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun. 2009;387:149–152. doi: 10.1016/j.bbrc.2009.06.142. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 13.Elliott DA, Tsoi K, Holinkova S, Chan SL, Kim WS, Halliday GM, Rye KA, Garner B. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol Aging. 2011;32:257–271. doi: 10.1016/j.neurobiolaging.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Pungaliya PP, Bai Y, Lipinski K, Anand VS, Sen S, Brown EL, Bates B, Reinhart PH, West AB, Hirst WD, et al. Identification and characterization of a leucine-rich repeat kinase 2 (LRRK2) consensus phosphorylation motif. PLoS One. 2010;5:e13672. doi: 10.1371/journal.pone.0013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Broe M, Huang Y, Anderson JP, Gai WP, Milward EA, Porritt M, Howells D, Hughes AJ, Wang X, et al. Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol. 2011;121:695–704. doi: 10.1007/s00401-011-0815-1. [DOI] [PubMed] [Google Scholar]
- 16.Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 17.Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun. 2006;351:631–638. doi: 10.1016/j.bbrc.2006.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greggio E, Bisaglia M, Civiero L, Bubacco L. Leucine-rich repeat kinase 2 and alpha-synuclein: intersecting pathways in the pathogenesis of Parkinson’s disease? Mol Neurodegener. 2011;6:6. doi: 10.1186/1750-1326-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alegre-Abarrategui J, Ansorge O, Esiri M, Wade-Martins R. LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol Appl Neurobiol. 2008;34:272–283. doi: 10.1111/j.1365-2990.2007.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Babar A, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Perry G, Chen SG. LRRK2 in Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2006;1:17. doi: 10.1186/1750-1326-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devine MJ, Lewis PA. Emerging pathways in genetic Parkinson’s disease: tangles, Lewy bodies and LRRK2. FEBS J. 2008;275:5748–5757. doi: 10.1111/j.1742-4658.2008.06707.x. [DOI] [PubMed] [Google Scholar]
- 24.Perry G, Zhu X, Babar AK, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Chen SG. Leucine-rich repeat kinase 2 colocalizes with alpha-synuclein in Parkinson’s disease, but not tau-containing deposits in tauopathies. Neurodegener Dis. 2008;5:222–224. doi: 10.1159/000113708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neuol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 26.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 27.Wakabayashi K, Engelender S, Yoshimoto M, Tsuji S, Ross CA, Takahashi H. Synphilin-1 is present in Lewy bodies in Parkinson’s disease. Ann Neurol. 2000;47:521–523. doi: 10.1002/1531-8249(200004)47:4<521::AID-ANA18>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 28.Shirakashi Y, Kawamoto Y, Tomimoto H, Takahashi R, Ihara M. alpha-Synuclein is colocalized with 14-3-3 and synphilin-1 in A53T transgenic mice. Acta Neuropathol. 2006;112:681–689. doi: 10.1007/s00401-006-0132-2. [DOI] [PubMed] [Google Scholar]
- 29.Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McFarland MA, Ellis CE, Markey SP, Nussbaum RL. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteom. 2008;7:2123–2137. doi: 10.1074/mcp.M800116-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- 32.Duka T, Duka V, Joyce JN, Sidhu A. Alpha-synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan Z, Agarwal-Mawal A, Paudel HK. 14-3-3 binds to and mediates phosphorylation of microtubule-associated tau protein by Ser9-phosphorylated glycogen synthase kinase 3beta in the brain. J Biol Chem. 2004;279:26105–26114. doi: 10.1074/jbc.M308298200. [DOI] [PubMed] [Google Scholar]
- 34.Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Goncalves SA, Pera M, Suarez-Calvet M, Clarimon J, Outeiro TF, Lleo A. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One. 2011;6:e26609. doi: 10.1371/journal.pone.0026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal. 2010;22:821–827. doi: 10.1016/j.cellsig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westerlund M, Ran C, Borgkvist A, Sterky FH, Lindqvist E, Lundstromer K, Pernold K, Brene S, Kallunki P, Fisone G, et al. Lrrk2 and alpha-synuclein are co-regulated in rodent striatum. Mol Cell Neurosci. 2008;39:586–591. doi: 10.1016/j.mcn.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, Bandopadhyay R, Lashley T, Renton AE, Kingsbury AE, Kumaran R, Kallis C, Vilarino-Guell C, O’Sullivan SS, Lees AJ, et al. LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: a morphological and quantitative study. Neuropathol Appl Neurobiol. 2011;37:777–790. doi: 10.1111/j.1365-2990.2011.01187.x. [DOI] [PubMed] [Google Scholar]
- 38.Simunovic F, Yi M, Wang Y, Macey L, Brown LT, Krichevsky AM, Andersen SL, Stephens RM, Benes FM, Sonntag KC. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain. 2009;132:1795–1809. doi: 10.1093/brain/awn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu G, Aliaga L, Cai H. Alpha-synuclein, LRRK2 and their interplay in Parkinson’s disease. Future Neurol. 2012;7:145–153. doi: 10.2217/fnl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 2212 kb)