

The plant genus Aglaia produces a number of secondary metabolites including the cyclopenta[b]benzofurans[1] rocaglamide 1, silvestrol 2, and cyclopenta[b,c]benzopyrans[2] (aglains) including ponapensin 3 (Figure 1). Cyclopenta[b]benzofuran natural products possess potent anticancer properties due to modulation of the activity of the RNA helicase eukaryotic initiation factor 4A (eIF4A), which is involved in loading ribosomes onto mRNA templates during translation initiation, a step frequently deregulated in cancer.[3] Due to its unusual structure and important biological activity, rocaglamide 1 has been targeted by many research groups and has inspired a number of elegant synthetic strategies.[4] We have reported a biomimetic approach to cyclopenta[b]benzofuran natural products involving a photocycloaddition/ketol shift rearrangement/reduction sequence using 3-hydroxyflavone (3-HF) derivatives such as 4 and methyl cinnamate 5a. This strategy enabled total syntheses of both 1 and 2 (Figure 1 and Scheme 1)[5] utilizing excited state intramolecular proton transfer (ESIPT) of 3-HF’s. Photoirradiation of 4 affords the oxidopyrylium intermediate 6 which undergoes [3+2] photocycloaddition with methyl cinnamate 5a to provide the corresponding aglain core 7a, which was converted to methyl rocaglate 8a in two steps (Scheme 1). Herein, we describe the scope of the photocycloaddition with various dipolarophiles, mechanistic and photophysical studies, and evaluation of the rocaglates produced as inhibitors of eukaryotic protein translation.
Figure 1.
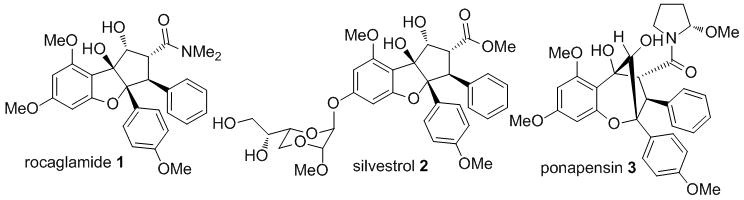
Representative aglain and rocaglate metabolites from Aglaia
Scheme 1.
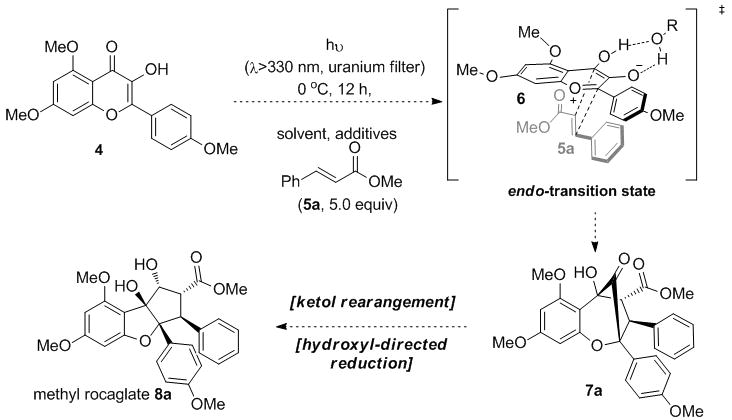
[3+2] Photocycloaddition approach to the cyclopenta[b]benzofurans
Although extensive photophysical studies concerning ESIPT of 3-HF derivatives have been conducted,[6] the reactivity of the resulting oxidopyrylium species (e.g. 6) still remains largely unexplored.[7] We first examined achiral photocycloadditions between 3-HF 4 and methyl cinnamate 5a in order to study the role of proton transfer in the ESIPT process. We initiated an extensive screening of H-bond donors, Brønsted acids and additives to optimize the cycloaddition of 3-HF 4 and methyl cinnamate 5a (0 °C, 12 h, λ > 330 nm to avoid photodimerization[8] of 5a).[9] Results of additive screening indicated that trifluoroethanol (TFE) improved both the isolated yield of cycloadduct 7a (55%) and endo-exo diastereoselectivity (5:1 dr).[9,10] Protic polar solvents may facilitate proton tunneling of the 3-HF excited state (N*), thereby stabilizing and increasing the population of the oxidopyrylium phototautomers (T*) 6/6′ (Figure 2).[6b–e] Indeed, excitation of 3-HF 4 in the presence of TFE resulted in a large ESIPT fluorescence emission band (λ (T*) = 530 nm) corresponding to oxidopyrylium phototautomers 6/6′.[9] The presence of a shoulder emission band (λ (N*) = 440 nm) may be attributed to excited state intramolecular charge transfer (ESICT) of 3-HF 4 by superposition with the emission band of the corresponding 3-methoxyflavone 9 (Figure 2).[9] After excitation of 3-HF 4, the normal excited form (N*) may undergo charge transfer to stabilize the accumulated dipole moment through ESICT resulting in phototautomers 6″/6‴ and a second λ(N*) emission.[11] Our photophysical data indicates that both excited state phototautomers 6/6′ and 6″/6‴ generated via ESICT and ESIPT, respectively, are present upon irradiation and may be of relevance in the photocycloaddition.
Figure 2.
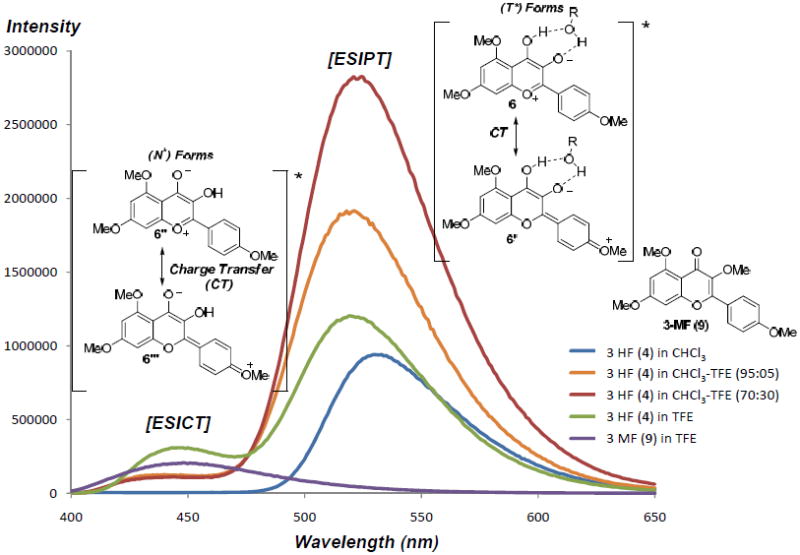
Fluorescence mission of 3-HF 4 and 3-MF 9 indicating both ESICT and ESIPT processes with presumed charge transfer in both excited states from phototautomers 6/6′ and 6″/6‴.
Using the optimum reaction conditions (CHCl3/TFE, 70:30), we next turned our attention to an evaluation of over 40 dipolarophiles 5 for their reactivity in the [3+2] photocycloaddition.[9] The set of dipolarophiles tested contained cinnamate derivatives modified at both termini (commercially available or readily prepared) and unactivated alkenes. We were pleased to find that a broad range of dipolarophiles were workable in photocycloadditions. Unfortunately, all β–alkylacrylate derivatives[9] proved to be unreactive under the reaction conditions (yield <10%), likely due to the lack of positive charge or radical stabilization at the β-position of the α,β-unsaturated ester (vide infra). As the use of TFE as cosolvent significantly increases the population of oxidopyrylium species 6/6′ (cf. Figure 2), less reactive dipolarophiles such as 5b–g were found to participate in the photocycloaddition. Accordingly, we were able to obtain novel cycloadducts including thioester 7b, imide 7d, Weinreb amide 7e, amide 7f, and cyanide 7g in moderate to good yields (Scheme 2). Use of electron withdrawing groups for LUMO lowering of dipolarophiles 5b–g appears to impact the yield of the reaction (e.g. 26% for 7d; 59% for 7g). Different reactivity was observed for ethyl and phenyl thioesters 5b and 5c (40% and 0% yield, 7b/7c respectively, which was in agreement with fluorescence quenching experiments[9]) indicating that steric factors may greatly influence photocycloadditions. Steric hindrance at the terminus of α,α-disubstituted dipolarophiles 5s–t may also be responsible for their lack of reactivity in photocycloadditions (Scheme 2, see inset). On the other hand, the substitution pattern of the aryl substituents (5h–r) appears to be very flexible. Interestingly, the ortho substituent present in 5j did not lead to favorable cycloaddition, whereas the para-substituted dipolarophile 5k afforded the desired aglain cycloadduct 7k (71%). Other electronic effects for reaction partners (5k–r) did not affect the course of the reaction leading to isolation of cycloadducts 8i–r.
Scheme 2.
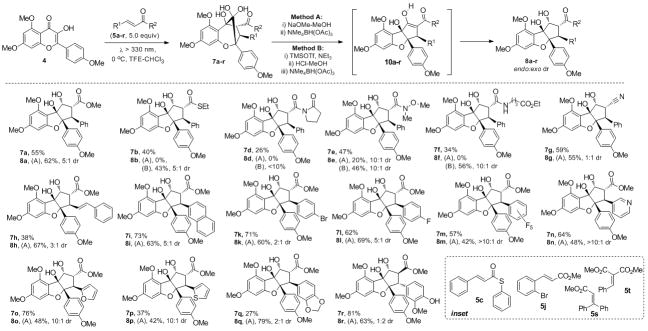
Scope of the [3+2] photocycloaddition to produce aglains 7a–r and rocaglates 8a–r (major diastereomer shown, dr obtained by 1H NMR integration)
Comparison of dipolarophile reactivity also revealed new insights concerning photocycloaddition diastereoselection. Unexpectedly, reaction of cinnamate 5r bearing a free phenol gave an inverse endo-exo diastereoselectivity (1:2 dr) of rocaglate 8r. In comparison, cinnamate 5q lacking a free phenol led to the formation of rocaglate 8q (2:1 dr) favoring the endo diastereomer. Interestingly, reaction of dienoate 5h resulted in complete chemo- and regioselectivity in the [3+2] photocycloaddition for the α,β-unsaturated moiety with moderate endo-exo diastereoselectivity (3:1 dr) affording cycloadduct 7h (38%). Examination of the diastereoselectivity outcome of rocaglate adducts 8k–m (from 2:1 to 5:1 to 10:1 dr) revealed an interesting trend for the dipolarophiles. These results suggest that electron poor aryl substituents at the β-position of the cinnamate (cf. Figure 3, 5m) may be involved in donor-acceptor (π-stacking) interactions[13] with the electron rich aromatic ring of the oxidopyrylium 6 in the excited state, thereby reinforcing substrate interactions in the endo transition state. The corresponding acceptor-donor interactions may also be considered wherein the charged delocalized oxidopyrylium phototautomer 6′ may interact with electron rich cinnamates (cf. Figure 3, 5o).[13] An equilibrium between phototautomers 6 and 6′ may explain the high diastereoselectivity observed for both electron-deficient (5l–m, 5–10:1 dr) and electron-rich cinnamates (5i, 5o–p, 10:1 dr).
Figure 3.
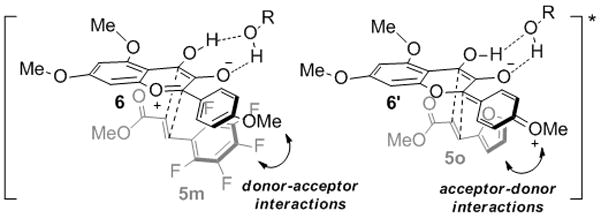
Proposed endo transition state arrangements from phototautomers 6/6′ in the excited state with dipolarophilese 5m and 5o
During our study, aglain derivative 7a was isolated as a solid material, encouraging us to separate the major endo-diastereomer by crystallization. Interestingly, the aglain core was found to exist preferentially as a hydrated bridgehead ketone (also observed for other aglain derivatives[9]) which may result from double hydrogen bond stabilization of the hydrate with the benzopyran ether oxygen and α-hydroxyl moieties (Figure 4).[14] Such stabilization highlights the high degree of electrophilicity of the bridgehead ketone and may be of importance for the further manipulation such as stereocontrolled reduction to access aglain natural products including ponapensin 3.[2]
Figure 4.

X-ray structure showing the hydrated form of the aglain derivative 7a
All aglain cycloadducts bearing a methyl ester moiety 7h–r were readily converted to the corresponding rearranged cyclopenta[b,c]benzofurans and further reduced to afford the desired rocaglates 8h–r accordingly to Method A (Schemes 2 and 3). As shown in Scheme 3 (Method A, Eq.1), alkaline conditions[5] may be used to convert aglain 7a to alkoxide 11a which may undergo α-ketol rearrangement to afford β-ketoester enolate 12a prior to workup and reprotonation to afford cyclopenta[b]benzofuran 10a. Further reduction produced the desired rocaglates 8a in 62% yield (two steps). Unfortunately, upon similar treatment aglain thioester 7b, imide 7d, and amides 7e–f were found to undergo retro-cycloaddition leading to regeneration of 3-HF 4 and the corresponding dipolarophiles 5b–f (Schemes 2 and 3, Method A, Eq. 2). Apparently the electron poor thioester, amide, and imide moieties of aglains 7b–f favored retro-cycloaddition rather than the expected ketol-shift rearrangement. Accordingly, we evaluated alternative conditions for ketol rearrangement and found that Lewis acids such as trimethylsilyltrifluoromethanesulfonate (TMSOTf) mediated the desired transformation and afforded the cyclopenta[b]benzofuran 8b after protodesilylation and hydroxyl-directed reduction (43% yield, three steps, Scheme 2 and 3, Method B, Eq. 3). Lewis acid-mediated ketol shift may occur through a concerted mechanism thus avoiding retro-cycloaddition.[15] Hydrated ketone 7b may be silylated by TMSOTf to afford hemiketal 13b, which may undergo pinacol-type rearrangement[16] involving a [1,2]-aryl shift to deliver the corresponding β-ketothioester. Using the TMSOTf protocol, aglain thioester 7b, Weinreb amide 7e, and amide 7f were successfully rearranged and transformed into rocaglates 8b, 8e, and 8f (Scheme 2, Method B). Notably, the Weinreb amide derivative 8d could be prepared using both methods (Method A: 20% yield (two steps); Method B: 46% (three steps)). Despite our efforts, aglain imide 7d could not be rearranged in a satisfactory manner. Finally, the aglain nitrile derivative 7g was readily converted to the desired rocaglate 8g under alkaline conditions followed by reduction (Method A, 55% yield, 1:1 dr).
Scheme 3.
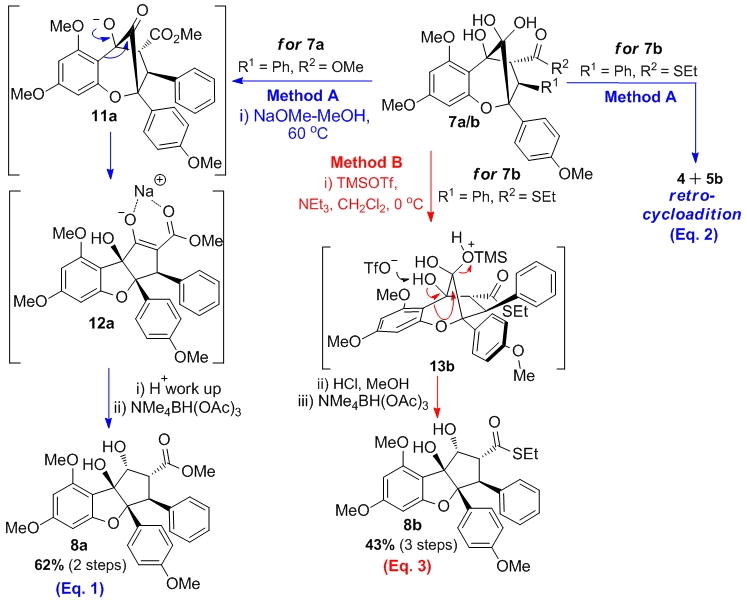
Basic vs. Lewis acid-mediated ketol rearrangements.
Given the effectiveness of dipolarophiles bearing β-aryl and related substituents, we next investigated trans-methylstyrene 5u and trans-stilbene 5v in the photocycloaddition with 3-HF 4.[9] Use of styrene 5u as dipolarophile afforded a complex mixture of regioisomeric and diastereoisomeric products. On the other hand, when trans-stilbene 5v was employed in the [3+2] photocycloaddition, a clean reaction was observed and furnished the aglain cycloadduct 7v in 40% yield (5:1 dr) (Scheme 4). The structure of 7v was determined by single x-ray crystal structure analysis and indicated the presence of a bridgehead ketone moiety.[9] The utility of stilbene as dipolarophile and our results from the overall dipolarophile screening suggest the involvement of the triplet biradicaloid[17] form of phototautomer 6 in the photocycloaddition.[18] Indeed, photocycloaddition of methyl cinnamate 5a and 3-HF 4 in the presence of the triplet quencher 9,10-dibromoanthracene did not lead to cycloadduct formation.[9,20] In another experiment, addition of benzophenone (triplet sensitizer, ET = 68.8 kcal/mol) to the reaction between trans-stilbene 5v and 3-HF 4 significantly increased the yield (from 40 to 56%) of the cycloadduct 7v[9] which supports involvement of the photoexcited triplet 14.[21] Based on our current studies, a radical ion mechanism involving photoinduced electron transfer (PET) from triplet excited state 14 to the dipolaraphile cannot be excluded.[19] Treatment of aglain 7v under alkaline conditions (Method A) did not effect ketol rearrangement. Under TMSOTf conditions (Method B), derivative 7v smoothly rearranged to a mixture of cyclopenta[b,c]benzofuran isomers and silylated products (vide infra). Protodesylilation of this mixture afforded an oxidized enone product which was further reduced to rocaglate 8v (56% yield, three steps).
Scheme 4.
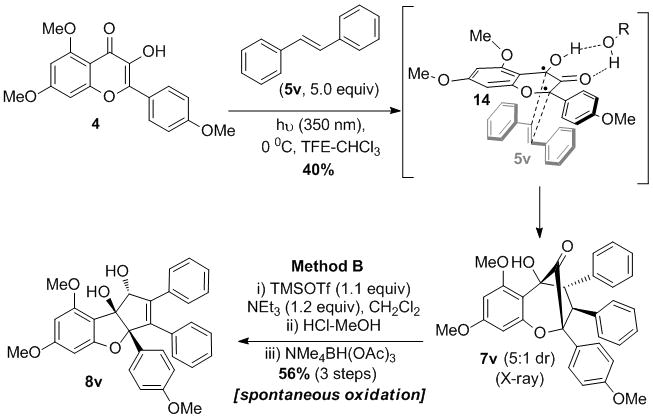
Use of stilbene 5v as dipolarophile in the [3+2] photocycloaddition
Having an efficient access to various rocaglate derivatives in racemic form, we evaluated their potencies as inhibitors of eukaryotic translation in comparison to enantiopure silvestrol 2.[3b] When tested for potency in vitro, 6 out of 25 compounds showed >50% inhibition of protein translation at 10 μM, all endo cycloaddition diastereomers (for a complete list of derivatives tested and % inhibition see Supporting information).[9] Titration of the six compounds (Figure 5A)[9] revealed that 8e and 8f were the most potent inhibitors with IC50’s of 300–400 nM. Silvestrol 2 showed an IC50 of approximately 100 nM in the same experiment (Figure 5A). We further tested the potency of these analogues in vivo for their ability to inhibit protein synthesis (Figure 5B). In this case, hydroxymate 8e[22] was the most potent analogue, inhibiting 85% of protein synthesis over the course of an hour, similar to silvestrol 2.
Figure 5.
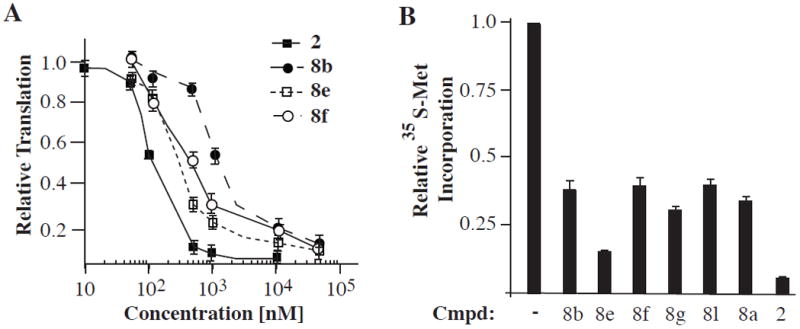
Evaluation of rocaglate derivatives as inhibitors of eukaryotic translation. A. Dose-dependent inhibition of in vitro translation. B. In vivo inhibition of protein synthesis in HeLa cells by rocaglate derivatives.[9]
In conclusion, we have expanded the biomimetic ESIPT photocycloaddition methodology to a broad range of dipolarophiles and further validated the [3+2] photocycloaddition pathway for concise access to a range of aglain and rocaglate structures. Evaluation of dipolarophiles revealed that donor-acceptor interactions of phototautomers 6/6′ may result in increased diastereocontrol. Our results also strongly support that photocycloadditions may proceed via a photoexcited triplet biradicaloid derived from 3-HF 4. A pinacol-type rearrangement provided an alternative to the base-mediated ketol shift protocol and enabled expedient access to hitherto inaccesible rocaglate derivatives. Finally, evaluation of rocaglates as inhibitors of eukaryotic translation led to the identification of a modified rocaglate derivative with potency similar to silvestrol 2 in vitro and in vivo. Further studies regarding ESIPT-mediated photocycloadditions and applications to complex molecule synthesis are currently in progress and will be reported in due course.
Supplementary Material
Footnotes
Financial support from the National Institutes of Health (R01 GM073855) is gratefully acknowledged. We thank Professors R. Johnson (UNH), F. D. Lewis (Northwestern University), and Dr. Baudouin Gerard (Broad Institute) for extremely helpful and stimulating discussions.
Dedicated to the memory of Pierre Potier and Christian Marazano for their outstanding contributions in biomimetic synthesis
Contributor Information
Dr. Stéphane P. Roche, Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston MA, 02215, USA, Fax: (+1) 617-358-2847
Dr. Regina Cencic, McGill University, Department of Biochemistry and The Rosalind and Morris Goodman Cancer Research Centre, Room 810, 3655 Drummond St., Montreal, QC, H3G 1Y6, Canada, Fax: (+1) 514-398-7384
Prof. Dr. Jerry Pelletier, Email: jerry.pelletier@mcgill.ca, McGill University, Department of Biochemistry and The Rosalind and Morris Goodman Cancer Research Centre, Room 810, 3655 Drummond St., Montreal, QC, H3G 1Y6, Canada, Fax: (+1) 514-398-7384
Prof. Dr. John A. Porco, Jr., Email: porco@bu.edu, Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston MA, 02215, USA, Fax: (+1) 617-358-2847
References
- 1.a) Proksch P, Edrada R, Ebel R, Bohnenstengel FI, Nugroho BW. Curr Org Chem. 2001;5:923–938. [Google Scholar]; b) Kim S, Salim AA, Swanson SM, Kinghorn AD. Anti-Cancer Agents Med Chem. 2006;6:319–345. doi: 10.2174/187152006777698123. [DOI] [PubMed] [Google Scholar]
- 2.Salim AA, Pawlus AD, Chai HB, Farnsworth NR, Kinghorn AD, Carcache-Blanco EJ. Bioorg Med Chem Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. Nat Chem Biol. 2006;2:213–220. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]; b) Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SMH, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA, Jr, Pelletier J. J Clin Invest. 2008;118:2651–2660. doi: 10.1172/JCI34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Trost BM, Greenspan PD, Yang BV, Saulnier MG. J Am Chem Soc. 1990;112:9022–9024. [Google Scholar]; b) Davey AE, Schaeffer MJ, Taylor RJK. J Chem Soc, Chem Commun. 1991:1137–1139. [Google Scholar]; f) Li HS, Fu B, Wang MA, Li N, Liu WJ, Xie ZQ, Ma YQ, Qin ZH. Eur J Org Chem. 2008:1753–1758. [Google Scholar]; g) Malona JA, Cariou K, Frontier AJ. J Am Chem Soc. 2009;131:7560–7561. doi: 10.1021/ja9029736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Gerard B, Jones G, II, Porco JA., Jr J Am Chem Soc. 2004;126:13620–13621. doi: 10.1021/ja044798o. [DOI] [PubMed] [Google Scholar]; b) Gerard B, Sangji S, O’Leary D, Porco JA., Jr J Am Chem Soc. 2006;128:7754–7756. doi: 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]; c) Gerard B, Cencic R, Pelletier J, Porco JA., Jr Angew Chem. 2007;119:7977–7980. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:7831–7834. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]
- 6.a) McMorrow D, Kasha M. J Phys Chem. 1984;88:2235–2243. [Google Scholar]; b) Klymchenko AS, Demchenko AP. J Am Chem Soc. 2002;124:12372–12379. doi: 10.1021/ja027669l. [DOI] [PubMed] [Google Scholar]; c) Klymchenko AS, Pivovarenko VG, Demchenko AP. J Phys Chem A. 2003;107:4211–4216. [Google Scholar]; d) Shynkar VV, Mély Y, Duportail G, Piémont E, Klymchenko AS, Demchenko AP. J Phys Chem A. 2003;107:9522–9529. [Google Scholar]; e) Bader AN, Pivovarenko VG, Demchenko AP, Ariese F, Gooijer C. J Phys Chem A. 2004;106:10589–10595. [Google Scholar]; f) Tomin VI, Javorski R. Optics Spectr. 2007;5:745–749. [Google Scholar]
- 7.a) Owen DJ, Rizzacasa MA, El Sous M, Spiniello M, Scammells PJ, Holloway G. WO 2006007634 PCT Int Appl. 2006 [Google Scholar]; b) El Sous M, Khoo ML, Holloway G, Owen D, Scammells PJ, Rizzacasa MA. Angew Chem. 2007;119:7981–7984. doi: 10.1002/anie.200702700. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:7835–7838. doi: 10.1002/anie.200702700. [DOI] [PubMed] [Google Scholar]; c) Thuaud F, Bernard Y, Turkeri G, Dirr R, Aubert G, Cresteil T, Baguet A, Tomasetto C, Svitkin Y, Sonenberg N, Nebigil CG, Désaubry L. J Med Chem. 2009;52:5176–5187. doi: 10.1021/jm900365v. [DOI] [PubMed] [Google Scholar]; d) Adams TE, El Sous M, Hawkins BC, Hirner S, Holloway G, Khoo ML, Owen DJ, Savage GP, Scammells PJ, Rizzacasa MA. J Am Chem Soc. 2009;131:1607–1616. doi: 10.1021/ja808402e. [DOI] [PubMed] [Google Scholar]
- 8.a) Lewis FD, Quillen SL, Hale PD, Oxman JD. J Am Chem Soc. 1988;110:1261–1267. [Google Scholar]; b) Sharma CVK, Panneerselvam K, Shimoni L, Katz H, Camell QHL, Desiraju GR. Chem Mater. 1994;6:1282–1292. [Google Scholar]; c) Khan M, Brunklaus G, Enkelmann V, Spiess HW. J Am Chem Soc. 2008;130:1741–1748. doi: 10.1021/ja0773711. [DOI] [PubMed] [Google Scholar]
- 9.See Supporting Information for complete experimental details.
- 10.For fluorinated alcohols in ESIPT and chemical synthesis, see: Park SY, Jang DJ. J Am Chem Soc. 2010;132:297–302. doi: 10.1021/ja907491u.For reviews, see: Bégué JP, Bonnet-Delpon D, Crousse B. Synlett. 2004:18–29.Shuklov IA, Dubrovina NV, Börner A. Synthesis. 2007:2925–2943.
- 11.a) Sytnik A, Gormin D, Kasha M. Proc Natl Acad Sci USA. 1994;91:11968–11972. doi: 10.1073/pnas.91.25.11968. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chou P-T, Huang C-H, Pu S-C, Cheng Y-M, Liu Y-H, Wang Y, Chen Co-T. J Phys Chem A. 2004;108:6452–6454. [Google Scholar]; c) Shynkar VV, Klymchenko AS, Piémont E, Demchenko AP, Mély Y. J Phys Chem A. 2004;108:8151–8159. [Google Scholar]; d) Chou PT, Pu SC, Cheng YM, Yu WS, Yu YC, Hung FT, Hu WP. J Phys Chem A. 2005;109:3777–3787. doi: 10.1021/jp044205w. [DOI] [PubMed] [Google Scholar]
- 12.El-Batta A, Jiang C, Zhao W, Anness R, Cooksy AL, Bergdahl M. J Org Chem. 2007;72:5244–5259. doi: 10.1021/jo070665k. [DOI] [PubMed] [Google Scholar]
- 13.For recent applications of donor-acceptor interactions, see: DeMartino MP, Chen K, Baran PS. J Am Chem Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y.Yamada S, Tokugawa Y. J Am Chem Soc. 2009;131:2098–2099. doi: 10.1021/ja809906c.For the pentafluorophenyl as acceptor, see: Irngartinger H, Escher T. Tetrahedron. 1999;55:10753–10760.Spitler EL, Monson JM, Haley MM. J Org Chem. 2008;73:2211–2223. doi: 10.1021/jo701740n.For the furan moiety as donor, see: Pac C, Nakasone A, Sakurai H. J Am Chem Soc. 1977;99:5806–5808.Albert IDL, Marks TJ, Ratner MA. J Am Chem Soc. 1997;119:6575–6582.
- 14.Cochran T, Abraham D, Martin L. J Chem Soc Chem Commun. 1972:494–495.Conroy JL, Sanders TC, Seto CT. J Am Chem Soc. 1997;119:4285–4291.and references therein; Yang D, Yip YC, Jiao GS, Wong MK. J Org Chem. 1998;63:8952–8956.Troast DM, Porco JA., Jr Org Lett. 2002;6:991–994. doi: 10.1021/ol025558l.
- 15.Ooi T, Ohmatsu K, Maruoka K. J Am Chem Soc. 2007;129:2410–2411. doi: 10.1021/ja063051q.Ohmatsu K, Tanaka T, Ooi T, Maruoka K. Tetrahedron. 2009;65:7516–7522.For a review on ketol rearrangements, see: Paquette LA, Hofferberth JE. Org React. 2003;62:477–567.
- 16.a) Bischofberger N, Walker KAM. J Org Chem. 1985;50:3604–3609. [Google Scholar]; b) Cabeza M, Heuze I, Bratoeff E, Ramírez E, Martínez R. Chem Pharm Bull. 2001;49:525–530. doi: 10.1248/cpb.49.525. [DOI] [PubMed] [Google Scholar]; c) Hou HF, Peddinti RK, Liao CC. Org Lett. 2002;4:2477–2480. doi: 10.1021/ol020087o. [DOI] [PubMed] [Google Scholar]
- 17.For the triplet state of the oxyallyl diradical, see: Bettinger HF. Angew Chem. 2010;122:680–682.Angew Chem Int Ed. 2010;49:670–671. doi: 10.1002/anie.200905482.
- 18.For biradical intermediates in [3+2] photocycloadditions, see: Kubo Y, Kusumoto K, Nakajima S, Inamura I. Chem Lett. 1999:113–114.Mizuno K, Sugita H, Hirai T, Maeda H, Otsuji Y, Yasuda M, Hashiguchi M, Shima K. Tetrahedron Lett. 2001;42:3363–3366.
- 19.a) Pac C, Miyauchi Y, Ishitani O, Ihama M, Yasuda M, Sakurai H. J Org Chem. 1984;49:26–34. [Google Scholar]; b) Miranda MA, García H. Chem Rev. 1994;94:1063–1089. [Google Scholar]; c) Warzecha KD, Demuth M, Görner H. J Chem Soc, Faraday Trans. 1997;93:1523–1530. [Google Scholar]; d) Hoffmann N. J Photochem Photobiol, C: Photochem Rev. 2008;9:43–60. [Google Scholar]
- 20.Tokumura K, Yagata N, Fujiwara Y, Itoh M. J Phys Chem. 1993;97:6656–6663. [Google Scholar]
- 21.For the triplet excited-state of 3-hydroxyflavones, see: Brewer WE, Studer SL, Standiford M, Chou PT. J Phys Chem. 1989;93:6088–6094.Martinez ML, Studer SL, Chou PT. J Am Chem Soc. 1990;112:2427–2429.Dick B. J Phys Chem. 1990;94:5152–5156.Tokumura K, Kurauchi M, Yagata N, Itoh M. Chem Phys Lett. 1996;258:495–500.
- 22.For the synthesis of related hydroxamates as potential insecticides, see: Dobler MR, Bruce I, Cederbaum F, Cooke NG, Diorazio LJ, Hall RG, Irving E. Tetrahedron Lett. 2001;42:8281–8284.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.