

Abstract
Cyanosporasides are marine bacterial natural products containing a chlorinated cyclopenta[a]indene core of suspected enediyne polyketide biosynthetic origin. Herein, we report the isolation and characterization of novel cyanosporasides C–F (3–6) from the marine actinomycetes “Salinispora pacifica” CNS-143 and Streptomyces sp. CNT-179, highlighted by the unprecedented C-2' N-acetylcysteamine functionalized hexose group of 6. Cloning, sequencing, and mutagenesis of homologous ~50 kb cyanosporaside biosynthetic gene clusters from both bacteria afforded the first genetic evidence supporting cyanosporaside's enediyne, and thereby p-benzyne biradical, biosynthetic origin and revealed the molecular basis for nitrile and glycosyl functionalization. This study provides new opportunities for bioengineering of enediyne derivatives and expands the structural diversity afforded by enediyne gene clusters.
Enediyne natural products have drawn substantial interest amongst chemists and pharmacologists owing to their structurally fascinating molecular scaffolds, potent DNA damaging activity, and unique biosynthetic assembly.1 The study of this fascinating class of bacterial compounds has been complicated by the lability of most 9-membered enediyne scaffolds in the absence of enediyne-binding apoproteins.2 This is one likely reason why remarkably few enediyne chemical structures have been fully elucidated, despite observations that genes encoding modular, iteratively-acting enediyne polyketide synthases (PKSEs) are relatively widespread among select genera of actinomycetes.3 Several known natural products, including the sporolides, cyanosporasides and fijiolides, have been postulated to represent spontaneous enediyne degradation products.4 Such products are of interest because they may offer insight into the divergence of biosynthetic pathways between those enediyne gene clusters that encode apoproteins and those that do not, and may afford access to unique biosynthetic genes that hold potential for engineering designer molecules with functional groups beyond the aromatic and glycosyl moieties common amongst most known enediynes. Herein, we report four new natural products of the cyanosporaside structure class from two phylogenetically distinct marine actinomycetes and suggest that their chlorinated cyclopenta[a]indene cores are derived from enediyne polyketide precursors based on the discovery and interrogation of their biosynthetic genes.
We previously reported that the marine bacterium “Salinispora pacifica” CNS-103 produces cyanosporasides A–B (1–2), novel chloro- and cyano-cyclopenta[a]indene glycosides, and hypothesized that their tricyclic aglycones were cyclized products of an enediyne polyketide precursor (Fig. 1b).4b Support for this proposal was later provided by Perrin et al. who demonstrated facile addition of nucleophilic halides to model enediyne substrates to yield halogen-substituted aromatic products via a proposed p-benzyne biradical intermediate (Fig. 1b).5 This halogenation mechanism contrasts common synthetic and biosynthetic mechanisms for aromatic halogen substitution that rather invoke electrophilic halogens and represents a new cyclization outcome of an enediyne substrate.
Figure 1.
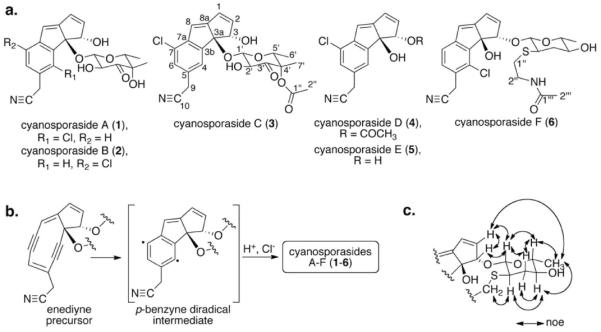
(a) Structures of cyanosporasides A–F (1–6); (b) Proposed enediyne origin and abiotic cycloaromatization via a diradical intermediate to yield cyanosporasides;4b,5 (c) Key NOESY correlations for determination of the relative stereochemistry of the pyranohexose unit in 6.
Chemical analysis of “S. pacifica” CNS-143 revealed several unreported cyanosporaside analogues most closely related to previously described 2. Through a combination of high-resolution mass spectrometry, detailed NMR analysis (Table S2, Figs. S2–S17), CD measurements (Fig. S1), and advanced Mosher's analysis (Fig. S18), structures of cyanosporasides C–E (3–5) were elucidated. Cyanosporaside C (3) is the C-4'-acetate analogue of 2, with the position of the acetate group of 3 established by NMR spectral differences at C-4'. The relative stereochemistry and absolute configuration of 3 were deduced by ROESY experiments and Mosher analysis, respectively (Fig. S18). Cyanosporasides 4 and 5 featured the aglycone of 2 without the deoxysugar moiety and differed from one another in the presence or absence, respectively, of an acetyl group at C-3 as revealed by long-range HMBC correlations.
Fermentation of the distantly related marine actinomycete Streptomyces sp. CNT-179 coincidentally revealed that it too biosynthesizes the cyanosporaside aglycones 4 and 5 with the same absolute configuration as those isolated from “S. pacifica” CNS-143 (Fig. S1). Further analysis of the S. sp. organic extract yielded a novel thioether analogue, cyanosporaside F (6), with the molecular formula C24H2735ClN2O5S based on the HRESIMS pseudomolecular ion m/z 513.1219 [M+Na]+. The main feature differentiating 6 from 14b was the attachment of an unusual pyranohexose sugar (C-1' to C-6') with an N-acetyl ethanamine unit (C-1” to C-2”) at C-3 of 6, as established by COSY and HMBC data. Analysis of the CD spectroscopic data of the aglycone of 6 and comparison to CD spectra for 3–5 (Fig. S1) revealed their identical absolute configurations. The relative stereochemistry of pyranohexose ring was determined by 1H-1H coupling constants and NOE correlations (Fig. 1c); attempts to determine the absolute configuration of the deoxysugar residue by Mosher ester derivatization were unsuccessful. From these data, the absolute and relative configurations of 6 were determined as 3S, 3aR, 1'R*, 2'S*, 4'R* and 5'S*. To the best of our knowledge, the hexose resi due of 6 represents the first natural example of an N-acetylcysteamine functionalized sugar.
With the discovery of cyanosporasides from two taxonomically distinct actinomycetes, we had the opportunity to explore the molecular basis for the biosynthesis of these unique nitrile and sugar functionalized molecules of hypothesized enediyne origin (Fig. 1b). From the draft genome of “S. pacifica” CNS-143,6 we identified just one enediyne biosynthetic gene cluster (cya), sharing homology with the previously established sporolide (spo) pathway from S. tropica.7 Sequence gaps within the putative cyanosporaside gene cluster (cya) were filled by sequencing portions of three overlapping cosmids encompassing the complete cluster. In total, a contiguous region of ~56 kb of DNA was established, with 34 putative open reading frames (orfs) spanning the ~49 kb proposed cya locus (Fig. 2a, Table S3).
Figure 2.
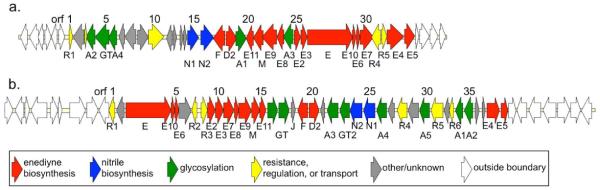
Organization and functional assignment of cyanosporaside biosynthetic gene clusters from (a) “Salinispora pacifica” CNS-143 (cya cluster, ~49 kb) and (b) Streptomyces sp. CNT-179 (cyn cluster, ~52 kb).
In the case of Streptomyces sp. CNT-179, PCR screening3 similarly established a single enediyne-associated gene cluster in this strain. Construction and screening of a cosmid library for these enediyne-encoding genes yielded the putative S. sp. CNT-179 cyanosporaside gene cluster (cyn) of 39 proposed orfs spanning ~52 kb (Fig. 2b, Table S4). Although the cya and cyn cyanosporaside clusters are rearranged and nonsyntenic, the majority of their shared gene products are >50% sequence identical (Table S4). Both the S. sp. and “S. pacifica” cyanosporaside clusters contain a predicted PKSE-encoding gene as well as other putative orfs highly conserved across previously reported biosynthetic pathways for 9-membered enediynes (Tables S3–S4).3,7,8 Further analysis of the KS domains from these PKSE genes using the online tool NapDos (http://napdos.ucsd.edu/)6 revealed a shared evolutionary history with other nine-membered enediynes.
To establish functional linkage of the “S. pacifica” cya locus with cyanosporaside production, we inactivated the conserved enediyne epoxide hydrolase cyaF by employing PCR-targeted gene replacement methods that we previously established in S. tropica.9 Chemical analysis of the resultant mutant bacterium revealed the abolished production of all known cyanosporasides, thereby providing the first direct support linking the production of these molecules to an enediyne biosynthetic pathway. We postulate that the enediyne polyketide precursor rapidly decomposes abiotically under the saline laboratory fermentation conditions to give the chlorinated cyanosporaside products, since we did not detect activity in the biochemical induction assay (BIA) for DNA-interfering compounds10 or observe evidence for enediynes in LC-MS or NMR screens of crude chemical extracts (data not shown). This rapid decomposition may be explained by the lack of predicted proteins with homology to known enediyne-binding apoproteins that function to stabilize labile 9-membered enediyne groups.1,2 While it is plausible that an apoprotein may be encoded but lacks homology with characterized enediyne-stabilizing proteins, as found with the maduropeptin-binding apoprotein,11 this scenario appears unlikely as the cyanosporaside clusters do not share short orfs consistent with the length of characterized enediyne-binding apoproteins.
Of >150,000 known natural products, fewer than 0.1% feature a nitrile moiety,12 making the cyanosporaside nitrile group particularly fascinating. As the construction of 9-membered enediyne skeletons proceeds from eight malonate units,1,13 cyanosporaside nitrile functionalization does not require formation of a new C–C bond but rather a new C–N bond at C-10. Pursuing the hypothesis of a unified cyanosporaside nitrile functionalization mechanism, sequences of putative proteins from both cyanosporaside gene clusters were compared (Table S4). Proteins with strong homology to those for enediyne core biosynthesis, glycosylation, pathway regulation, and antibiotic resistance were eliminated from consideration, leaving three candidates for nitrile biosynthesis – the aminotransferases CyaN1 and CynN1 (82% similarity), the oxidoreductases CyaN2 and CynN2 (78% similarity), and the cytochrome P450 monooxygenases Orf9 and Orf29 (80% similarity) from “S. pacifica” and Streptomyces sp., respectively. To explore their role in cyanosporaside biosynthesis, cyaN1, cyaN2, and orf9 were individually inactivated via PCR-targeted gene replacement. Chemical profiling of resulting “S. pacifica” mutants revealed the loss of cyanosporaside production in the cyaN1∷aprR aminotransferase and cyaN2∷aprR oxidoreductase mutants, while deletion of orf9 did not abolish cyanosporaside production (Fig. S19), suggesting its non-involvement in nitrile functionalization.
Thus, we propose that nitrile functionalization minimally occurs via the highly conserved two gene N1–N2 operon (Fig. 2). CynN1 and CyaN1 are pyridoxal phosphate-dependent family III aminotransferases (Fig. S21),14 with limited homology to BorJ (~35% identity), an aminotransferase implicated in the nitrile functionalization of borrelidin.15 BorJ and related aminotransferases typically act upon carbonyl groups, catalyzing conversion to amines.15 Thus, a C-10 aldehyde intermediate is plausible in cyanosporaside assembly.
CynN2 and CyaN2 are homologous to flavin-dependent oxidoreductases and dehydrogenases and are candidates for formation of the aldehyde intermediate on which the aminotransferase may act. Another possibility is that CynN2 and CyaN2 catalyze oxidation of the amine group, afforded by aminotransferases CynN1 and CyaN1, to yield the nitrile moiety. Borrelidin provides a biosynthetic literature precedent for bacterial conversion of a primary amine to a nitrile group, and a multifunctional cytochrome P450 was proposed for the conversion via a putative aldoxime intermediate.15 Neither CynN2 or CyaN2 exhibited significant similarity to the oxidoreductase proposed in nitrile functionalization of borrelidin, suggesting that nitrile functionalization of cyanosporasides proceeds through a divergent route. With two candidate genes now identified for nitrile functionalization of cyanosporasides, the stage is set for biochemical investigations to unveil the unique functionality expected for the N1 and N2 enzymes.
Cyanosporasides A–C (1–3) represent the only known natural products featuring a 3'-oxo-4'-methyl-β-fucopyranose group. Methylation at C-4' is particularly intriguing, as methyl branching at this position is rare in comparison to C-3' and C-5'.16 Bioinformatics analysis of the cya gene cluster revealed six genes (cyaA1–A5; cyaGT) encoding proteins homologous to characterized glycosylation enzymes (Fig. 2a, Table S3). CyaA1 and CyaA2 are most closely related to characterized glucose-1-phosphate thymidylyltransferases and 4,6-dehydratases, respectively, and are together proposed to catalyze conversion of D-glucose-1-phosphate to TDP-4-keto-6-deoxy-α-D-glucose, the committed pathway intermediate to 6-deoxy sugar units.16 CyaA3 exhibits 47% similarity to a biochemically characterized SAM-dependent C-methyltransferase that carries out regioselective C-3' methylation in mycarose biosynthesis.17 We propose that CyaA3 deprotonates H-3' of TDP-4-keto-6-deoxy-α-D-glucose to give an enolate intermediate poised for nucleophilic methylation at C-4' and afford the unique regiochemistry observed for sugar moieties 7–8 (Fig. 3). The essential role of the CyaA3 methyltransferase was probed by deleting the corresponding gene to abolish production of glycosylated cyanosporasides. In cyanosporaside C (3), the deoxysugar residue is acylated via acyltransferase CyaA4 to yield 8, as supported by PCR-directed gene elimination of cyaA4, in which production of 3 was selectively lost in the corresponding deletion mutant (Fig. S20). Finally, cyaGT is proposed to act as a glycosyltransferase, based on its high similarity to known O-glycosyltransferases, as supported by the elimination of glycosylated cyanosporasides among cyaGT∷aprR mutants. Interestingly, elimination of the putative epimerase-encoding gene, orf24, did not alter cyanosporaside biosynthesis (Fig. S20), refuting an essential role of this gene in cyanosporaside biosynthesis.
Figure 3.
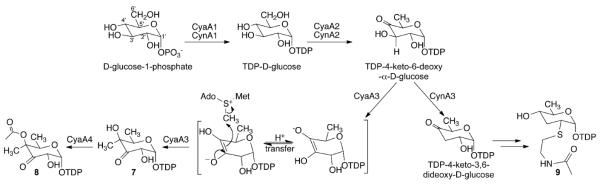
Proposed pathway for biosynthesis of unprecedented sugar groups 7–8 of cyanosporasides A–C (1–3) from “S. pacifica” and 9 of cyanosporaside F (6) from Streptomyces. sp.
Cyanosporaside F (6) is structurally distinguished from the “S. pacifica”cyanosporasides A–C (1–3) by the glycosyl group, which to our knowledge represents the first example of an N-acetylcysteamine functionalized secondary metabolic sugar. Based on examination of the putative cyn sugar biosynthetic gene cluster that shares close homologs to cyaA1 and cyaA2 (Table S4), biosynthesis of the sugar unit in 6 is proposed to diverge from the “S. pacifica”sugar pathway following assembly of TDP-4-keto-6-deoxy-α-d-glucose (Fig. 3). Based on the proposed functions of glycosylation-associated proteins deduced from the cyn gene cluster (Table S4), sulfur functionalization of C-2' is anticipated to proceed through substitution of the C-2' hydroxyl group (Fig. 3), potentially through a nucleophilic substitution mechanism. However, the biosynthetic details of this functionalization remain enigmatic.
In summary, we discovered new members of the cyanosporaside natural product family and showed that these chloro- and cyano-cyclopenta[a]indene glycosides are indeed enediyne polyketide biosynthetic products as previously hypothesized.4b We moreover identified a two-gene operon implicated in nitrile functionalization that distinguishes the cyanosporasides from other enediyne products. The relatively simple structures and small gene clusters of the cyanosporasides (~50 kb) in comparison with other characterized enediynes (~70−100+ kb) may render the cyanosporaside clusters as promising model systems for exploring hypotheses regarding the bio chemistry of enediyne biosynthesis.
Supplementary Material
ACKNOWLEDGMENT
Funding was graciously provided by the NIH through an IRACDA postdoctoral fellowship to A.L.L. (GM068524) and research grants to B.S.M and P.R.J. (GM085770) and W.F. (CA044848).
Footnotes
Supporting Information. Experimental section, supporting tables and figures, and detailed compound characterization are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Liang ZX. Nat. Prod. Rep. 2010;27:499–528. doi: 10.1039/b908165h. [DOI] [PubMed] [Google Scholar]
- (2).Jean M, Tomasi S, van de Weghe P. Org. Biomol. Chem. 2012;10:7453–7456. doi: 10.1039/c2ob26033f. [DOI] [PubMed] [Google Scholar]
- (3).Zazopoulos E, Huang KX, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM. Nat. Biotechnol. 2003;21:187–190. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]; Liu W, Ahlert J, Gao QJ, Wendt-Pienkowski E, Shen B, Thorson JS. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11959–11963. doi: 10.1073/pnas.2034291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]; (b) Oh DC, Williams PG, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2006;8:1021–1024. doi: 10.1021/ol052686b. [DOI] [PubMed] [Google Scholar]; (c) Nam S-J, Gaudencio SP, Kauffman CA, Jensen PR, Kondratyuk TP, Marler LE, Pezzuto JM, Fenical W. J. Nat. Prod. 2010;73:1080–1086. doi: 10.1021/np100087c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Perrin CL, Rodgers BL, O'Connor JM. J. Am. Chem. Soc. 2007;129:4795–4799. doi: 10.1021/ja070023e. [DOI] [PubMed] [Google Scholar]
- (6).Ziemert N, Podell S, Penn K, Badger J, Allen E, Jensen PR. PloS One. 2012;7:e34064. doi: 10.1371/journal.pone.0034064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McGlinchey RP, Nett M, Moore BS. J. Am. Chem. Soc. 2008;130:2406–2407. doi: 10.1021/ja710488m. [DOI] [PubMed] [Google Scholar]; Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu W, Christenson S, Standage S, Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]; Liu W, Nonaka K, Nie LP, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. Chem. Biol. 2005;12:293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- (9).Eustáquio AS, Pojer F, Noel JP, Moore BS. Nat. Chem. Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Elespuru RK, White RJ. Cancer Res. 1983;43:2819–2830. [PubMed] [Google Scholar]
- (11).Van Lanen SG, Oh T-J, Liu W, Wendt-Pienkowski E, Shen B. J. Am. Chem. Soc. 2007;129:13082. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fleming F. Nat. Prod. Rep. 1999;16:597–606. [Google Scholar]
- (13).Van Lanen S, Shen B. Curr. Top. Med. Chem. 2008;8:448–459. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yonaha K, Nishie M, Aibara S. J. Biol. Chem. 1992;267:12506–12510. [PubMed] [Google Scholar]
- (15).Olano C, Moss SJ, Brana AF, Sheridan RM, Math V, Weston AJ, Mendez C, Leadlay PF, Wilkinson B, Salas JA. Mol. Microbiol. 2004;52:1745–1756. doi: 10.1111/j.1365-2958.2004.04090.x. [DOI] [PubMed] [Google Scholar]
- (16).Thibodeaux CJ, Melançon CE, III, Liu H-W. Angew. Chem. Int. Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen H, Zhao Z, Hallis TM, Guo Z, Liu H-W. Angew. Chem. Int. Ed. 2001;40:607–610. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.