

Abstract
Background
Vascular calcification (VC) is prevalent in patients suffering from chronic kidney disease. Factors promoting calcification include abnormalities in mineral metabolism, particularly high phosphate levels. Inorganic phosphate (Pi) is a classical inducer of in vitro VC. Recently, an inverse relationship between serum magnesium concentrations and VC has been reported. The present study aimed to investigate the effects of magnesium on Pi-induced VC at the cellular level using primary HAVSMC.
Methods
Alive and fixed HAVSMC were assessed during 14 days in the presence of Pi with increasing concentrations of magnesium (Mg2+) chloride. Mineralization was measured using quantification of calcium, von Kossa and alizarin red stainings. Cell viability and secretion of classical VC markers were also assessed using adequate tests. Involvement of transient receptor potential melastatin (TRPM) 7 was assessed using 2-aminoethoxy-diphenylborate (2-APB) inhibitor.
Results
Co-incubation with Mg2+ significantly decreased Pi-induced VC in live HAVSMC, no effect was found in fixed cells. At potent concentrations in Pi-induced HAVSMC, Mg2+ significantly improved cell viability and restored to basal level increased secretions of osteocalcin and matrix gla protein, whereas a decrease in osteopontin secretion was partially restored. The block of TRPM7 with 2-APB at 10−4 M led to the inefficiency of Mg2+ to prevent VC.
Conclusions
Increasing Mg2+ concentrations significantly reduced VC, improved cell viability and modulated secretion of VC markers during cell-mediated matrix mineralization clearly pointing to a cellular role for Mg2+ and 2-APB further involved TRPM7 and a potential Mg2+ entry to exert its effects. Further investigations are needed to shed light on additional cellular mechanism(s) by which Mg2+ is able to prevent VC.
Keywords: chronic kidney disease, human aortic vascular smooth muscle cells, magnesium, phosphate
INTRODUCTION
Vascular calcification (VC) is commonly observed in patients with atherosclerosis, diabetes and chronic kidney disease (CKD) [1–3]. In adults with CKD, VC leads to increased mortality rates compared with the general population, even after adjusting for age and diabetic status [4]. The high incidence of cardiovascular mortality is partially credited to increased intimal and medial calcifications of the large arteries, including the aorta [5, 6]. VC is not only due to passive precipitation of calcium phosphate, but is also described as a tightly regulated process sharing similarities with bone formation [7]. The mechanism of this active process has been evaluated in in vitro studies on isolated cell-like vascular smooth muscle cells (VSMC). Exposure of VSMC to high phosphate and calcium concentrations leads to a dose-dependent increase in mineralization implying a transdifferentiation of VSMC to osteoblast-like cells [8, 9]. Changes in the expression of bone-associated (bone morphogenetic protein 2 and 7, osteocalcin) and mineralization-regulating [osteopontin, matrix gla protein (MGP)] proteins are classically reported in the course of VC [10, 11]. Nowadays, it is acknowledged that non-traditional cardiovascular risk factors such as abnormalities in bone and mineral metabolism as well as the uraemic status might increase the prevalence of VC and cardiovascular disease in CKD patients.
Despite its involvement as a co-factor of many enzymes, its function for maintaining vascular tone and in heart rhythm and finally its role in skeletal and mineral metabolism, magnesium has been generally overlooked as a potential modulator in the calcification process. Recently, an inverse relationship between serum magnesium concentrations and VC was reported in observational clinical studies [12, 13]. A limited number of clinical studies investigated the influence of serum magnesium on VC and cardiovascular mortality in uraemic or non-uraemic populations. Data from these studies are largely discussed in [14] and are clearly pointing towards a potential beneficial role of magnesium to improve VC and survival in CKD. Few experimental studies in animal models, mostly performed in rodents, confirmed these findings [14]. At the cellular level, the effect of magnesium on calcification has not been extensively investigated yet. Data on the prechondrogenic cell line ATDC5 suggest that excess Mg2+ might inhibit the excess Ca2+-promoted mineralization mediated by MGP [15]. Later, Montezano et al. [16] studied trans-differentiation and calcification in isolated VSMC and aortas of rodents in the presence of magnesium. Results showed that magnesium negatively regulates VC and osteogenic differentiation through transient receptor potential melastatin (TRPM)7 activity and increased expression of anti-calcification proteins (osteopontin, bone morphogenetic protein 7 and MGP). More recently, Salem et al. [17] revealed existing relationships between magnesium, inhibition of VC on calcification-induced aortic rings of rats and clinical biomarkers. Kircelli et al. [18] showed that increasing magnesium concentrations reduced the calcium deposition in calcification-induced bovine VSMC and modulated calcification markers.
As previously mentioned in vitro studies suggest magnesium to be a potent inhibitor of the VC process. To our knowledge, the effect of magnesium on induced calcification was not tested on primary VSMC from human origin. Therefore, the main goal of this study was to investigate increasing magnesium concentrations on Pi-induced calcification of human aortic VSMC (HAVSMC), and whether the expected effect of magnesium is bound to cellular activities or rather an extracellular passive phenomenon.
MATERIALS AND METHODS
Chemicals
All chemicals were purchased from Sigma unless otherwise stated.
Cell culture of HAVSMC
Primary HAVSMC were isolated in our laboratory from explants of human aortic tissue (obtained with appropriate ethical approval #2009/19), as described previously [19]. Briefly, medial tissue was separated from a segment of the human aorta after removal of the endothelium. Small pieces of tissue (1–2 mm2) were placed in a culture dish in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% fetal bovine serum (FBS, Dominique Dutscher), 4.5 g of glucose, 1 mmol/L of pyruvate, 100 U/mL of penicillin and 100 µg/mL of streptomycin in a 5% CO2 incubator at 37°C. Cells that migrated from explants were collected when confluent. The cells were maintained in DMEM supplemented with 15% FBS, the medium was replaced twice a week. HAVSMC were identified by their typical hill and valley morphology and purity of the primary cell culture was further checked by immunocytochemistry using a monoclonal antibody against the α-smooth muscle actin protein 1A4 (Acta 2) (Santa Cruz Biotechnology). α-Smooth muscle actin 1A4 is an isoform typical of smooth muscle cells (SMC) and is present in high amounts in vascular SMC [20]. The cells were used between passages 6 and 12, during which time they were able to calcify. Cells isolated from five independent donors were tested alternatively depending on the availability of cells requested for the various experiments.
For calcification assays, cells were seeded at 7500 cells/well in 48-well plates. Cells were treated for the indicated times (i.e. 7, 10 and 14 days) with various media conditions from 2 to 3 days after plating. For cell viability assays, cells were seeded at 3200 cells/well in 96-well plates. For cell layer staining assays, cells were seeded at 15 000 cells/well in 24-well plates. For all experiments, cells were treated as described in the calcification assays section.
In some experiments, HAVSMC were exposed to the TRPM7 inhibitor 2-aminoethoxy-diphenylborate (2-APB) (10−6 to 10−4 M). 2-APB was added to the medium from the beginning and at each medium renewal of the experiment.
Cell fixation
After seeding, the cells were fixed using a 3.7% formaldehyde solution at room temperature for 15 min. After removal of formaldehyde, cells were washed with PBS and dried. Calcification was then performed as it was for live cells.
Calcification assays
DMEM medium initially contains 0.9 mM of Pi, 1.8 mM of Ca2+ and 0.8 mM of Mg2+. Calcifications assays were conducted in 1% FBS DMEM and Pi concentration was increased to reach 3 mM. Mg2+ effect on calcification was observed at various concentrations. When indicated, the media Pi and Mg2+ concentrations were increased using NaH2PO4 and MgCl2 supplementation, respectively. Both live and fixed cells were calcified in an identical way, and the same calcification medium was used. The medium was replaced twice a week and Mg2+ was added from the beginning and at each medium renewal of the experiment. Supernatants were collected for further investigations.
For precise biochemical Ca2+ measurements, cells were washed with PBS without Ca2+ and Mg2+ and then decalcified with 0.6 N HCl overnight. The calcium content was determined colourimetrically with the o-cresolphthalein complexone method (OCP). Briefly, the principle of this method is based on the purple-coloured complex formed by calcium with OCP in an alkaline medium. The optical density (OD) of the samples were measured with a spectrophotometer at 565 nm and compared with a curve calibrated with calcium standards [21]. The protein content was measured using Bio-Rad protein assay reagent (Bio-Rad) according to the manufacturer's protocol. For live cells, the calcium content of the cell layer was normalized to protein content.
For alizarin red staining (AR), cells were washed with PBS and fixed with ethanol 95%. Then, samples were exposed to alizarin red 40 mM (pH 4.2). After two washing steps, wells were photographed showing the presence of induced mineralization.
For von Kossa staining (VK), cells were fixed with ethanol 95% for 15 min, rinsed and incubated with a 5% AgNO3 solution for 30 min. Cells were further rinsed with water, incubated for 5 min in a photographic revelation solution, washed with water, then incubated 5 min with a 5% sodium thiosulfate solution to remove unreacted silver, washed again in water and finally dried. Following their acquisition with a Photometrics CH250 CCD camera, well pictures were processed using the Software ImageJ (NIH). Cell layers were pictured after a classical von Kossa staining followed by a haematoxylin counterstaining according to Mayer. After washing with water and mounting, the cell layer was pictured using an Axioskop 40 (Carl Zeiss) coupled to Histolab® (Microvision Instruments).
Cell viability
The cell viability was indirectly assessed through measuring the mitochondrial activity by using the water-soluble tetrazolium salt WST-1 (Roche Applied Science). WST-1 was added to each well at a final concentration of 10% in medium according to the manufacturer's protocol. The absorbance was measured at 450 nm with a reference wavelength at 630 nm using a scanning multiwell microplate reader after 2 h of incubation with WST-1. Results are expressed as the percentage of the control (Ct).
Enzyme-linked immunosorbent assay measurements
After 10 and 14 days of incubation with the various media conditions, supernatants of indicated conditions were collected and kept at –80°C. Elisa kits for human MGP and osteocalcin (OCN) were purchased from Uscn Life Science. Human bone morphogenetic protein-2 (BMP-2) and 7 (BMP-7) were assessed using Elisa Quantikine® (R&D Systems). Human osteopontin (OPN) was assessed using Elisa DuoSet® from R&D systems. Supernatant measurements were performed according to the manufacturer's protocol.
Specific controls
Media from the various experimental conditions were assessed for correct calcium, phosphorus and magnesium levels using an ADVIA 1800 Siemens autoanalyser (Siemens Healthcare Diagnostics). No change in pH was observed at Pi 3 mM with or without addition of maximum Mg2+ concentrations. This excludes a potential role of medium acidification in the observed decrease of mineralization.
We checked whether the effects of MgCl2 on calcification reduction were due to the chloride ions in the MgCl2 salt used for these experiments, but addition of 2.4 mM NaCl (the maximum concentration of Cl− corresponding to the addition of MgCl2 to reach a total concentration of 2 mM of Mg2+) did not inhibit calcification after 10 and 14 days of culture in the presence of Pi 3 mM using AR, VK and OCP methods (data not shown).
Statistical analysis
All results are expressed as mean ± standard deviation (SD). Statistical significance was determined in an analysis of variance (ANOVA) multigroup analysis using the Fisher PLSD post test. A P-value of <0.05 was considered to be significant. Alternatively, the non-parametric Mann–Whitney test was used when ANOVA failed to reach significance because of the huge standard deviation observed between the different donors.
RESULTS
Mg2+ inhibits Pi-induced calcification in HAVSMC
In our model, deposition of calcified matrix in HAVSMC occurs by rising Pi concentration in the medium up to 3 mM. The amount of calcium per milligram protein was quantified in a variety of conditions using the OCP method at Days 10 and 14 of incubation with the indicated conditions. AR and VK stainings were also performed at Days 10 and 14 as quality tests to further confirm occurrence of calcium/phosphate (Ca/P) deposition in our samples (data not shown). HAVSMC from a total of five different donors were used during the whole study. As pointed out in Figure 1A, the accumulation of calcium at Day 14 was very different from one donor to another. The huge biological variations between the donors concerning calcium deposition did not allow us to directly compare the amounts of calcium between conditions. For the following calcium deposition experiments, all data were normalized to the 3 mM Pi condition, the 3 mM Pi condition was set to 1 to highlight any reduction in calcium deposition. In a preliminary series of experiments, cells from three different donors were tested to identify the most potent Mg2+ concentrations to prevent mineralization. In addition, we tested the effect of MgCl2 alone on calcification. MgCl2 by itself neither induced calcification at Day 10 (data not shown), nor at Day 14, as shown in Figure 1B. In the presence of 3 mM Pi, magnesium at a total concentration of 1.5 and 2 mM in medium was effective to significantly inhibit calcium deposition at Day 14 (Figure 1C). A similar trend was observed at Day 10, but with a less pronounced calcium deposition reduction and without reaching statistical significance (data not shown). As a result of the preliminary investigations, total magnesium concentrations of 1.5 and 2 mM were kept for the remaining experiments. In addition to the biochemical analysis, Ca/P deposits were also identified by von Kossa staining. The inhibition of Ca/P deposition by adding extracellular magnesium concentrations was optically confirmed (Figure 1D). Haematoxylin counterstaining according to Mayer imparts a light transparent red stain to the nuclei of cells, whereas von Kossa darkens the Ca/P depositions. Both size and amount of the granular calcified formations seem to decrease with the addition of magnesium, in comparison with Pi alone. No deposits were seen in conditions without supplemental phosphate. Figure 2A summarizes the results of calcium deposition for HAVSMC of the five tested donors after 14 days of Pi-induced calcification. Calcium deposition was significantly less in HAVSMC with increasing concentrations of Mg2+ compared with the Pi only condition (P < 0.0001) at Day 14. At Day 10, however, even if the results were following a similar trend, only a total concentration of 2 mM Mg2+ significantly decreased the Pi-induced deposition of calcium (P < 0.05).
FIGURE 1:

Magnesium reduces Pi-induced mineral deposition in HAVSMC. HAVSMC were cultured for 14 days at the indicated concentrations of Mg2+ and in the presence or absence of 3 mM Pi. (A) Calcium deposition from HAVSMC of five different donors was assessed using OCP. Filled square, filled circle, star, filled triangle and filled diamond represent means of the Ca2+ content of each of the five donors for each indicated condition. (B) Calcium deposition from HAVSMC of three different donors was assessed using OCP with increasing concentration of Mg2+ without induction of Pi. Data are quoted as a ratio of Pi and represent the mean ± SD of each condition (n = 6). Ct was significantly different from Pi, ****P < 0.0001 versus Pi. No significant differences were found between Ct and Mg2+ conditions. (C) Calcium deposition from HAVSMC of three different donors was assessed using OCP with increasing concentration of Mg2+ and induction of calcification by Pi. Data are represented as a ratio of Pi and represent the mean ± SD of each condition (n = 6). Ct was significantly different from Pi. **P < 0.01 versus Pi and ****P < 0.0001 versus Pi. (D) Von Kossa followed by a haematoxylin/Mayer counterstaining was performed on the cell layer; nuclei are depicted in red while granulated calcifications are coloured in black (×25 magnification).
FIGURE 2:
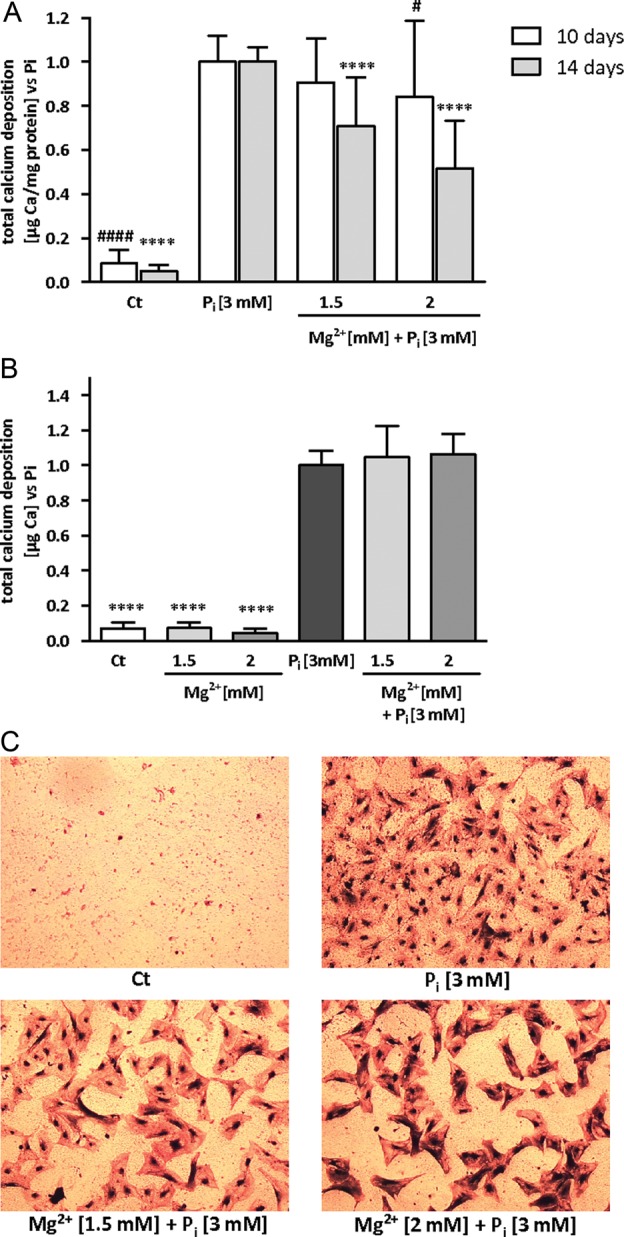
Magnesium is efficient in live but not in fixed HAVSMC. HAVSMC were cultured for 14 days at the indicated concentrations of Mg2+ and in the presence of 3 mM Pi. (A) Calcium deposition from Pi-induced HAVSMC of five different donors was assessed using OCP with increasing concentration of Mg2+. Data are quoted as a ratio of Pi and represent the mean ± SD of each condition (n = 16). Ct was significantly different Pi. #P < 0.05 versus Pi and ####P < 0.0001 at Day 10. ****P < 0.0001 versus Pi at Day 14. (B) Calcium deposition on fixed HAVSMC of three different donors was assessed using OCP with increasing concentration of Mg2+ in the presence of Pi. Data are quoted as a ratio of Pi and represent the mean ± SD of each condition (n = 12). Ct was significantly different from Pi, ****P < 0.0001 versus Pi. No significant differences were found between Ct and Mg2+ conditions. No significant differences were found between Pi and Pi + Mg2+ conditions. (C) Von Kossa followed by a haematoxylin/Mayer counterstaining was performed on the cell layer of fixed cells (×25 magnification).
Mg2+ is inefficient in preventing calcification in fixed HAVSMC
In a recent paper, Villa-Bellosta et al. [22] analysed the involvement of cellular activity during the initial calcium phosphate deposition of Pi-induced calcification by working with live and fixed cells. They underlined the role of the cell culture medium composition, especially salts and pH, in passive mineral deposition. We assessed the role of Mg2+ in a fixed HAVSMC setting (Figure 2B) and observed no decrease in calcium deposition after 14 days in the presence of the potent magnesium concentrations, suggesting that live cells are necessary for Mg2+ to exert its protective effect. The results were similar at Days 7 and 10 of incubation in the presence of Pi and Mg2+ (data not shown). It is of note that calcium deposition occurred slightly differently in fixed compared with live cells. Mineralization occurred faster in fixed cells when comparing absolute levels of calcium deposition. With the Pi 3 mM condition at Day 7 of incubation, we obtained far higher amounts of calcium for each of the three tested donors for the fixed cells compared with the three donors of the preliminary experiment of the live cells. However, at Day 14, absolute levels of calcium were comparable between fixed and live cells (data not shown). No change in pH was observed at Pi 3 mM with or without addition of maximum Mg2+ concentrations (data not shown). A potential role of medium acidification in the observed decrease of mineralization was thus excluded. As shown in Figure 2C, the initial shape of HAVSMC was kept with fixation. In the presence of 3 mM of Pi, mineral deposition was occurring mainly on the cell body, whereas we did not observe big granular calcified formations as described above for live cells. Addition of higher doses of magnesium did not lower the amount of calcification deposits.
Secretion of calcification markers under calcifying conditions
The calcification process results from an imbalance between pro-osteogenic factors, degradation of osteogenesis inhibitors and calcium phosphate product deposition. We chose to assess levels of OPN, MGP and BMP-7 as anti-calcification markers. OCN and BMP-2 were tested as pro-osteogenic markers. Unfortunately, we were unable to detect BMP-2 and BMP-7 in our supernatants; therefore, only data on OPN, MGP and OCN can be reported. As the same trend of results was observed in supernatants for each calcification marker at 10 days of incubation in a preliminary experiment (data not shown), only results after 14 days of incubation with the various indicated conditions will be presented. As expected, OCN levels were significantly increased after 14 days in the presence of 3 mM Pi (Figure 3A). At 1.5 and 2 mM magnesium concentrations, OCN levels fell back to the control level (Ct). No changes were found between Ct and Mg2+ conditions without addition of Pi. As reported in a previous work [23] with HAVSMC, Pi increased total MGP concentration in the culture medium. This effect was reversed by higher concentrations of magnesium in the medium. No statistically significant difference was found between Ct and high magnesium conditions without addition of Pi (Figure 3B). Finally, OPN, a known classical inhibitor of calcification, was investigated in supernatants (Figure 3C). In the presence of Pi, OPN level was drastically reduced and an addition of Mg2+ was not able to completely reverse this decrease. In this measurement, standard deviation between the different donors was higher than for the previous mentioned markers. The lack of a complete reversion of Pi-induced decrease of OPN by the addition of Mg2+ as well as the high inter-individual variability of HAVSMC OPN secretion from the different donors might partly explain the lack of statistical significance between Pi alone and Pi with magnesium conditions. By applying a Mann–Whitney test on the Pi and Pi + 1.5 mM Mg2+ conditions, a significance of 0.058 was reached.
FIGURE 3:

Secretion of calcification markers in Pi-induced HAVSMC in the presence of magnesium. Protein concentrations of OCN, MGP and OPN were quantified in the supernatants of HAVSMC by ELISA. Three different donors including three replicates were assessed. HAVSMC were cultured for 14 days at the indicated concentrations of Mg2+ and Pi. (A) OCN secretion from Pi-induced HAVSMC. Data are quoted as a ratio of Ct and represent the mean ± SD of each condition (n = 9). *P < 0.05 and ***P < 0.001 versus Pi. No significant differences were found between Ct and Mg2+ conditions. (B) MGP secretion from Pi-induced HAVSMC. Data are quoted as a ratio of Ct and represent the mean ± SD of each condition (n = 9). **P < 0.01, ***P < 0.001 and ****P < 0.0001 versus Pi. No significant differences were found between Ct and Mg2+ conditions. (C) OPN secretion from Pi-induced HAVSMC. Data are quoted as a ratio of Ct and represent the mean ± SD of each condition (n = 9). **P < 0.01, ***P < 0.001 versus Pi. No significant differences were found between Ct and Mg2+ conditions.
Cell viability in the presence of Pi and Mg2+
We decided to evaluate the effects of magnesium on cell viability of Pi-induced HAVSMC (Figure 4A). Cells were treated as for the calcification assays. After 14 days of incubation, the addition of Pi did not affect cell viability even if the larger observed SD is reflecting a heterogeneous behaviour of HAVSMC to face Pi among the tested donors. However, higher magnesium levels significantly improved cell viability in the presence of Pi. An increase of around 10% in viability in the presence of magnesium was observed, although only 1% of FBS was present in the medium, which per se considerably limits the cellular growth. This result is pointing out a substantial beneficial effect of magnesium on cell viability. In the absence of Pi, higher concentrations of magnesium enhanced cell viability a little, but not significantly.
FIGURE 4:

Magnesium effects on mineralization are cell-mediated. HAVSMC were cultured for 14 days at the indicated concentrations of Mg2+ and Pi. (A) Cell viability from Pi-induced HAVSMC of three different donors was assessed. Data are quoted as a ratio of Ct (%) and represent the mean ± SD of each condition (n = 12). *P < 0.05 versus Pi. (B) Calcium deposition from Pi-induced HAVSMC of three different donors was assessed using OCP with increasing concentration of Mg2+. 2-APB at 10−4 M was added to block TRPM7. Data are quoted as a ratio of Pi and represent the mean ± SD of each condition (n = 12). Ct was significantly different from Pi. *P < 0.05 and **P < 0.01 versus Pi.
Use of 2-APB demonstrates involvement of TRPM7 in observed Mg2+ effects
To gain insight into the intracellular molecular mechanisms underlying the prevention of HAVSMC calcification by magnesium, we decided to assess Mg2+ entry by inhibiting TRPM7 using 2-APB. Indeed, TRPM7 was recently shown to be functionally involved in magnesium's modulation of VSMC differentiation to an osteogenic phenotype [16]. As shown in Figure 4B, addition of 2-APB at 10−4 M abolished the previously shown, positive effect of Mg2+ on calcification after 14 days of combined Pi and Mg2+ exposure. Lower concentrations of 2-APB (10−6 and 10−5 M) were not efficient in blocking the reduction of calcification by Mg2+ (data not shown). Hamaguchi et al. [24] extensively studied Na+-independent Mg2+ transport sensitive to 2-APB in VSMC and found that Mg2+ transport through TRPM7 was efficiently blocked with concentrations of 2-APB around 10−4 M comforting us in the use of 2-APB at this concentration. It is of note that 2-APB at 10−4 M significantly enhanced calcification in the presence of Mg2+ comparing with the Pi 3 mM alone condition. 2-APB itself did not induce mineralization without the presence of Pi. The same trend of results was observed at 10 days of incubation at the various indicated conditions (data not shown). We also checked the cell viability in the presence of 2-APB 10−4 M with or without addition of Pi or Mg2+. After 14 days of incubation, a slight, but statistically not significant decrease in cell viability in APB 10−4 M conditions was observed. No change was found in the presence of 2-APB 10−4 M after 10 days of exposure (data not shown).
DISCUSSION
Our study is the first to provide in vitro evidence for a protective role of magnesium on Pi-induced calcification in a primary cell culture model of HAVSMC. We used Pi at a total concentration of 3 mM which is a concentration observed in patients in late stages of CKD. Two magnesium concentrations (1.5 and 2 mM of total Mg2+) were found to be efficient and strongly attenuated Ca/P deposition in spite of a high inter-individual variability observed between different HAVSMC donors. The attenuating effect of magnesium on the calcification process was already significant after 10 days but was accentuated after 14 days of exposure of cells with Pi and Mg2+. Recent publications support that matrix calcification could be initiated through passive Ca/P deposition and that it could be the formation of Ca/P nanocrystals that triggers the osteogenic changes which are associated with the accumulation of extracellular, well-organized, crystalline structures [22, 25]. As it was already known that magnesium is able to impair hydroxyapatite crystal growth in vitro [26, 27], we decided to test whether the observed effect of Mg2+ also occurred during passive Ca/P deposition. Consequently, mineral deposition was studied in fixed HAVSMC. As expected, Ca/P deposition was increased in the presence of Pi 3 mM, but higher concentration of Mg2+ failed to inhibit this passive calcification process. Thus, live cells seem to be necessary for Mg2+ to exert its protective effect, suggesting a potentially active cellular role. This finding does not exclude a potentially passive role for Mg2+ ions in the onset of calcification, because in the presence of Mg2+, several ion substitutions were described in the context of hydroxyapatite formation, thereby altering crystal composition and structure [22].
Apart from passive mineral deposition, it is well established that VC is resulting from an active cell-mediated process. Under normal phosphate conditions, VSMC express smooth muscle lineage markers characterizing the contractile phenotype such as smooth muscle α-actin. After treatment with elevated phosphate, there is a dramatic loss of the contractile markers, and a simultaneous modulation of osteogenic markers and/or calcification inhibitors such as OPN, OCN, MGP and BMP-2 and 7 is observed [7–9, 28]. We decided to assess the secretion of these markers in cell culture supernatants from our experiments. We were unable to detect BMP-2 and 7, even though BMP-2 has already been quantified in supernatants of mouse aortic SMC [25]. OCN is the most abundant non-collagenous protein of the bone extracellular matrix and is found in the matrix of calcified cells. OCN is specifically synthesized by osteoblasts in bone and osteoblast-like cells in VC [8, 29]. Despite structural similarities with MGP, such as gla residues, OCN does not display MGP’s anti-mineralization properties. Until now, loss- and gain-of-function experiments have failed to identify a function for OCN in extracellular matrix mineralization in vivo [30]. Our results show a strong increase in secreted OCN in the presence of supplemental Pi. This indicates that a cell-mediated matrix mineralization effectively occurred. Increasing concentrations of Mg2+ completely reversed this phenomenon, suggesting that Pi-induced calcification importantly decreased. These results confirmed data from [16]. If OCN correlates to the degree of calcium deposition, an inhibition of OCN secretion through the addition of magnesium in Pi-induced HAVSMC is indicating a potential involvement of a cellular mechanism.
MGP is a vitamin K-dependent, γ-carboxylated protein present in bone and vascular smooth muscle. The mechanism by which MGP inhibits VC is not fully understood. The MGP-fetuin complex in serum appears to prevent propagation of Ca/P precipitation, but the precise role of MGP is still not clear [31]. MGP binds hydroxyapatite in vitro [32], and this is consistent with the fact that in calcified aortas, the MGP content is proportional to hydroxyapatite content, and that MGP is concentrated around hydroxyapatite deposits [33, 34]. Our data showed an important increase in total MGP secretion in Pi-induced HAVSMC. Similar data have been described previously [23], and we postulate that the concentration of total MGP in the supernatant is linked to the degree of VC as a secondary, compensatory mechanism, rather than a primary effect. This is in agreement with previous studies showing that cultured VSMC constitutively express MGP, and that MGP mRNA expression was found to be up-regulated in calcifying VSMC nodular cultures [35]. Moreover, immunohistochemical studies showed that MGP was abundant at sites of calcification in human arteries [36]. More recently, post-translational modifications of MGP, such as c-glutamyl carboxylation and serine phosphorylation of MGP, were implicated in its function as a calcification inhibitor [37]. Novel data showed similar increases for both carboxylated and uncarboxylated forms of MGP in calcified aortas from rats with renal failure [38]. Finally, the increase in the circulating, diphosphorylated and uncarboxylated form of MGP was proposed as a marker for the progression of VC in CKD patients [39]. Our data showed that supplementation with magnesium restores total MGP secretion to its basal level in Pi-induced HAVSMC. This finding is in concordance with a previous work on the prechondrogenic cell line ATDC5 [15].
Lastly, OPN secretion was investigated. OPN is a secreted, glycosylated phosphoprotein normally found in mineralized tissues or in VC. The combination of electronegative glutamic and aspartic acid residues, serine/threonine kinase substrate sites and the putative calcium-binding motifs endow OPN with the ability to bind significant amounts of Ca2+ (50 mol Ca2+ to 1 mol OPN) [40, 41]. These properties likely contribute to OPN's ability to bind and regulate apatite crystal growth. The first study to assess OPN expression in HAVSMC showed that OPN mRNA was detectable but did not appear to be up-regulated in the calcifying nodular cells; subsequently, HAVSMC secrete basically very low levels of OPN protein which were not detectable in calcifying cells [35]. As well, murine SMC from OPN−/− mice showed enhanced susceptibility to calcification in vitro [42]. Our results are in accordance with these studies, despite other studies reported that OPN is abundant at sites of calcification in human atherosclerotic plaques and in calcified aortic valves, for review, see Giachelli et al. [28]. In our model, Pi-induced calcification led to a severe decrease in OPN secretion. The loss of this VC inhibitor might have a preponderant importance in the development of matrix mineralization. The addition of Mg2+ partially, but not significantly, reverses the decrease in OPN. In rat VSMC, Mg2+ supplementation during VC significantly increased OPN content supporting our results [16].
Apoptosis has been reported in the process of human VC and seems necessary because apoptotic bodies derived from VSMC may act as nucleating structures for Ca/P crystal formation [43]. Proudfoot et al. reported that apoptosis occurs in a human model of VC in which post-confluent VSMC cultures spontaneously form nodules and calcify after 28 days. The large SD observed in the presence of Pi might explain why cell viability was not affected in our experimental setup. Comparing with [43], our cells were not post-confluent and were incubated only 14 days under calcification-inducing conditions. Thus, this specific setup might not be the most suitable one to study apoptosis. However, increased concentrations of Mg2+ significantly improved cell viability in our setting of Pi-induced HAVSMC. This beneficial effect is in accordance with the study on bovine VSMC [18]. Further investigations on cell viability and apoptosis in calcifying HAVSMC are needed to elucidate the exact mechanisms and to further strengthen the protective role of magnesium in this context.
In the last few years, magnesium homeostasis was shown to be regulated by TRPM7 in osteoblast differentiation, proliferation and migration [44, 45] as well as in VSMC in the context of hypertension and transdifferentiation to an osteogenic phenotype [16, 46]. TRPM7 contains a magnesium-permeable pore fused to a kinase domain at the COOH terminus. 2-APB is classically reported to be a potent inhibitor of TRPM7. Our data show that a concentration of 10−4 M of 2-APB was able to abolish the protective effect of Mg2+ on Pi-induced calcification. At this concentration, 2-APB was shown to efficiently block Mg2+ entry through TRPM7 [24]. Thus, for the first time, TRPM7 activity can be directly linked to mineral deposition. In a previous study [16], the effect of 2-APB was investigated on protein expression of OCN, MGP and OPN in calcifying rat VSMC. The authors also found a loss of the beneficial effects of Mg2+. Taken together, these results strongly suggest an active, intracellular role for Mg2+ in attenuating the VC process, likely through entering the cell via TRPM7. Of course, deeper investigations are needed to clarify the role of TRPM7 in this context.
The use of magnesium as a drug to lower serum calcium and phosphorus in CKD was recently reviewed [47]. The clinical relevance of the effective concentrations of magnesium in reducing HAVSMC-induced mineralization (i.e. 1.5 and 2 mM of Mg2+) is not far from the serum levels found in CKD patients with magnesium carbonate supplementation. Recently, a pilot study compared combined magnesium carbonate plus calcium carbonate with calcium acetate monotherapy for 12 weeks [48]. Serum magnesium levels were elevated in patients receiving magnesium carbonate with the highest serum magnesium reported being 1.95 mM, but no patients experienced symptoms related to hypermagnesaemia. The combined treatment succeeded to efficiently control serum phosphorus levels as well as to safely and significantly reduce calcium load as reflected by serum calcium levels compared with calcium monotherapy. In addition, it has been reported that patients being treated with high magnesium dialysate concentrations (1.00 mM) also have elevated levels around 1.34 mM without apparent clinical symptoms [49]. Moreover, a recent investigation of serum magnesium levels in CKD patients of stages 3–5 showed a positive correlation of flow-mediated dilatation to serum magnesium levels up to 1.64 mM [50].
In conclusion, increased concentrations of magnesium efficiently reduced Pi-induced calcification. Our study, together with previous findings, strongly supports considering magnesium as a potential candidate for the therapy of VC now. Whether the in vitro data collected in the present study also have relevance in clinical setting is matter of additional works. Likewise, further investigations are certainly needed to gain additional mechanistic insights into magnesium's mode of action to prevent VC.
CONFLICT OF INTEREST STATEMENT
J.P.-D. is a consultant to Fresenius Medical Care Deutschland GmbH. S.S. and J.B. are employees of Fresenius Medical Care Deutschland GmbH. Z.A.M. has received research grants and honorarium from Fresenius Medical Care Deutschland GmbH.
ACKNOWLEDGEMENTS
The authors thank the ‘Laboratoire de Biochimie’, ‘Laboratoire de Biologie Endocrinienne et Osseuse’ and ‘Service de Chirurgie Cardiaque’ du ‘Centre Hospitalier Universitaire d'Amiens' for their respective contributions in this work. L.L. especially thanks M.A. Conte for her valuable technical help with calcium measurements. This study was supported by Fresenius Medical Care Deutschland GmbH, Germany.
REFERENCES
- 1.Takasu J, Budoff MJ, Katz R, et al. Relationship between common carotid intima-media thickness and thoracic aortic calcification: the multi-ethnic study of atherosclerosis. Atherosclerosis. 2010;2:142–146. doi: 10.1016/j.atherosclerosis.2009.09.013. doi:10.1016/j.atherosclerosis.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishiyama M, Suzuki E, Katsuda J, et al. Associations of coronary artery calcification and carotid intima-media thickness with plasma concentrations of vascular calcification inhibitors in type 2 diabetic patients. Diabetes Res Clin Pract. 2009;2:189–196. doi: 10.1016/j.diabres.2009.04.023. doi:10.1016/j.diabres.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Braun J, Oldendorf M, Moshage W, et al. Electron beam computed tomography in the evaluation of cardiac calcification in chronic dialysis patients. Am J Kidney Dis. 1996;2:394–401. doi: 10.1016/s0272-6386(96)90363-7. doi:10.1016/S0272-6386(96)90363-7. [DOI] [PubMed] [Google Scholar]
- 4.Foley RN, Parfrey PS. Cardiovascular disease and mortality in ESRD. J Nephrol. 1998;2:239–245. [PubMed] [Google Scholar]
- 5.London GM, Guérin AP, Marchais SJ, et al. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;2:1731–1740. doi: 10.1093/ndt/gfg414. doi:10.1093/ndt/gfg414. [DOI] [PubMed] [Google Scholar]
- 6.Noordzij M, Cranenburg EM, Engelsman LF, et al. NECOSAD Study Group. Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol Dial Transplant. 2011;2:1662–1669. doi: 10.1093/ndt/gfq582. doi:10.1093/ndt/gfq582. [DOI] [PubMed] [Google Scholar]
- 7.Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res. 2004;2:560–567. doi: 10.1161/01.RES.0000141775.67189.98. doi:10.1161/01.RES.0000141775.67189.98. [DOI] [PubMed] [Google Scholar]
- 8.Giachelli CM. Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol. 2003;14(9 Suppl 4):S300–S304. doi: 10.1097/01.asn.0000081663.52165.66. doi:10.1097/01.ASN.0000081663.52165.66. [DOI] [PubMed] [Google Scholar]
- 9.Shanahan CM, Crouthamel MH, Kapustin A, et al. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;2:697–711. doi: 10.1161/CIRCRESAHA.110.234914. doi:10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyson KL, Reynolds JL, McNair R, et al. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol. 2003;2:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. doi:10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- 11.Hruska KA, Mathew S, Saab G. Bone morphogenetic proteins in vascular calcification. Circ Res. 2005;2:105–114. doi: 10.1161/01.RES.00000175571.53833.6c. doi:10.1161/01.RES.00000175571.53833.6c. [DOI] [PubMed] [Google Scholar]
- 12.Ishimura E, Okuno S, Kitatani K, et al. Significant association between the presence of peripheral vascular calcification and lower serum magnesium in hemodialysis patients. Clin Nephrol. 2007;2:222–227. doi: 10.5414/cnp68222. doi:10.2215/CJN.01790506. [DOI] [PubMed] [Google Scholar]
- 13.Turgut F, Kanbay M, Metin MR, et al. Magnesium supplementation helps to improve carotid intima media thickness in patients on hemodialysis. Int Urol Nephrol. 2008;2:1075–1082. doi: 10.1007/s11255-008-9410-3. [DOI] [PubMed] [Google Scholar]
- 14.Massy ZA, Drüeke TB. Magnesium and outcomes in patients with chronic kidney disease: focus on vascular calcification, atherosclerosis and survival. Clin Kidney J. 2012;5(Suppl 1):i52–i61. doi: 10.1093/ndtplus/sfr167. doi:10.1093/ndtplus/sfr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakatani S, Mano H, Ryanghyok IM, et al. Excess magnesium inhibits excess calcium-induced matrix-mineralization and production of matrix gla protein (MGP) by ATDC5 cells. Biochem Biophys Res Commun. 2006;2:1157–1162. doi: 10.1016/j.bbrc.2006.07.180. doi:10.1016/j.bbrc.2006.07.180. [DOI] [PubMed] [Google Scholar]
- 16.Montezano AC, Zimmerman D, Yusuf H, et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension. 2010;2:453–462. doi: 10.1161/HYPERTENSIONAHA.110.152058. doi:10.1161/HYPERTENSIONAHA.110.152058. [DOI] [PubMed] [Google Scholar]
- 17.Salem S, Bruck H, Bahlmann FH, et al. Relationship between magnesium and clinical biomarkers on inhibition of vascular calcification. Am J Nephrol. 2012;2:31–39. doi: 10.1159/000334742. doi:10.1159/000334742. [DOI] [PubMed] [Google Scholar]
- 18.Kircelli F, Peter ME, Sevinc Ok E, et al. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol Dial Transplant. 2012;2:514–521. doi: 10.1093/ndt/gfr321. doi:10.1093/ndt/gfr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross R. The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol. 1971;2:172–186. doi: 10.1083/jcb.50.1.172. doi:10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skalli O, Pelte MF, Peclet MC, et al. Alpha-smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J Histochem Cytochem. 1989;2:315–321. doi: 10.1177/37.3.2918221. doi:10.1177/37.3.2918221. [DOI] [PubMed] [Google Scholar]
- 21.Gitelman HJ. An improved automated procedure for the determination of calcium in biological specimens. Anal Biochem. 1967;2:521. doi:10.1016/0003-2697(67)90110-8. [Google Scholar]
- 22.Villa-Bellosta R, Millan A, Sorribas V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am J Physiol Cell Physiol. 2011;2:C210–C220. doi: 10.1152/ajpcell.00229.2010. doi:10.1152/ajpcell.00229.2010. [DOI] [PubMed] [Google Scholar]
- 23.Ivanovski O, Nikolov IG, Joki N, et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE−/−) mice. Atherosclerosis. 2009;2:55–62. doi: 10.1016/j.atherosclerosis.2008.10.043. doi:10.1016/j.atherosclerosis.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 24.Hamaguchi Y, Matsubara T, Amano T, et al. Na+-independent Mg2+ transport sensitive to 2-aminoethoxydiphenyl borate (2-APB) in vascular smooth muscle cells: involvement of TRPM-like channels. J Cell Mol Med. 2008;2:962–974. doi: 10.1111/j.1582-4934.2008.00157.x. doi:10.1111/j.1582-4934.2008.00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sage AP, Lu J, Tintut Y, et al. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;2:414–422. doi: 10.1038/ki.2010.390. doi:10.1038/ki.2010.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ennever J, Vogel JJ. Magnesium inhibition of apatite nucleation by proteolipid. J Dent Res. 1981;2:838–841. doi: 10.1177/00220345810600041301. [DOI] [PubMed] [Google Scholar]
- 27.Boskey AL, Posner AS. Effect of magnesium on lipid-induced calcification: an in vitro model for bone mineralization. Calcif Tissue Int. 1980;2:139–143. doi: 10.1007/BF02408533. doi:10.1007/BF02408533. [DOI] [PubMed] [Google Scholar]
- 28.Giachelli CM, Speer MY, Li X, et al. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ Res. 2005;2:717–722. doi: 10.1161/01.RES.0000161997.24797.c0. doi:10.1161/01.RES.0000161997.24797.c0. [DOI] [PubMed] [Google Scholar]
- 29.Ducy P, Desbois C, Boyce B, et al. Increased bone formation in osteocalcin deficient mice. Nature. 1996;2:448–452. doi: 10.1038/382448a0. doi:10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- 30.Murshed M, Schinke T, McKee MD, et al. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;2:625–630. doi: 10.1083/jcb.200402046. doi:10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price PA, Thomas GR, Pardini AW, et al. Discovery of a high molecular weight complex of calcium, phosphate, fetuin, and matrix gamma-carboxyglutamic acid protein in the serum of etidronate-treated rats. J Biol Chem. 2002;2:3926–3934. doi: 10.1074/jbc.M106366200. doi:10.1074/jbc.M106366200. [DOI] [PubMed] [Google Scholar]
- 32.Roy ME, Nishimoto SK. Matrix Gla protein binding to hydroxyapatite is dependent on the ionic environment: calcium enhances binding affinity but phosphate and magnesium decrease affinity. Bone. 2002;2:296–302. doi: 10.1016/s8756-3282(02)00821-9. doi:10.1016/S8756-3282(02)00821-9. [DOI] [PubMed] [Google Scholar]
- 33.Price PA, Faus SA, Williamson MK. Warfarin-induced artery calcification is accelerated by growth and vitamin D. Arterioscler Thromb Vasc Biol. 2000;2:317–327. doi: 10.1161/01.atv.20.2.317. doi:10.1161/01.ATV.20.2.317. [DOI] [PubMed] [Google Scholar]
- 34.Shanahan CM, Cary NR, Salisbury JR, et al. Medial localization of mineralization-regulating proteins in association with Mönckeberg's sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;2:2168–2176. doi: 10.1161/01.cir.100.21.2168. doi:10.1161/01.CIR.100.21.2168. [DOI] [PubMed] [Google Scholar]
- 35.Proudfoot D, Skepper JN, Shanahan CM, et al. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler Thromb Vasc Biol. 1998;2:379–388. doi: 10.1161/01.atv.18.3.379. doi:10.1161/01.ATV.18.3.379. [DOI] [PubMed] [Google Scholar]
- 36.Spronk HM, Soute BA, Schurgers LJ, et al. Matrix Gla protein accumulates at the border of regions of calcification and normal tissue in the media of the arterial vessel wall. Biochem Biophys Res Commun. 2001;2:485–490. doi: 10.1006/bbrc.2001.5996. doi:10.1006/bbrc.2001.5996. [DOI] [PubMed] [Google Scholar]
- 37.Schurgers LJ, Spronk HM, Skepper JN, et al. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;2:2503–2511. doi: 10.1111/j.1538-7836.2007.02758.x. doi:10.1111/j.1538-7836.2007.02758.x. [DOI] [PubMed] [Google Scholar]
- 38.Lomashvili KA, Wang X, Wallin R, et al. Matrix Gla protein metabolism in vascular smooth muscle and role in uremic vascular calcification. J Biol Chem. 2011;2:28715–28722. doi: 10.1074/jbc.M111.251462. doi:10.1074/jbc.M111.251462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schurgers LJ, Barreto DV, Barreto FC, et al. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: a preliminary report. Clin J Am Soc Nephrol. 2010;2:568–575. doi: 10.2215/CJN.07081009. doi:10.2215/CJN.07081009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jono S, Peinado C, Giachelli CM. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J Biol Chem. 2000;2:20197–20203. doi: 10.1074/jbc.M909174199. doi:10.1074/jbc.M909174199. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Bal BS, Gorski JP. Calcium and collagen binding properties of osteopontin, bone sialoprotein, and bone acidic glycoprotein-75 from bone. J Biol Chem. 1992;2:24871–24878. [PubMed] [Google Scholar]
- 42.Speer MY, Chien YC, Quan M, et al. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc Res. 2005;2:324–333. doi: 10.1016/j.cardiores.2005.01.023. doi:10.1016/j.cardiores.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 43.Proudfoot D, Skepper JN, Hegyi L, et al. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;2:1055–1062. doi: 10.1161/01.res.87.11.1055. doi:10.1161/01.RES.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 44.Abed E, Martineau C, Moreau R. Role of melastatin transient receptor potential 7 channels in the osteoblastic differentiation of murine MC3T3 cells. Calcif Tissue Int. 2011;2:246–253. doi: 10.1007/s00223-010-9455-z. doi:10.1007/s00223-010-9455-z. [DOI] [PubMed] [Google Scholar]
- 45.Abed E, Moreau R. Importance of melastatin-like transient receptor potential 7 and magnesium in the stimulation of osteoblast proliferation and migration by platelet-derived growth factor. Am J Physiol Cell Physiol. 2009;2:C360–C368. doi: 10.1152/ajpcell.00614.2008. doi:10.1152/ajpcell.00614.2008. [DOI] [PubMed] [Google Scholar]
- 46.Callera GE, He Y, Yogi A, et al. Regulation of the novel Mg2+ transporter transient receptor potential melastatin 7 (TRPM7) cation channel by bradykinin in vascular smooth muscle cells. J Hypertens. 2009;2:155–166. doi: 10.1097/hjh.0b013e3283190582. doi:10.1097/HJH.0b013e3283190582. [DOI] [PubMed] [Google Scholar]
- 47.Hutchison AJ, Wilkie M. Use of magnesium as a drug in chronic kidney disease. Clin Kidney J. 2012;5(Suppl 1):i62–i70. doi: 10.1093/ndtplus/sfr168. doi:10.1093/ndtplus/sfr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spiegel DM, Farmer B, Smits G, et al. Magnesium carbonate is an effective phosphate binder for chronic hemodialysis patients: a pilot study. J Ren Nutr. 2007;2:416–422. doi: 10.1053/j.jrn.2007.08.005. doi:10.1053/j.jrn.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 49.de Francisco AL, Leidig M, Covic AC, et al. Evaluation of calcium acetate/magnesium carbonate as a phosphate binder compared with sevelamer hydrochloride in haemodialysis patients: a controlled randomized study (CALMAG study) assessing efficacy and tolerability. Nephrol Dial Transplant. 2010;2:3707–3717. doi: 10.1093/ndt/gfq292. doi:10.1093/ndt/gfq292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanbay M, Yilmaz MI, Apetrii M, et al. Relationship between serum magnesium levels and cardiovascular events in chronic kidney disease patients. Am J Nephrol. 2012;2:228–237. doi: 10.1159/000341868. doi:10.1159/000341868. [DOI] [PubMed] [Google Scholar]