

Abstract
Sacrococcygeal location of myxopapillary ependymoma (MPE) is uncommon. Local recurrence and metastases are on record inspite of its benign characteristics. We report a rare case of sacrococcygeal MPE in an 11-month-old female child who showed typical myxopapillary ependymal histology along with anaplastic ependymal component. Ki-67 labeling index in the myxopapillary component was 4-5% and in the anaplastic component was 70%. Six weeks after gross total resection of the tumor, the child presented with local recurrence and metastasis in the right inguinal lymph nodes and was treated with chemotherapy. The present case of sacrococcygeal MPE with anaplastic ependymoma component is the second case on record in the medical literature, and the first case without any syndromic features. Metastasis in this case can be explained because of the anaplastic component, with mitotic count of 5-6/high power field and high Ki-67 labeling index.
Keywords: Anaplastic ependymoma, Ki-67, myxopapillary ependymoma, sacrococcyx
Introduction
Myxopapillary ependymoma (MPE) – WHO Grade I, has been primarily described in cauda equina-filum terminale region and elsewhere in the cranio-spinal axis along the ventricular system.[1] Subcutaneous sacrococcygeal location of this benign tumor is very uncommon and unusual.[2] Metastatic deposits of sacrococcygeal MPE in the regional lymph nodes, lungs, liver and bone have been reported.[2] The present case of sacrococcygeal MPE with anaplastic ependymoma component and inguinal lymph node metastasis in an infant is the second case on record in the medical literature, and the first case without any syndromic features.[3]
Case Report
An 11-month-old female child, with appropriate for age social and motor milestones, presented with a subcutaneous sacrococcygeal mass of 1 month duration. The skin over the lesion was stretched but not fixed to it. The mass was firm in consistency with restricted mobility. There was no bladder bowel disturbance. Ultrasound examination showed a large, solid mass with cystic component having mixed echogenicity, measuring 6.5 × 6 × 5.5 cm in the sacrococcygeal region. Magnetic resonance imaging (MRI) of the pelvis was limited only to the axial and coronal planes in T2-weighted sequence, which exhibited a 6 × 5.8 × 5 cm solid mass located in the midline posterior pelvis. The mass straddled across the sacrococcyx and was dumbbell shaped, with almost equal pre- and post-sacral components [Figure 1a–c]. The mass within the pelvis was insinuating around the rectum, and a small part was seen extending posterior and lateral to the urinary bladder. No pelvic lymphadenopathy was noted. Provisional clinical and radiological diagnosis was sacrococcygeal teratoma of Altman type II based on anatomical location and extension.
Figure 1.
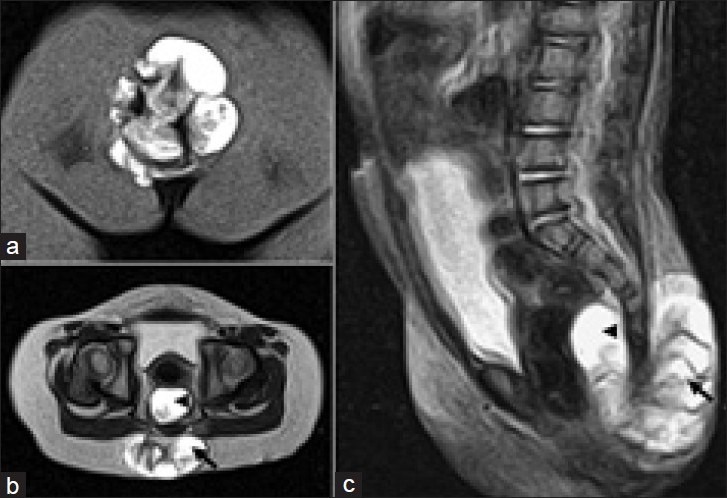
(a) Coronal fat suppressed image showing a rounded hyperintense mass lesion in the posterior lower midline; (b) Axial T2 weighted image shows the hyperintense pelvic component (arrowhead) and post-sacral component of the lesion (arrow); (c) Sagittal T2 weighted image shows that the lesion has both pre-sacral (arrowhead) and post-sacral (arrow) components straddling across the sacrococcyx, and insinuating around the rectum
The excision of sacrococcygeal mass was performed under general anesthesia after placing the patient in a prone (jackknife) position. The rectum was packed with gauze soaked in povidone iodine to prevent contamination of surgical field. Using the posterior sacral approach, a chevron skin incision was made. The post-sacral component of tumor was mobilized, but there was difficulty in dissection of the pre-sacral component of the tumor. The patient's position was changed to laparotomy lithotomy position. On abdominal exposure, the tumor's distal margin was mobilized from the posterior rectal wall, and lateral margins from the urinary bladder, and delivered by sacral incision. During surgery, care was taken to prevent injuries to the urinary bladder, sphincter, posterior rectal wall, pre-sacral venous plexus and sacral nerve roots. Hence, the tumor was excised en block with the combined approach and coccygectomy was performed. Post-operative period was uneventful.
Morphological examination showed an irregular mass with peripherally attached fat and skeletal muscle. Cut-surface was solid-cystic with focal papillae, myxoid areas and enclosed coccyx [Figure 2a]. Microscopy revealed a tumor composed of solid and cystic areas delineated by fibrocollagenous septae. The predominant component of tumor had characteristic perivascular rosettes and papillae with vascular cores lined by cuboidal to low columnar ependymal cells having eccentric nucleus and cytoplasmic processes abutting the vessel walls, and without discernible mitotic figures. The vascular cores showed myxoid change with focal hyalinization [Figure 2b]. This predominant myxopapillary component was continuous with highly cellular areas [Figure 2c] composed of perivascular rosettes and canals, lined by cells having oval to elongated nuclei with coarse chromatin [Figure 2d and e]. Nuclear overlapping and atypia, 5-6 mitotic figures/high power field (HPF) and punctate foci of necrosis were seen in this anaplastic ependymal component of the tumor. Ki-67 labeling index in the myxopapillary component was 4-5% [Figure 2f] and in the anaplastic component was 70% [Figure 2d and f]. The tumor was seen infiltrating the fibrocollagenous stroma and skeletal muscles.
Figure 2.
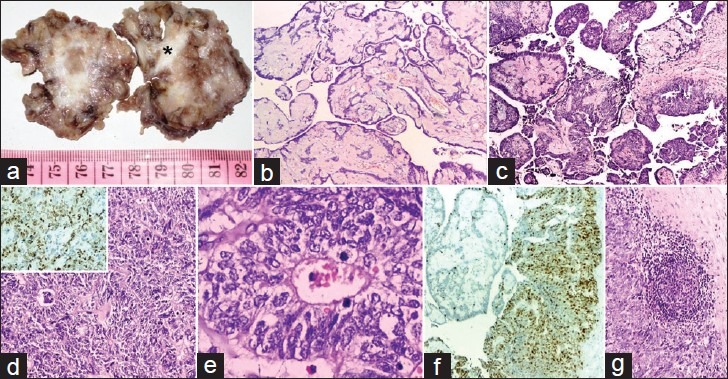
(a) Cut-surface with solid-cystic areas and enclosed coccyx (*); (b) papillae with myxoid fibrovascular cores lined by benign ependymal cells (H and E, ×100); (c) myxopapillary component in continuity with anaplastic ependymoma component (H and E, ×100); (d) composed of perivascular rosettes and canals (H and E, ×100; inset: High Ki-67 labeling index); (e) lined by pleomorphic cells with nuclear atypia and mitotic figures (H and E, ×400); (f) contrasting low and high Ki-67 labeling index in myxopapillary and anaplastic ependymal component (H and E, ×100); (g) Metastasis in lymph node (H and E, ×100)
The child presented 6 weeks later with a small recurrent pre-sacral deposit and palpable right-sided inguinal lymph nodes measuring 0.5-1 cm in diameter. Serum estimation of α-feto protein (AFP) and β-human chorionic gonadotropin (β-HCG) was within normal limits. Microscopy of the excised lymph nodes revealed metastasis of the anaplastic ependymoma component of the sacrococcygeal tumor [Figure 2g]. The patient received six cycles of Cisplatin and Etoposide. Currently, 1 year after completion of chemotherapy, there is no evidence of recurrence or further metastasis.
Discussion
Till date, 75 cases of subcutaneous sacrococcygeal MPE have been described in the medical literature.[2] The reasons for occurrence in this unusual site are either metastasis or direct extension to the sacrococcygeal soft tissues from a primary in the cauda equina-filum terminale, pre-sacral, pelvic or abdominal tumor. Rarely, primary MPE in skin or soft tissue of the sacrococcygeal area, without any demonstrable connection with the spinal cord, has been documented.[4] They probably originate from the coccygeal medullary vestige, heterotopic ependymal cell rests, extradural remnants of the filum terminale or extension of the intradural filum terminale.[2] The present case was probably a direct extension of tumor from the cauda equina-filum terminale because it had a dumbbell configuration, with almost equal pre-sacral and post-sacral (subcutaneous) components and the coccyx was part of the excised tumor specimen.
The age of presentation of sacrococcygeal MPE is 2 months to 67 years, with no sex predilection,[2] which is distinct from cauda equina MPE with 6-82 years as age of presentation and male: female ratio of 2.2:1.[1] Clinically, the differential diagnoses of a lesion in the sacrococcyx are pilonidal sinus, epidermal inclusion cyst, meningocele, lipoma, sacrococcygeal teratoma and neurogenic tumors.[2,4] Radiologically, sacrococcygeal MPE are hypointense on T1, heterogenously hyperintense on T2-weighted MRI and have heterogenous contrast enhancement. Most of them are circumscribed but others exhibit invasion into adjacent soft tissue or sacral bone.[2]
Hashish et al. recorded complications of sacrococcygeal teratoma resections such as post-operative constipation and fecal incontinence, bladder dysfunction and recurrence in 14.7%, 5.9% and 1.8%, respectively,[5] although varied incidences have been reported in different studies. Recurrence can occur due to incomplete resection with the presence of microscopic residues, non-resection of the entire coccyx, tumor spillage or nature of tumor per se.[5] The most common post-operative complication is wound infection in 15-20% of the cases, and other uncommon complications are draining sinus, wound dehiscence and rectoperineal fistula.[6] Although the present case had recurrence, there was no feature of bowel dysfunction and neuropathic bladder.
Spinal MPEs have a tendency for slow growth and local recurrence,[7] but multifocality,[8] local spinal dissemination,[9] intracranial[10] and extraneural metastasis[7] have also been described. Secondary intracranial lesions were detected at presentation to 13 years after the resection of the primary spinal MPE.[10] Similarly, metastasis to regional lymph nodes, lungs, liver or bone has been found in 20% of subcutaneous sacrococcygeal MPE, with metastases developing 10-20 years after the initial presentation.[2] According to Helwig et al., presence of 1-2 mitotic figures/HPF in occasional cases did not correlate with the metastatic behavior of sacrococcygeal MPE.[4]
Disseminated spinal MPEs had a mean Ki-67 labeling index of 2.4%, which does not signify malignant transformation.[11] Histologically, MPE with intracranial metastasis does not show features of malignancy.[10] The mean proliferation index for disseminated anaplastic ependymomas was 21%.[11] In comparison, our study showed 5-6 mitotic figures/HPF and Ki-67 labeling index of 70% in the anaplastic ependymoma component, which explains the metastasis to the right inguinal lymph nodes.
Singular case reports of sacrococcygeal teratoma with MPE[12] and anaplastic ependymoma[13] components are on record. Extensive sampling of our specimen did not reveal any germ cell or teratomatous component, which was further substantiated by normal serum AFP and β-HCG levels. Extradural subcutaneous ependymal tumor with myxopapillary and ependymoblastic differentiation in a case of Schinzel-Geidion syndrome[3] depicted solid and cystic areas, foci of necrosis, high mitosis and Ki-67 labeling index (40%) in the anaplastic component comparable to the present case.
Gross total resection is the best therapeutic option for MPE, and subtotal resection needs to be followed by adjuvant radiotherapy to prevent recurrence. Because the present case behaved aggressively, chemotherapy was instituted in treating this child. Long-term, regular follow-up with cranio-spinal MRI is recommended to detect recurrence or secondary deposits.[7]
Sacrococcygeal tumors in the pediatric age group should be extensively sampled to look for germ cell or teratomatous or anaplastic tumor components to institute proper management and should also be regularly followed-up with cranio-spinal MRI.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.McLendon RE, Rosenblum MK, Schiffer D, Weistler OD. Myxopapillary ependymoma. In: Louis DN, Ohgaki H, Weistler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. Lyon: International Agency of Research on Cancer; 2007. pp. 72–3. [Google Scholar]
- 2.Louis DN, Reifenberger G, Brat DJ, Ellison DW. Tumours: Introduction and neuroepithelial tumours. In: Love S, Louis DN, Ellison DW, editors. Greenfield's Neuropathology. 8th ed. II. London: Hodder Arnold; 2008. pp. 1821–2000. [Google Scholar]
- 3.Beschorner R, Wehrmann M, Ernemann U, Bonin M, Horber V, Oehl-Jaschkowitz B, et al. Extradural ependymal tumor with myxopapillary and ependymoblastic differentiation in a case of Schinzel-Giedion syndrome. Acta Neuropathol. 2007;113:339–46. doi: 10.1007/s00401-006-0179-0. [DOI] [PubMed] [Google Scholar]
- 4.Helwig EB, Stern JB. Subcutaneous sacrococcygeal myxopapillary ependymoma. A clinicopathologic study of 32 cases. Am J Clin Pathol. 1984;81:156–61. doi: 10.1093/ajcp/81.2.156. [DOI] [PubMed] [Google Scholar]
- 5.Hashish AA, Fayad H, El-attar AA, Radwan MM, Ismael K, Ashour MH, Elhalaby E. Sacrococcygeal teratoma: Management and outcomes. Ann Paed Surg. 2009;5:119–25. [Google Scholar]
- 6.Ein SH. Resection of Sacrococcygeal Teratoma. In: Fessler RG, Sekhar LN, editors. Atlas of Neurosurgical Techniques: Spine and Peripheral Nerves. New York: Thieme; 2006. pp. 723–8. [Google Scholar]
- 7.Tzerakis N, Georgakoulias N, Kontogeorgos G, Mitsos A, Jenkins A, Orphanidis G. Intraparenchymal myxopapillary ependymoma: Case report. Neurosurgery. 2004;55:981. doi: 10.1227/01.neu.0000137278.84588.06. [DOI] [PubMed] [Google Scholar]
- 8.Andoh H, Kawaguchi Y, Seki S, Asanuma Y, Fukuoka J, Ishizawa S, et al. Multi-focal Myxopapillary Ependymoma in the Lumbar and Sacral Regions Requiring Cranio-spinal Radiation Therapy: A Case Report. Asian Spine J. 2011;5:68–72. doi: 10.4184/asj.2011.5.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fassett DR, Pingree J, Kestle JR. The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg. 2005;102:59–64. doi: 10.3171/ped.2005.102.1.0059. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborti S, Govindan A, Alapatt JP, Radhakrishnan M, Santosh V. Primary myxopapillary ependymoma of the fourth ventricle with cartilaginous metaplasia: A case report and review of the literature. Brain Tumor Pathol. 2012;29:25–30. doi: 10.1007/s10014-011-0059-8. [DOI] [PubMed] [Google Scholar]
- 11.Rezai AR, Woo HH, Lee M, Cohen H, Zagzag D, Epstein FJ. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618–24. doi: 10.3171/jns.1996.85.4.0618. [DOI] [PubMed] [Google Scholar]
- 12.Hany MA, Bouvier R, Ranchère D, Bergeron C, Schell M, Chappuis JP, et al. Case report: A preterm infant with an extradural myxopapillary ependymoma component of a teratoma and high levels of alpha-fetprotein. Pediatr Hematol Oncol. 1998;15:437–41. doi: 10.3109/08880019809016573. [DOI] [PubMed] [Google Scholar]
- 13.Busse C, Nazeer T, Kanwar VS, Wolden S, LaQuaglia MP, Rosenblum M. Sacrococcygeal immature teratoma with malignant ependymoma component. Pediatr Blood Cancer. 2009;53:680–1. doi: 10.1002/pbc.22079. [DOI] [PubMed] [Google Scholar]