

Abstract
Purpose
To identify the disease-causing mutation(s) in a Chinese family with autosomal recessive Usher syndrome type 1 (USH1).
Methods
An ophthalmic examination and an audiometric test were conducted to ascertain the phenotype of two affected siblings. The microsatellite marker D11S937, which is close to the candidate gene MYO7A (USH1B locus), was selected for genotyping. From the DNA of the proband, all coding exons and exon-intron boundaries of MYO7A were sequenced to identify the disease-causing mutation(s). Restriction fragment length polymorphism (RFLP) analysis was performed to exclude the alternative conclusion that the mutations are non-pathogenic rare polymorphisms.
Results
Based on severe hearing impairment, unintelligible speech, and retinitis pigmentosa, a clinical diagnosis of Usher syndrome type 1 was made. The genotyping results did not exclude the USH1B locus, which suggested that the MYO7A gene was likely the gene associated with the disease-causing mutation(s) in the family. With direct DNA sequencing of MYO7A, two novel compound heterozygous mutations (c.3742G>A and c.6051+1G>A) of MYO7A were identified in the proband. DNA sequence analysis and RFLP analysis of other family members showed that the mutations cosegregated with the disease. Unaffected members, including the parents, uncle, and sister of the proband, carry only one of the two mutations. The mutations were not present in the controls (100 normal Chinese subjects=200 chromosomes) according to the RFLP analysis.
Conclusions
In this study, we identified two novel mutations, c.3742G>A (p.E1248K) and c.6051+1G>A (donor splice site mutation in intron 44), of MYO7A in a Chinese non-consanguineous family with USH1. The mutations cosegregated with the disease and most likely cause the phenotype in the two affected siblings who carry these mutations compound heterozygously. Our finding expands the mutational spectrum of MYO7A.
Introduction
Usher syndrome (USH) is an autosomal recessive disease characterized by the association of retinitis pigmentosa (RP) and sensorineural hearing loss (SNHL), with or without vestibular dysfunction. The syndrome is the most common cause of combined blindness and deafness, accounting for more than 50% of individuals who are both deaf and blind, about 18% of RP cases, and 5% of all cases of congenital deafness [1]. According to previous reports, the overall prevalence of USH ranges from 3.2 per 100,000 to 6.2 per 100,000 individuals [2].
Clinically, USH is divided into three types based on disease severity and progression of the clinical course. Usher type 1 (USH1) is characterized by profound congenital deafness, vestibular areflexia, and early onset RP. Patients with type 2 (USH2) display moderate to severe hearing loss and later onset of RP. Patients with Usher syndrome type 3 (USH3) show progressive postlingual hearing loss, variable onset of RP, and variable vestibular dysfunction. Genetically, Usher syndrome is highly heterogeneous. To date, mutations in 12 loci and ten genes have been found to be responsible for USH. For USH1, six genes—MYO7A (USH1B) [3], USH1C (USH1C) [4], CDH23 (USH1D) [5], PCDH15 (USH1F) [6], USH1G (USH1G) [7], and CIB2 (USH1J) [8]—have been cloned. Mutations in three genes—USH2A (USH2A) [9,10], GPR98 (USH2C) [11], and WHRN (USH2D) [12]—have been identified as disease-causing for USH2. Mutations in the CLRN1 (USH3A) gene have been found in cases of USH3 [13].
Among the three types, USH1 is the most severe form. Patients typically lose their hearing at birth or in the first year of life and usually do not develop normal speech. The vestibular function may also be impaired, which causes a delay in motor development and walking in those affected compared to normal children. Onset of RP often occurs in the first decade of life, with a progressively constricted visual field and impaired visual acuity. In USH1 families, MYO7A is the most commonly mutated gene, accounting for approximately 50% [14,15], followed by CDH23, PCDH15, USH1C, and USH1G.
The MYO7A gene is located on the long arm of chromosome 11 at position 13.5 (11q13.5). The biggest transcript of MYO7A consists of 49 exons and encodes a 2,215 amino acid unconventional myosin named myosin VIIA. Thus far, more than 250 MYO7A mutations have been reported. Most of these mutations (more than 95%) cause Usher syndrome type 1, while the rest of the mutations are responsible for autosomal recessive or dominant non-syndromic deafness, according to the Human Gene Mutation Database Professional 2012.2.
Here we report on the investigation of a Chinese family with early onset of combined blindness and deafness, key features of USH1. Genotyping results suggested that MYO7A was likely the gene associated with the disease in this family. Through direct DNA sequence analysis of the 48 coding exons of MYO7A, we identified two novel compound heterozygous mutations that likely cause the observed phenotype.
Methods
Study subjects
A Chinese family consisting of 11 individuals (seven participated in this study) from Hubei province was investigated in this study. Written informed consent was obtained from the participants, and the research was approved by the Ethics Committee of Huazhong University of Science and Technology. Detailed ophthalmologic examinations of the subjects, including visual acuity, slit-lamp biomicroscopy, applanation intraocular pressure, visual field, dilated funduscopic examination, and fundus photography, as well as audiometric tests, were performed at the Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. Whole peripheral blood samples were collected by clinicians in Venous Blood Vacuum Collection Tubes containing tripotassium EDTA as anticoagulant, from seven family members in the Union Hospital, Tongji Medical College, Wuhan, PRC. The blood samples were stored at -4 °C for up to 7 days before DNA was extracted using the DNA isolation kit for mammalian blood (Tiangen Biotech Co., Ltd., Beijing, China).
Genotyping
To date, six genes associated with USH1 have been cloned. Haplotype analysis was done in the order of their causal frequency: MYO7A, CDH23, PCDH15, USH1C, USH1G and CIB2 [8,16]. For MYO7A, the microsatellite marker D11S937 (77.8 Mb on chromosome 11, Applied Biosystems, Inc., Foster City, CA), which is tightly linked to the candidate gene MYO7A (76.8 Mb on chromosome 11), was selected for genotyping. The PCR products were separated by electrophoresis on an ABI 3100 Genetic Analyzer (Applied Biosystems), and genotypes were determined with Peak Scanner Software v1.0 (Applied Biosystems).
Mutation screening
Specific primers were designed to amplify all coding exons and intron-exon junctions of MYO7A (GenBank accession number NM_000260.3; primer sequences are available on request). Amplification conditions were 94 °C for 5 min, then 35 cycles of 94 °C for 30 s, 56–60 °C for 30 s, 72 °C for 30 s or 60 s, and a final extension time of 10 min at 72 °C. PCR products were separated on a 1.5% agarose gel. DNA fragments were purified from the gel with the QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA). DNA sequencing was performed with the BigDye Terminator Cycle Sequencing Kit on an ABI 3130 Genetic Analyzer (Applied Biosystems).
Restriction fragment length polymorphism analysis
For the c.3742G>A mutation, a 454 bp DNA fragment was amplified (forward: 5′-CGG CTG CCC TCA AAA TCC ACA T-3′; reverse: 5′-TGG CAG GTA AAG GCA TTG AGA CA-3′) and cut into 172 bp and 282 bp with BstXI (New England BioLabs Inc., Beverly, MA) at 37 °C for 4 h. For the c.6051+1G>A mutation, a 99 bp DNA fragment was amplified (forward: 5′-CCA CGG TGC CAG GGA AGG ATC-3′; reverse: 5′-CAA CGC TAG CTG TGC ACG AAG G-3′) and cut into 41 bp and 48 bp with ScrFI (New England BioLabs) at 37 °C for 4 h. Both mutations eliminated the respective restriction sites. Digested PCR products were run on 1.5% agarose gels. Samples from all available family members and 100 normal controls were used in the RFLP analysis.
Results
Clinical examinations and pedigree analysis
There are 11 individuals in the non-consanguineous family (Figure 1). The proband’s paternal grandparents and maternal grandparents were not enrolled in the genetic study. The 20-year-old proband had gradually declining night vision and hearing loss since childhood and so did his younger brother. The proband was emmetropic with Snellen visual acuity recorded as 0.6 in both eyes. Examination of the anterior segment showed normal cornea, clear anterior chamber, and transparent lens. Fundus photography revealed vessel attenuation and characteristic bone spicule-like pigment with macular involvement (Figure 2). Pure tone audiometry showed severe to profound bilateral sensorineural hearing impairment (Figure 3). The two affected individuals have completely lost their hearing and are practically unable to speak. Other members of the family are normal. According to the family history and results from the audiometric test and ophthalmic examinations, we concluded that the two patients display the typical presentation of autosomal recessive Usher syndrome type 1 (Figure 1).
Figure 1.

Pedigree of the Chinese family with autosomal recessive USH1. The filled symbols represent the affected individuals, while the empty symbols indicate the normal individuals. The proband (III1) is identified with an arrow. There is no consanguinity in this family.
Figure 2.
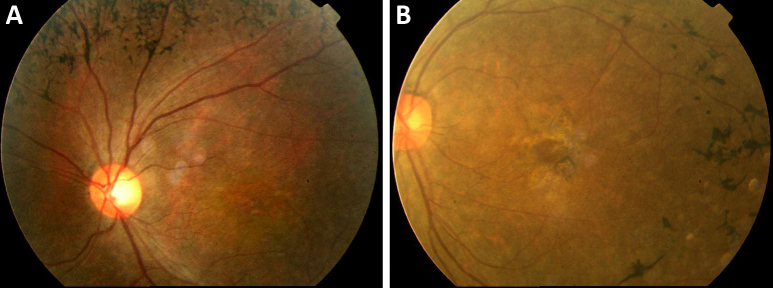
Fundus photographs of the proband at age 20 years. A: Retinal vessel attenuation and characteristic bone spicule pigment are shown, indicating a typical retinitis pigmentosa phenotype. B: The macula was involved.
Figure 3.

Audiometry results of the proband. Severe to profound bilateral hearing impairment was indicated.
Genotyping and mutational analysis
The genotyping results did not exclude MYO7A, suggesting that one or more mutation(s) in MYO7A likely caused the phenotype observed in the two patients. Sequence analysis of all 48 coding exons and exon-intron boundaries of MYO7A revealed in the proband two novel compound heterozygous mutations. One was a c.3742G>A mutation in exon 29, which resulted in a substitution of lysine for glutamic acid at codon 1248 (p.E1248K; Figure 4A). The other was a G to A change at the conserved donor splice site in intron 44 (c.6051+1G>A; Figure 4B). We performed DNA sequencing for other available family members. The results indicated that the two mutations completely cosegregated with the disease in this family.
Figure 4.

Identification of the two novel MYO7A mutations and their cosegregation with the disorder in the family. A: DNA sequencing profiles for regions around position C.3742. The upper panel represents the sequencing result of wild-type (wt) from individual II1. The lower panel represents the sequencing result of mutant (mt) from individual III1. The mutation c.3742G>A is indicated with an arrow on the mt trace. The affected codon is marked with an underline. B: DNA sequencing profiles for regions around position c.6051+1. The mutation c.6051+1G>A is indicated with an arrow on the mt trace. The exon-intron boundary is labeled. C: Representative restriction fragment length polymorphism (RFLP) analysis for position c.3742. For normal individuals II1, II3, and II4, restriction by BstXI resulted in two bands, while for individuals II2, III1, and III3, restriction by BstXI resulted in three bands, suggesting the presence of a mutation in the polymerase chain reaction (PCR) product. D: Representative RFLP analysis for position c.6051+1. For normal individuals II1, II2, and III3, restrictive digested by ScrFI resulted in just one mixed band with fragments 41 bp and 48 bp, while for individuals II3, II4, III1, and III2, digested by ScrFI resulted in two bands, suggesting the presence of a mutation in the PCR product. E: RFLP analysis for the two mutations in 100 normal controls out of the family. Only one representative image for each mutation (left panel for position c.3742G, right panel for position c.6051+1G) was selected to be shown in the paper. The asterisk in the right panel indicates the PCR product without adding the restriction endonuclease ScrFI, which serves as a control.
We further performed RFLP analysis for the family members as well as other 100 normal individuals. To identify the mutation c.3742G>A, a 454-bp fragment was amplified and digested by BstXI. As shown in Figure 4C, for homozygous normal individuals II1, II3, and II4, the restrictive digestion by BstXI resulted in two bands of 282 bp and 172 bp. In contrast, for the heterozygous individuals (II2, III1, III2, and III3) who carry the mutation c.3742G>A, the restrictive digestion by BstXI resulted in three bands of 454 bp, 282 bp, and 172 bp.
To identify the mutation c.6051+1G>A, a 99-bp fragment was amplified and digested by ScrFI. As shown in Figure 4D, for homozygous normal individuals II1, II2, and III3, the restrictive digestion resulted in a band of about 50 bp. In contrast, for the heterozygous individuals (II3, II4, III1, and III2) who carry the mutation c.6051+1G>A, the restrictive digestion resulted in two bands of 50 bp and 99 bp. These results confirmed that our patients III1 and III2 with USH carry both mutations of c.3742G>A and c.6501+1G>A.
RFLP analysis also indicated that these two mutations were not present in the 100 normal control individuals. The RFLP results for some of the normal controls are shown in Figure 4E.
The c.3742G>A mutation was predicted to cause a glutamic acid to lysine change at codon 1248. According to the alignment of the amino acid sequences of the first MyTH4 domain of myosin VIIA (Figure 5), the glutamic acid at position 1248 (E1248) is completely conserved, from Homo sapiens to Caenorhabditis elegans.
Figure 5.

Alignment of the amino acid sequences of the first MyTH4 domain of myosin VIIA from different species. The arrow indicates the highly conserved E1248 residue.
Discussion
In the present study, we identified two novel mutations of MYO7A, c.3742G>A and c.6051+1G>A, in a Chinese family. The two mutations cosegregated with the disorder in the family and were absent in the 100 normal controls. These results indicate that the two compound heterozygous mutations are likely pathogenic and are the cause of USH1 in this family.
The human myosin VIIA is composed of a N-terminal motor domain (1–729), a neck region containing five IQ motifs (745–857), a short predicted coiled coil domain (858–935), a MyTH4 domain (1017–1253), a 4.1-Ezrin-radixin-moesin (FERM) domain (1258–1602), an SH3 domain (1603–1672), and a second C-terminal MyTH4-FERM tandem domain (1747–2205). The motor domain is also known as the myosin head-like domain, and could bind F-actin and ATP [17].
Myosin VIIA is expressed in the hair cells of the inner ear, the retinal pigment epithelium (RPE), and the photoreceptor cells of the retina. In the photoreceptor cells, myosin VIIA is present at the connecting cilium and regulates opsin transport [18]. In the RPE cells, myosin VIIA participates in the light cycle–dependent movement of melanosome transportation [19] and the normal functioning of the visual retinoid cycle [20] and is associated with lysosomes [21]. In the hair cells of the inner ear, myosin VIIA and the other four USH1-related proteins (harmonin encoded by USH1C, Sans encoded by USH1G, CDH23 encoded by USH1D, PCDH15 encoded by USH1F) participate in the formation of the mechanotransduction complex [22], which is critical for detecting sound.
Several proteins have been identified to interact with myosin VIIA. In addition to interacting with actin, harmonin, Sans, CDH23, and PCDH15, myosin VIIA also interacts with MYRIP [19], RPE65 [20], and WHRN (USH2D) [23]. Myosin VIIA is predominantly monomeric in cells, but can be induced to dimerize via the predicted coiled-coil-domain after cargo binding and functions as a cargo transporter [24].
The c.3742G>A mutation was predicted to cause a glutamic acid change to lysine at the highly conserved codon 1248 in the first MyTH4-FERM tandem domain, which mediates the interaction between myosin VIIA and the scaffold protein Sans (USH1G) [25]. The E1248K mutation may prevent this interaction and thus could disrupt the normal function of myosin VIIA.
The c.6051+1G>A mutation is a donor splice site mutation in intron 44 of the MYO7A gene. Using the online software NetGene2 v2.4, we predicted an impact of the mutation on pre-mRNA splicing of MYO7A. The mutation would destroy that splice site, leading to a truncated protein lacking the last approximate 200 amino acid residues. The C-terminal tail of myosin VIIA, containing the SH3, MyTH4, and FERM domains (1605–2215), interacts with harmonin (USH1C) [26]. Therefore, the mutant myosin VIIA protein may lose the ability to bind harmonin, which acts as a scaffold protein linking the proteins in the cell membrane to the proteins in the cytoskeleton. The additive effects of the two identified compound heterozygous mutations could cause a complete dysfunction of myosin VIIA in the two patients, leading to the observed phenotype.
Applying genotyping microarray and next-generation sequencing will allow for large-scale screening in patients with USH. These methods will become the standard approaches in clinical diagnosis and scientific research in the future. Although numerous mutations in USH genes have been found, much remains to be done to understand the pathogenesis of Usher syndrome and facilitate development of potential treatments for the disease.
Acknowledgments
We are grateful to the family members for their participation in this study. This study is supported by grants from the National Natural Science Foundation of China [No. 81270983, 31071106, and 30771199].
References
- 1.Yan D, Liu XZ. Genetics and pathological mechanisms of Usher syndrome. J Hum Genet. 2010;55:327–35. doi: 10.1038/jhg.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. An update on the genetics of usher syndrome. J Ophthalmol. 2011;2011:417217. doi: 10.1155/2011/417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Et A. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–1. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 4.Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26:51–5. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- 5.Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, Del CCM, Vila MC, Molina OP, Gal A, Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27:108–12. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–71. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- 8.Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, Yousaf R, Sinha GP, Lee S, Terrell D, Hegde RS, Ali RA, Anwar S, Andrade-Elizondo PB, Sirmaci A, Parise LV, Basit S, Wali A, Ayub M, Ansar M, Ahmad W, Khan SN, Akram J, Tekin M, Riazuddin S, Cook T, Buschbeck EK, Frolenkov GI, Leal SM, Friedman TB, Ahmed ZM. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. 2012;44:1265–71. doi: 10.1038/ng.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–7. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 10.van Wijk E, Pennings RJE, Te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FPM, Cremers CWRJ, Kremer H. Identification of 51 Novel Exons of the Usher Syndrome Type 2A (USH2A) Gene That Encode Multiple Conserved Functional Domains and That Are Mutated in Patients with Usher Syndrome Type II. Am J Hum Genet. 2004;74:738–44. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weston MD, Luijendijk MWJ, Humphrey KD. M O Ller C, Kimberling WJ. Mutations in the VLGR1 Gene Implicate G-Protein Signaling in the Pathogenesis of Usher Syndrome Type II. Am J Hum Genet. 2004;74:357–66. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebermann I, Scholl HP, Charbel IP, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121:203–11. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- 13.Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001;69:673–84. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaijo T, Aller E, Beneyto M, Najera C, Graziano C, Turchetti D, Seri M, Ayuso C, Baiget M, Moreno F, Morera C, Perez-Garrigues H, Millan JM. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J Med Genet. 2007;44:e71. doi: 10.1136/jmg.2006.045377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Quesne Stabej P, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, Emmerson J, Haralambous E, Hughes Y, Steel KP, Luxon LM, Webster AR, Bitner-Glindzicz M. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012;49:27–36. doi: 10.1136/jmedgenet-2011-100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolz HJ, Roux AF. Clinical utility gene card for: Usher syndrome. Eur J Hum Genet. 2011;19 doi: 10.1038/ejhg.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue A, Ikebe M. Characterization of the motor activity of mammalian myosin VIIA. J Biol Chem. 2003;278:5478–87. doi: 10.1074/jbc.M210489200. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19:6267–74. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Amraoui A, Schonn JS, Kussel-Andermann P, Blanchard S, Desnos C, Henry JP, Wolfrum U, Darchen F, Petit C. MyRIP, a novel Rab effector, enables myosin VIIa recruitment to retinal melanosomes. EMBO Rep. 2002;3:463–70. doi: 10.1093/embo-reports/kvf090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopes VS, Gibbs D, Libby RT, Aleman TS, Welch DL, Lillo C, Jacobson SG, Radu RA, Steel KP, Williams DS. The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet. 2011;20:2560–70. doi: 10.1093/hmg/ddr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soni LE, Warren CM, Bucci C, Orten DJ, Hasson T. The unconventional myosin-VIIa associates with lysosomes. Cell Motil Cytoskeleton. 2005;62:13–26. doi: 10.1002/cm.20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grati M, Kachar B. Myosin VIIa and Sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci USA. 2011;108:11476–81. doi: 10.1073/pnas.1104161108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, Perfettini I, Legrain P, Richardson G, Hardelin JP, Petit C. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet. 2005;14:401–10. doi: 10.1093/hmg/ddi036. [DOI] [PubMed] [Google Scholar]
- 24.Sakai T, Umeki N, Ikebe R, Ikebe M. Cargo binding activates myosin VIIA motor function in cells. Proc Natl Acad Sci USA. 2011;108:7028–33. doi: 10.1073/pnas.1009188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu L, Pan L, Wei Z, Zhang M. Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science. 2011;331:757–60. doi: 10.1126/science.1198848. [DOI] [PubMed] [Google Scholar]
- 26.Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–99. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]