

Abstract
Introduction
Fatty acid amide hydrolase (FAAH) is responsible for the enzymatic degradation of the fatty acid amide family of signaling lipids, including the endogenous cannabinoid (endocannabinoid) anandamide. The involvement of the endocannabinoid system in pain and other nervous system disorders has made FAAH an attractive target for drug development. Companion molecular imaging probes are needed, however, to assess FAAH inhibition in the nervous system in vivo. We report here the synthesis and in vivo evaluation of [18F]PF-9811, a novel PET ligand for non-invasive imaging of FAAH in the brain.
Methods
The potency and selectivity of unlabeled PF-9811 were determined by activity-based protein profiling (ABPP) both in vitro and in vivo. [18F]PF-9811 was synthesized in a 3-step, one-pot reaction sequence, followed by HPLC purification. Biological evaluation was performed by biodistribution and dynamic PET imaging studies in male rats. The specificity of [18F]PF-9811 uptake was evaluated by pre-administration of PF-04457845, a potent and selective FAAH inhibitor, 1 h prior to radiotracer injection.
Results
Biodistribution studies show good uptake (SUV∼0.8 at 90 min) of [18F]PF-9811 in rat brain, with significant reduction of the radiotracer in all brain regions (37%–73% at 90 min) in blocking experiments. Dynamic PET imaging experiments in rat confirmed the heterogeneous uptake of [18F]PF-9811 in brain regions with high FAAH enzymatic activity, as well as statistically significant reductions in signal following pre-administration of the blocking compound PF-04457845.
Conclusions
[18F]PF-9811 is a promising PET imaging agent for FAAH. Biodistribution and PET imaging experiments show that the tracer has good uptake in brain, regional heterogeneity, and specific binding as determined by blocking experiments with the highly potent and selective FAAH inhibitor, PF-04457845.
Keywords: FAAH, Positron emission tomography, Rats, Fluorine-18, PET ligand, Molecular imaging
1. Introduction
Endogenous cannabinoids (endocannabinoids) are lipid transmitters that activate the cannabinoid G-protein coupled receptors CB1 and CB2 [1–4]. N-Arachidonoyl ethanolamine (anandamide, AEA) is one of two endocannabinoids identified in mammals [5] and its biological actions are terminated by the integral membrane serine hydrolase fatty acid amide hydrolase (FAAH) [6,7]. FAAH, in addition to hydrolyzing AEA, regulates other fatty acid amide signaling molecules [8] with anti-inflammatory and analgesic properties such as N-palmitoyl ethanolamine (PEA), which exerts its biological activity via non-cannabinoid pathways [9]. Genetic or pharmacological inactivation of FAAH leads to elevated endogenous levels of fatty acid amides and a range of analgesic, anti-inflammatory, anxiolytic and anti-depression effects in animal models [10,11]. Importantly, these behavioral phenotypes occur in the absence of the undesirable side effects (i.e., disruptions in motility, cognition, or body temperature) observed with direct cannabinoid receptor agonists, indicating that FAAH may represent an attractive therapeutic target for treatment of pain, inflammation, and other central nervous system (CNS) disorders.
Several classes of reversible and irreversible covalent FAAH inhibitors have been reported (Fig. 1), including electrophilic ketone inhibitors OL-135 (1) [12], carbamates such as URB597 (2) [13] and ureas such as PF-3845 (3) [14,15]. In addition, several groups have recently reported apparent noncovalent FAAH inhibitors with no obvious serine-interacting groups [16,17]. The carbamates and ureas have been shown to irreversibly inhibit FAAH by carbamylation of FAAH's catalytic Ser241 nucleophile [18,19]. Recently we have disclosed our efforts to optimize the potency, selectivity and physicochemical properties of the urea series leading to the identification of PF-04457845 (4) as a clinical candidate [20,21]. In this manuscript, we disclose the development of a fluorine-18 labeled analog of 4, [18F]PF-9811 (6), as a novel radiotracer for PET imaging of FAAH in the brain.
Fig. 1.
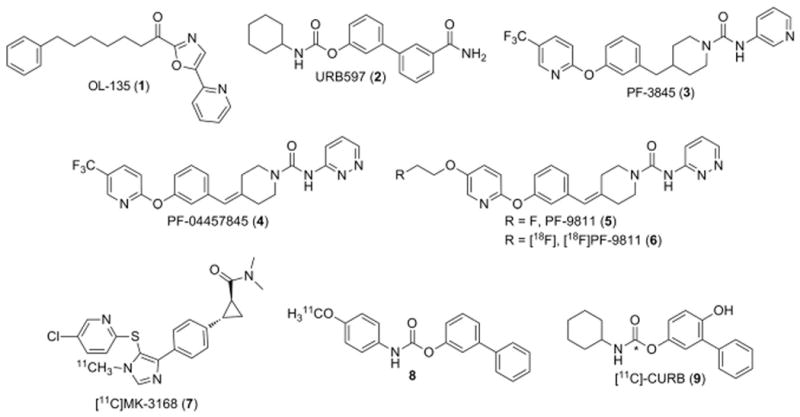
Chemical structures of FAAH inhibitors (1–4) and radiotracers (5–9) including an [18F]-labeled analog of PF-04457845 referred to as [18F]PF-9811.
4 has excellent pharmacokinetic properties including high oral bioavailability (88%) and high brain penetration (B/P=1.3–1.6) in rats [20,21], and displays exquisite selectivity for FAAH relative to other serine hydrolases as determined by competitive activity-based protein profiling (ABPP). A single oral administration of 4 at 1 mg/kg exhibited sustained inhibition of brain FAAH activity, maximal elevations of brain AEA, and in vivo efficacy in rat models of acute inflammatory pain for at least 24 h. Significantly, we also reported a tight relationship between in vivo efficacy and FAAH activity/AEA modulation in brain and blood leukocytes/plasma, where near complete inhibition of FAAH and a maximal sustained elevation of AEA appear to be needed for in vivo efficacy [15,20]. The ability to measure both FAAH activity and AEA levels from blood leukocytes and plasma, respectively, provides valuable peripheral target and mechanistic biomarkers for human clinical studies. However a similar central mechanistic biomarker is lacking in humans because it is challenging to measure AEA levels in cerebrospinal fluid [22]. It would therefore be highly desirable to have a PET tracer capable of measuring FAAH activity in human brain to serve as a central biomarker and guide selection of a dose that fully inhibits FAAH in the brain.
Recently three carbon-11 labeled inhibitors have been reported as potential PET radiotracers for imaging FAAH in the brain, with two of them based on the URB597 template. Researchers at Merck disclosed the results of PET scans performed under baseline and blocking conditions for [11 C]MK-3168 (7), a reversible FAAH inhibitor [23]. Heterogeneous, specific brain uptake consistent with known regional FAAH distribution was observed in rhesus monkeys, while human studies showed high brain uptake and a more homogeneous regional profile. Studies of compound URB597-based 8 resulted in low brain uptake, significant peripheral metabolism, and the existence of radiolabeled metabolites in the CNS [24]. In contrast, [11 C]-CURB (9) had good brain uptake in rats and was shown to irreversibly bind to FAAH which was blocked by pretreatment with 2 [25]. We set out to develop a radiotracer for PET imaging of FAAH based on the more potent and selective scaffold of 4, a FAAH inhibitor which has been advanced into clinical studies [20,21,26]. We also wanted to incorporate a fluorine-18 label so we could image for longer time periods if necessary. We found that the trifluoromethyl substituent of 4 could be replaced with a fluoroethoxy group while maintaining excellent FAAH inhibition potency and selectivity as well as favorable in vitro ADME properties, which set the stage for incorporating the 18F-radioisotope.
2. Results
2.1. Chemistry
As illustrated in Scheme 1, we started the synthesis of unlabeled PF-9811 from tert-butyl 4-(3-((5-bromopyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate 10, which was synthesized from commercially available 5-bromo-2-chloropyridine in four steps following the previously reported synthetic route for 4 [20]. Treatment of bromopyridine 10 with bis(pinacolato)diboron in the presence of catalytic amount of [1,1-bis(diphenylphosphine)ferrocene]dichloropalladium (II) afforded pinocolborane 11 in 75% yield, which was converted to the corresponding pyridyl alcohol 12 in 65% yield via oxidation mediated by peracetic acid. Mitsunobu reaction with 2-fluoroethanol gave the corresponding fluoroethoxy intermediate 13. Removal of the Boc protecting group using HCl in dioxane and subsequent coupling reaction with phenyl pyridazin-3-ylcarba-mate in the presence of triethylamine provided PF-9811 in 45% over three steps. In vitro ADME data indicated that PF-9811 had high passive permeability (MDCK PappAB=18.2 10−6 cm/s) and an acceptable efflux ratio (MDR1 BA/AB=2.52).
Scheme 1.
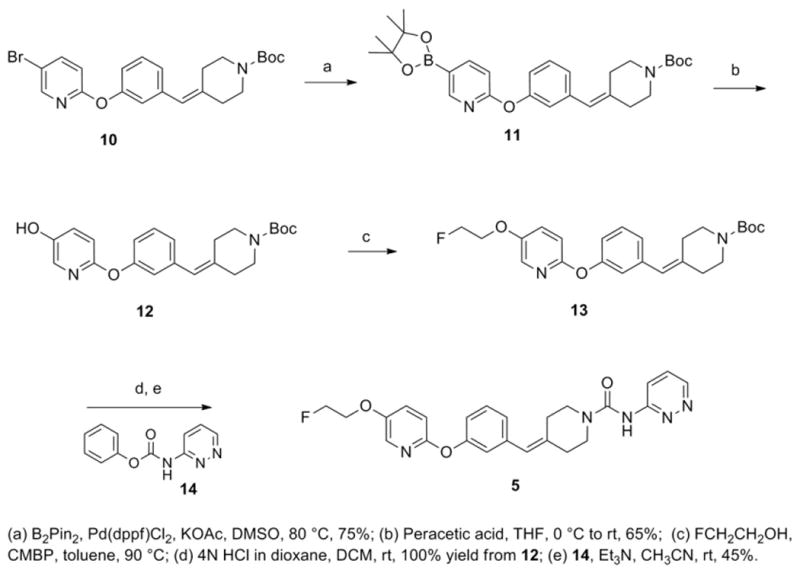
Synthetic scheme of 4-(3-((5-(2-fluoroethoxy)pyridin-2-yl)oxy)benzylidene)-N-(pyridazin-3-yl)piperidine-1-carboxamide (5).
We employed a 2-step method to prepare the tosylate precursor 16 for radiolabeling (Scheme 2). Treatment of the advanced pyridyl alcohol intermediate 12 with 2-bromoethoxy tert-butyl dimethylsilane and potassium carbonate in the presence of 18-crown-6, followed by silyl deprotection with TBAF in one-pot yielded the corresponding alcohol 15 in 83% yield, which upon tosylation afforded the desired radiolabeling precursor 16 in 57% yield.
Scheme 2.
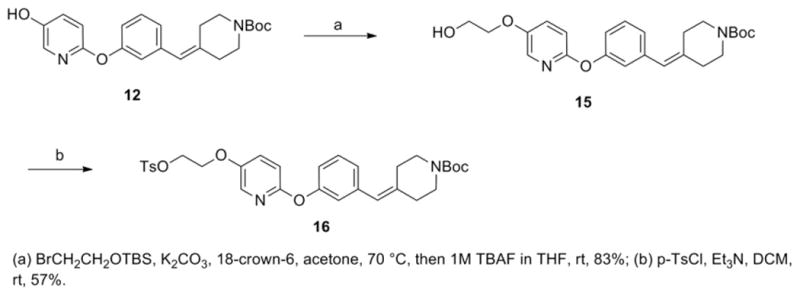
Synthetic scheme of tert-butyl 4-(3-((5-(2-(tosyloxy)ethoxy)pyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate (16).
2.2. Radiochemistry
We envisioned three possible routes to synthesize [18F]PF-9811. The most direct route is to fluorinate the tosylated starting material 17 (Scheme 3). All efforts to produce [18F]PF-9811 by this route proved unsuccessful. This included using either acetonitrile or 2-methyl-2-butanol as the solvent, temperatures from 80 to 100 °C, and 10– 30 min reaction times. Analysis of the reaction by both thin layer chromatography and liquid chromatography indicated starting material and polar by-products as the main components in the crude mixture. Similar results were observed in the chemistry using the unlabeled (fluorine-19) analogs, and no attempts were made to isolate and identify the by-products.
Scheme 3.
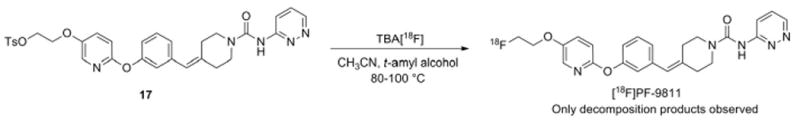
Direct fluorination scheme (Route A) to produce [18F]PF-9811.
The second route was a two-step method that first generated the 2-[18F]fluoroethyl tosylate synthon from the ditosyloxyethane precursor, followed by reaction with the phenol precursor 18 (Scheme 4). Initially efforts were made toward a one-pot reaction where the 2-[18F]fluoroethyl tosylate precursor was not isolated. After generation of the 2-[18F]fluoroethyl tosylate, the solvent was evaporated and it was reacted directly with the phenol precursor and base (tetra-butylammonium hydroxide), followed by HPLC purification. Unfortunately, the final product could not be separated from other chemical impurities cleanly using a number of different columns and mobile phases. It was envisioned that if the 2-[18F]fluoroethyl tosylate was isolated first, followed by reaction with the precursor in a separate vessel, the crude mixture would be cleaner and easier to purify. As a result, a two-pot procedure was attempted which first isolated the 2-[18F]fluoroethyl tosylate via semi-prep HPLC as a solution in dimethylacetamide. This solution was then reacted with the phenol precursor, using solid potassium carbonate as the base instead of tetrabutylammonium hydroxide. However, this method did not significantly improve the chemical purity of the final product, and thus this route was ultimately abandoned.
Scheme 4.
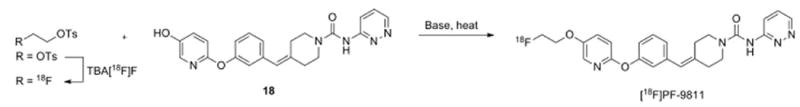
Two-step fluorination scheme (Route B) to produce [18F]PF-9811.
The route that ultimately proved successful and reliable was a three-step, one-pot route (Scheme 5). This route involved direct fluorination on a BOC-protected tosylated amine (16) to produce 19, followed by deprotection of the amine with trifluoroacetic acid, evaporation of the trifluoroacetic acid to generate 20, and coupling of 20 with phenyl pyrazine-3-yl-carbamate (14) to provide the final compound. Each individual step proceeds quite quickly and the reaction sequence is reliable. The key step is making sure all of the trifluoroacetic acid is evaporated following the deprotection, otherwise the coupling reaction will fail to proceed. Indeed, the major radioactive by-product in the final purification is the deprotected uncoupled amine 19. This can be minimized by using a large excess of the organic base in the final coupling step. Utilizing this procedure, 80–180 mCi of [18F]PF-9811 could be isolated from 1800–2000 mCi of [18F]fluoride in 87 min following end of beam (∼11% decay-corrected yield). Radiochemical and chemical purities were very good (>98% and>92%, respectively), with specific activities exceeding 12,000 Ci/mmol. The measured logP7.4 of [18F]PF-9811 was 3.14±0.02 (measured in quadruplicate).
Scheme 5.
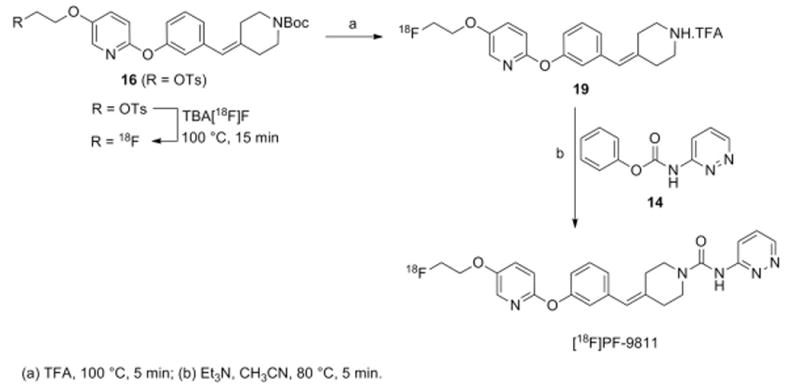
Three-step, one pot fluorination scheme (Route C) to produce [18F]PF-9811.
2.3. Potency and selectivity of the parent fluorine-containing FAAH analog 5 by ABPP
We employed competitive activity-based protein profiling (ABPP) to assess the potency and selectivity of each FAAH inhibitor in mouse brain proteomes [27–29]. This technique involves initial treatment of native proteomes with an inhibitor and then with a serine hydrolase-directed fluorophosphonate-rhodamine (FP-Rh) probe. Loss of target-associated fluorescent signals can then be used to quantitate the extent of inhibition. Using this platform, PF-9811 was found to maintain good FAAH potency (16 nM) albeit 5-fold less potent than its parent, 4 (Fig. 2A). We next determined whether PF-9811 could inhibit FAAH in vivo. C57Bl/6 mice were treated with either vehicle or inhibitor (10 mg/kg, i.p. or p.o.). After 1 h, each mouse was sacrificed, and their brains harvested for gel-based ABPP analysis (Fig. 2B). FAAH was found to be completely inhibited in PF-9811-treated mice by either i.p. or p.o. administration with no detectable off-targets.
Fig. 2.
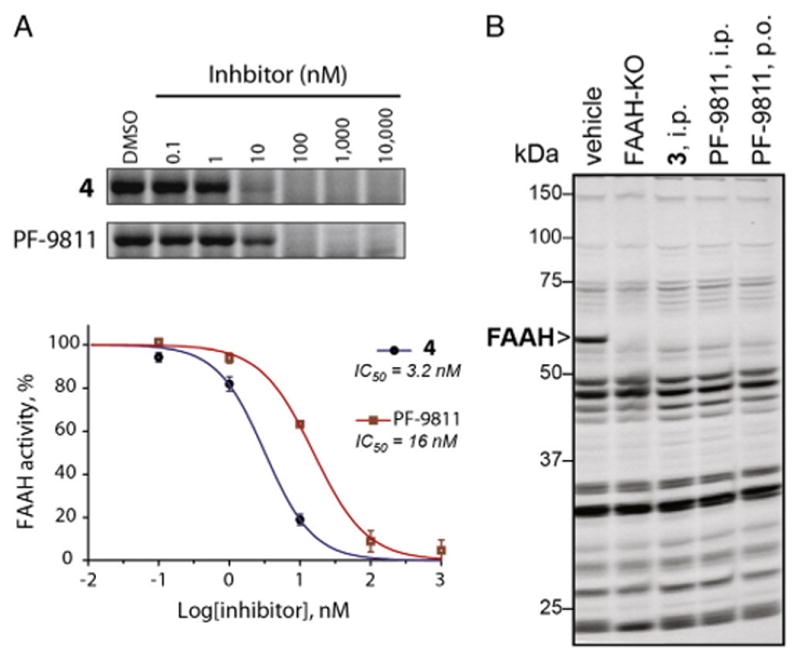
In vitro and in vivo inhibition of FAAH by 3, 4, and PF-9811 (5) as measured by competitive ABPP. (A) In vitro concentration-dependent inhibition of FAAH by 4 and PF-9811 in mouse brain homogenates. (B) Brain serine hydrolase activity profiles of mice treated with inhibitors (10 mg kg−1, i.p. or p.o.) for 1 h using competitive ABPP.
2.4. Brain biodistibution in rats
Results of rat brain biodistribution are presented in Table 1. [18F] PF-9811 showed localization by 60–90 min to brain regions known to express FAAH, such as hippocampus, cortex and cerebellum [25,30–32]. Blocking conditions compared to baseline at each time point showed statistical reduction in tracer binding in all brain regions.
Table 1.
Biodistribution (SUV±standard deviation) in rats at 10, 60 and 90 min post [18F]PF-9811 injection.
| Tissue | 10 min | 10 min blockeda | 60 min | 60 min blockeda | 90 min | 90 min blockeda |
|---|---|---|---|---|---|---|
| Blood | 0.224±0.070 | 1.031±0.069 ** | 0.063±0.012 | 0.747±0.191 ** | 0.061±0.009 | 0.703±0.117 ** |
| Striatum | 0.427±0.021 | 0.309±0.028 ** | 0.441±0.073 | 0.255±0.082 | 0.573±0.081 | 0.240±0.065 ** |
| Hippocampus | 0.579±0.018 | 0.282±0.014 ** | 0.673±0.118 | 0.233±0.076 ** | 0.770±0.126 | 0.234±0.061 ** |
| Cortex | 0.596±0.030 | 0.265±0.014 ** | 0.674±0.109 | 0.219±0.068 ** | 0.789±0.096 | 0.216±0.055 ** |
| Thalamus | 0.382±0.051 | 0.304±0.020 | 0.374±0.045 | 0.251±0.074 | 0.544± 0.063 | 0.238±0.058 ** |
| Hypothalamus | 0.410±0.027 | 0.351±0.024 | 0.426± 0.045 | 0.260± 0.088 | 0.545±0.126 | 0.257±0.079 * |
| Cerebellum | 0.666±0.017 | 0.291±0.008 ** | 0.751±0.094 | 0.248± 0.076 ** | 0.857±0.144 | 0.249± 0.065 ** |
| Brain stem | 0.319±0.029 | 0.313±0.011 | 0.331±0.041 | 0.264± 0.076 | 0.391± 0.040 | 0.246± 0.050 * |
| Rest of brain | 0.471±0.038 | 0.294±0.018 ** | 0.555±0.081 | 0.239±0.071 * | 0.612±0.088 | 0.231±0.055 ** |
| Bone | 0.180±0.001 | 0.332±0.010 | 0.133±0.058 | 0.391±0.088 | 0.159±0.001 | 0.472±0.133 |
| Liver | 14.179±3.336 | 7.725±0.701 * | 17.346±4.789 | 4.488±1.343 * | 16.116±4.670 | 4.165±0.664 * |
P<0.01 and
P<0.001 compared to baseline data at the same time points; N=4 except for bone N=2 (i.e. no statistical analysis t-test for bone tissues).
Animals were injected with an orally administered blocking dose of 1 mg/kg 4 1 h prior to tracer injection.
Fig. 3 shows a comparison of the standard uptake value (SUV) of [18F]PF-9811 at 90 min post-injection at baseline and under blocking conditions where 4 at 1 mg/kg was orally administered 1 h prior to tracer injection. All brain regions showed statistically significant reduction of tracer uptake, consistent with the distribution of FAAH activity in the rat brain. Upon blocking with 4 at 1 mg/kg, the radioactivity in the blood increased over baseline. We hypothesize that this is due to FAAH being blocked in the brain and peripheral tissues by 4 leading to an increase in the amount of unchanged radiotracer in the blood compartment, although no rigorous metabolite analyses were performed to substantiate this.
Fig. 3.
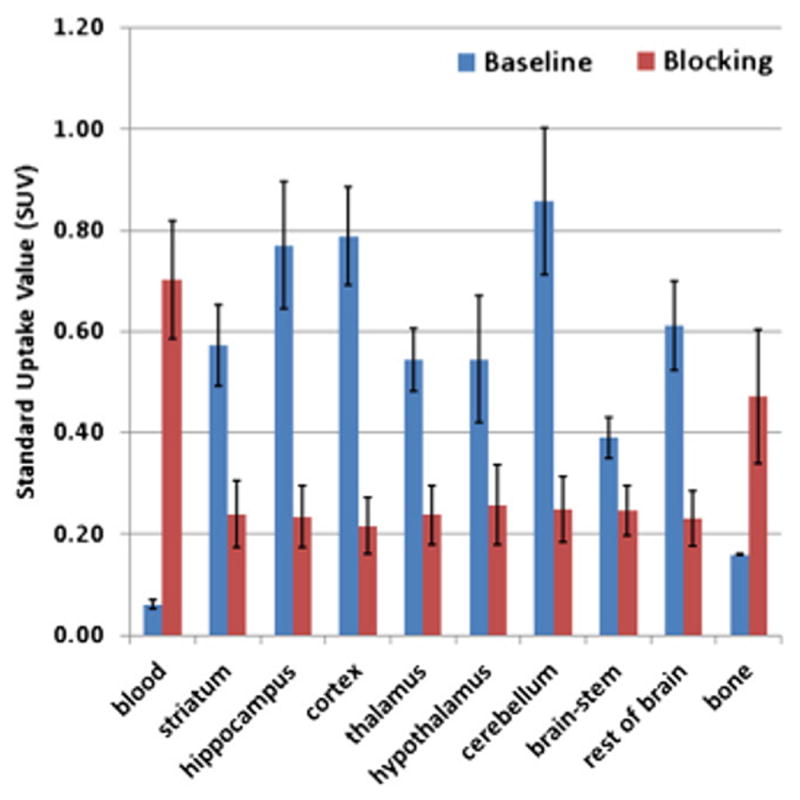
Bio-distribution (SUV data with standard deviation bars) in rats (N=4 except bone N=2) at 90 min post [18F]PF-9811 injection. Blocking was carried out by oral administration of 4 at 1 mg/kg 1 h prior to tracer injection. P<0.01 for hypothalamus and brain-stem and P<0.001 for blood and all other brain regions compared to baseline tissue data.
2.5. In vivo PET imaging in rats
Fig. 4A shows representative time activity curves (TACs) both at baseline and under blocking conditions with pretreatment of 4. In vivo PET imaging yielded comparable results to those observed by the biodistribution study. As shown in Fig. 4B, PET imaging showed the highest accumulation in hippocampus and cortex which correlated well with the known distribution of FAAH in the CNS. Tracer accumulation plateaued by ∼60 min post-injection and then remained steady up to 90 min and beyond. This is consistent with the irreversible mechanism of this class of urea FAAH inhibitors which have been shown to covalently modify the active site serine in FAAH [14]. PET images also showed blockade of the brain uptake of [18F]PF-9811 with pretreatment of 4 and preferential distribution of the tracer to the blood was observed by the increase in surrounding tissue activity (Fig. 4B).
Fig. 4.
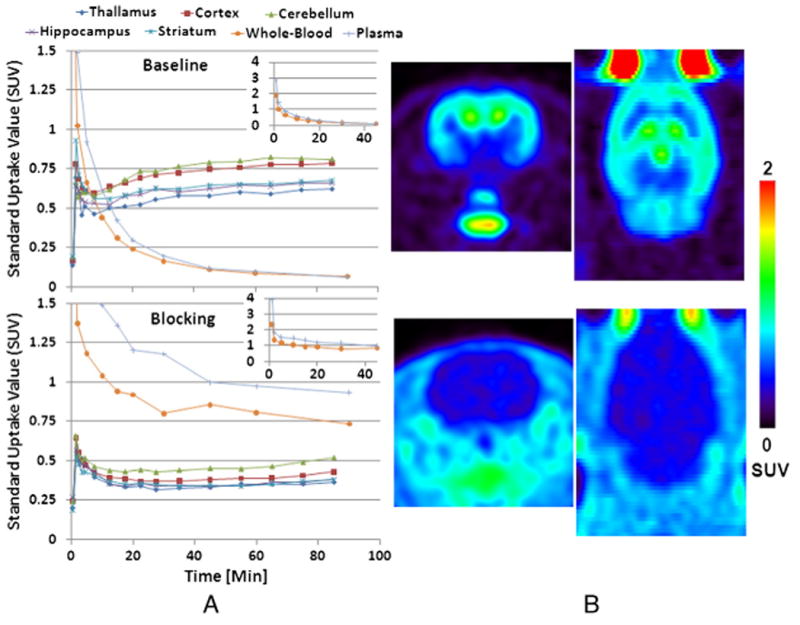
(A) Time activity curves (TACs) from [18F]PF-9811 at baseline (top) and blocking with 1 mg/kg 4 orally administered 1 h prior to PET tracer (bottom). The two insets show the full-scaled whole-blood and plasma TACs (between 0 and 40 min) for the baseline and blocking rats, respectively. The two rats were scanned under anesthesia of 2%∼2.5% isoflurane +oxygen for 90 min. The baseline rat was injected with 940 μCi of [18F]PF-9811 and the blocking rat with 1751 μCi of the hot tracer. (B) SUV images summed between 30 min and 90 min of PET imaging (transverse and coronal sections).
3. Discussion and conclusions
The interest in FAAH as a therapeutic target for pain, addiction, inflammation, and various CNS disorders has led not only to a number of highly potent and selective inhibitors such as 4, but also to a need for a clinically-validated central target biomarker. The high potency and exquisite specificity of 4 led us to use this benzylidene piperidine urea as a scaffold for designing the fluorine-18 based PET ligand [18F] PF-9811 for imaging FAAH. Previous SAR of this scaffold suggested that the para- position on the pyridine ring of 4 could be modified without a substantial loss in potency [21]. This proved to be the case, as replacing the trifluoromethyl moiety of 4 with a fluoroethoxy group resulted in PF-9811 which potently and selectively inhibited FAAH based on ABPP experiments (Fig. 2). Moreover, favorable ADME data for 4 provided added confidence that this compound would display characteristics necessary for a PET ligand designed for CNS targeting. PET radiochemistry efforts provided a robust three-step, one-pot synthesis that produced [18F]PF-9811 in good yield (11% decay-corrected from [18F]fluoride) and high specific activity (>12,000 Ci/mmol). A measured log P7.4 of 3.14 for [18F]PF-9811 suggested that its lipophilicity was in a favorable range for good blood–brain barrier permeability [33].
Biodistribution experiments of [18F]PF-9811 in rats showed good uptake in all regions of the brain, with higher SUVs in the cortex, hippocampus, and cerebellum (Fig. 3 and Table 1). These results correspond well with the known regional brain distribution of FAAH [31] and with [11C]CURB, another covalent irreversible PET ligand for FAAH [25]. Since this class of compounds inhibits FAAH by a covalent, irreversible mechanism, one would expect a steady accumulation of the radiotracer as a covalently bound adduct with FAAH in regions of the brain where FAAH is distributed. Indeed, a statistically significant (P<0.05) increase in signal was seen in all regions of the brain except the brain stem between the 10 and 90 min time points (Table 1). Defluorination (as determined by bone uptake) was minimal, and high liver uptake was observed—reflecting the high expression of FAAH enzyme in this organ [34]. Specificity of [18F]PF-9811 for FAAH was demonstrated by pretreatment with 4, which reduced uptake across all brain regions (37%–73% at 90 min). A complete inhibition of the FAAH enzyme in the blocking studies also resulted in a dramatic reduction of signal in the liver (74%) with a concomitant 12-fold increase in signal in the blood at 90 min. Presumably, the higher signal in the blood pool is a result of the irreversible inhibition of FAAH in the brain and peripheral organs which leads to an increased amount of unchanged [18F]PF-9811 in the circulation.
In vivo rat PET imaging studies largely mirror those of the biodistribution study, with baseline experiments showing good, heterogeneous uptake in the brain (SUV∼0.8 at 90 min for the hippocampus and cortex). The signal in the brain steadily increased and plateaued at about 70 min. This is consistent with the known mechanism of action of a tracer that covalently binds to its target. Blocking experiments resulted in decreased uptake (e.g. ∼40% for hippocampus) at the end of 90 min, with a concurrent increase in the blood and plasma activity. No true reference region exists in the brain for FAAH and no rigorous metabolite analyses were performed in this set of experiments to determine arterial input function values. However, it should be emphasized that FAAH is completely inhibited by 4 during the time course of the experiment based on our previous studies [20,21]. Biodistribution data at 90 min for baseline and blocking studies indicated that specific to non-specific binding ratios in tissues showing prominent uptake are 2.3 (hippocampus), 2.6 (cortex) and 2.4 (cerebellum), providing a reasonable window for measuring varying levels of FAAH inhibition. The kinetics of [18F]PF-9811 appear to be well-matched to the isotope choice, as the activity seems to plateau towards the end of the 90 min baseline experiment, and the end point could easily be extended if needed. Quantitative kinetic modeling of the PET data would require metabolite correction in plasma as the level of metabolite could likely be very different in baseline and displacement studies due to complete FAAH inhibition.
In conclusion, the physicochemical properties and potency of PF-9811 make it an attractive candidate for the radiosynthesis of an [18F]-labelled radiotracer for PET imaging of FAAH. The high brain uptake and specific binding of [18F]PF-9811 with the expected regional distribution suggest that [18F]PF-9811 represents a promising radiotracer for measuring cerebral FAAH in vivo using PET.
4. Materials and methods
4.1. General materials and methods
4.1.1. Chemistry
Solvents and reagents were of reagent grade and were used as supplied by the manufacturer. All reactions were run under a N2 atmosphere. Organic extracts were routinely dried over anhydrous Na2SO4. Concentration refers to rotary evaporation under reduced pressure. Chromatography refers to flash chromatography using disposable RediSep Rf 4 to 120 g silica columns or Biotage disposable columns on a CombiFlash Companion or Biotage horizon automatic purification system. Microwave reactions were carried out in a microwave reactor manufactured by Smithcreator of Personal Chemistry. All target compounds were analyzed using Ultra High Performance Liquid Chromatography/Ultra Violet/Evaporative Light Scattering Detection coupled to time of flight mass spectrometry (UHPLC/UV/ELSD/TOFMS). The UHPLC-MS was performed on a Waters ACQUITY UHPLC system (Waters, Milford, MA), which was equipped with a binary solvent delivery manager, column manager, and sample manager coupled to ELSD and UV detectors (Waters, Milford, MA). Detection was performed on a Waters LCT premier XE mass spectrometer (Waters, Milford, MA). The instrument was fitted with an Acquity BEH (Bridged Ethane Hybrid) C18 column (30 mm×2.1 mm, 1.7 μm particle size, Waters, Milford, MA) operated at 60 °C. Unless otherwise noted, all tested compounds were found to be >95% pure by this method.
4.1.2. Radiolabeling
Oxygen-18 enriched (97%) water was purchased from Huayi Isotopes (cat. # WT-98-5). All solvents were purchased from Sigma-Aldrich. Radiochemistry was performed on a GE TracerLab FXFN synthesis module. [18F]PF-9811 was purified by the HPLC available on the GE TracerLab FXFN module, consisting of a Sykam S-1021 pump, a Knauer K-2001 UV detector (λ=254 nm) in series with a Berthold β+-flow detector, on a Phenomenex Luna C-18(2) column (10×250 mm, 10 μm) equipped with a Phenomenex semi-prep Security Guard holder (cat. # AJ0-7220 with cartridge AJ0-7221) at 5 mL/min with 40% v/v acetonitrile/water (modified with 0.1% formic acid) as the mobile phase. Quality control analysis of [18F]PF-9811 was performed on an Agilent 1100 using a Phenomenex Luna C-18(2) column (4.6×150 mm, 3 μm) and 50% acetonitrile/0.1% formic acid (1.0 mL/min) as the mobile phase for radiochemical and chemical purity. The identity of the labeled compound was confirmed by co-injection of the authentic on HPLC. The specific activity was determined by injection of an aliquot of the final solution with known radioactivity on the analytical HPLC system described above. The area of the UV peak measured at 254 nm corresponding to the carrier product was measured and compared to a standard curve relating mass to UV absorbance. Radioactivity was measured with a Capintec CRC-15 PET dose calibrator.
4.1.3. Biodistribution studies
The Pfizer Institutional Animal Care and Use Committee approved the surgical and imaging procedures. The Groton division of Pfizer Worldwide Research and Development is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Brain biodistribution studies of [18F]PF-9811 were performed in 24 male Sprague–Dawley (SD) rats (Charles River) with a mean body weight 325 g (±40 g, SD). Animals were grouped into 6 groups of 4, with a baseline and blocking group at 10, 60 and 90 min post tracer injection. Blocking drug was 4 dosed p.o. at 1.0 mg/kg, a dose previously shown to fully inhibit FAAH [20]. [18F]PF-9811 was synthesized to high specific activity and radiochemical purity (vide infra). Rats were briefly anesthetized (2-3% isoflurane +O2) for tail vein injection of radiotracer (0.30–2.10 mCi), then allowed to recover for conscious uptake of [18F]PF-9811. Injected tracer mass was held relatively constant at 0.35–0.40 nmol/kg. For tissue collections for brain biodistribution, rats were anesthetized by isoflurane (5%+O2) followed by cervical dislocation. Blood, liver, bone (femur) and various brain regions were collected, placed in pre-weighed tubes and counted in an automated gamma counter (Wallac Wizard 1470, Perkin Elmer). The count results were corrected for decay to time of tracer injection and converted to a Standard Uptake Value (SUV).
SUV = (μCi/g tissue)/(injected tracer μCi/body weighting)
Mean and standard deviation (SD) were determined and baseline and blocked groups were compared by Student's t-test for each time point.
4.1.4. PET imaging
Six jugular vein cannulated (JVC) male Sprague–Dawley (Charles River) rats (345±28 g, SD) were used in PET imaging experiments. PET was conducted at baseline and blocking conditions (pre treatment with 4 p.o.) as described in the biodistribution studies. For PET imaging, rats were anesthetized and maintained with isoflurane (∼2%)+oxygen, and a tail vein cannula was placed for tracer injection. Respiration rates were monitored during PET to maintain 45–65 breaths/min and core body temperature was maintained using a heat lamp. The rat brain area was centered in the field of view of a Focus F220 microPET (Siemens Medical Solutions, Knoxville, TN) and a dynamic PET scan was conducted for 90 min, with scan acquisition starting immediately on IV injection (1.5462±0.4130 mCi, SD) of [18F]PF-9811. Blood was sampled during PET imaging at 1, 2, 5, 10, 15, 20, 30, 45, 60 and 90 min post-injection of tracer and analyzed for radioactivity in whole blood and plasma by gamma counter (Wallac Wizard 1470, Perkin Elmer).
Dynamic PET data was histogrammed into 16 time frames, 5 frames of 1 min, 5 frames of 5 min and 6 frames of 10 min. The Focus 220 scanner consists of 48 rings of detectors axially and 3D sinograms were created with correction for random coincidences and global dead-time, using a detector span of 3 and maximum ring difference of 47. PET images were reconstructed applying corrections for detector normalization, decay, attenuation and scattering using a hybrid 3D OSEM/MAP reconstruction algorithm (microPET Manager, Siemens Medical Solutions) to a 128×128×95(pixels)×16(frames) image of a voxel size of 0.237×0.237×0.796 mm3. PET images were then registered to a rat brain atlas for generation of time activity curves (TACs) as shown.
4.2. Synthesis of unlabeled compounds
4.2.1 Synthesis of tert-butyl 4-(3-((5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate (11)
To 10 (2.0 g, 4.5 mmol) [35] were added bis(pinacolato)diboron (1.5 g, 5.9 mmol), potassium acetate (1.3 g, 14 mmol) and [1,1-bis(diphenylphosphine)ferrocene]dichloropalladium (II) (0.19 g, 0.26 mmol) followed by DMSO (11 mL). The mixture was purged with nitrogen for 10 min and heated to 80 °C for 2 h.The reaction flask was then cooled and diluted with water and ethyl acetate (EtOAc). After filtering through a pad of Celite, the layers were separated and the aqueous phase extracted again with EtOAc (×3). The combined organic layers were washed with water and brine and dried over Na2SO4. Concentration under vacuum gave the crude residue which was purified by flash chromatography (EtOAc/heptane: 1/4) to yield 1.7 g of compound 11 as a white solid (75%). 1H NMR (400 MHz, CDCl3) : δ 1.29–1.34 (m, 12H), 1.43–1.48 (m, 9H), 2.29 (t, J=5.6 Hz, 2H), 2.44 (t, J=5.6 Hz, 2H), 3.37 (t, J=5.7 Hz, 2H), 3.47 (t, J=5.7 Hz, 2H), 6.31 (s, 1H), 6.83 (d, J=8.2 Hz, 1H), 6.92–7.02 (m, 3H), 7.28– 7.34 (m, 1H), 8.01 (dd, J=8.2, 2.0 Hz, 1H), 8.54 (d, J=2.0 Hz, 1H). LC/MS: 493.4 (M+H) and 411.2 for the boronic acid formed under the LC/MS conditions.
4.2.2. Synthesis of tert-butyl 4-(3-((5-hydroxypyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate (12)
A solution of 11 (1.4 g, 2.3 mmol) in THF (9 mL) was cooled to 0 °C and peracetic acid (available as 39% solution in acetic acid; 0.53 mL, 3.13 mmol) was added dropwise. The mixture was allowed to warm to rt and stirred overnight. The mixture was cooled again to 0 °C and quenched with aqueous sodium bisulfate solution. Product was extracted into EtOAc (×3) and the combined organic layers were washed with water and brine and dried over Na2SO4. Concentration under vacuum afforded the crude residue which was purified by flash chromatography (EtOAc/heptane: 1/3) to yield 0.7 g of compound 12 as a colorless gum (65%). 1H NMR (400 MHz, CDCl3): δ 1.45 (m, 9 H) 2.27 (t, J=5.5 Hz, 2 H), 2.40 (t, J=5.5 Hz, 2 H), 3.34 (t, J=5.7 Hz 2 H), 3.43–3.49 (m, 2 H), 6.29 (s, 1 H), 6.80 (d, J=9.0 Hz, 2 H), 6.87–6.95 (m, 2 H), 7.20–7.29 (m, 3 H), 7.81 (br. s., 1 H). LC/MS: 381.2 (M−H).
4.2.3. Synthesis of 4-(3-((5-(2-fluoroethoxy)pyridin-2-yl)oxy)benzylidene)-N-(pyridazin-3-yl)piperidine-1-carboxamide (13)
To a solution of 12 (190 mg, 0.5 mmol) in toluene (5 mL) was added 2-fluoroethanol (40.2 mg, 0.6 mmol) and cyanomethylenetri-butylphosphorane (CMBP) (151 mg, 0.6 mmol) at rt. The mixture was then heated to 90 °C for 2.5 h and then cooled to rt and stirred overnight. Concentration under vacuum afforded the crude residue which was purified by flash chromatography (EtOAc/heptane: 1/3) to yield 213 mg of compound 13 as a colorless gum. 1H NMR (400 MHz, CDCl3): δ 1.45 (s, 9H), 2.29 (t, J=5.27 Hz, 2H), 2.43 (t, J=5.66 Hz, 2H), 3.36 (t, J=5.9 Hz, 2H), 3.47 (t, J=5.9 Hz, 2H), 4.14–4.27 (m, 2H), 4.64–4.82 (m, 2H), 6.30 (s, 1H), 6.83–6.99 (m, 4H), 7.25–7.34 (m, 2H), 7.87 (d, J=2.93 Hz, 1H). LC/MS: 429.4 (M+H).
4.2.4. Synthesis of 4-(3-((5-(2-fluoroethoxy)pyridin-2-yl)oxy) benzylidene)-N-(pyridazin-3-yl)piperidine-1-carboxamide (5)
To a solution of 13 (226 mg, 0.52 mmol) in dichloromethane (1.5 mL) at rt was added 4 N HCl in dioxane (0.53 mL, 2.11 mmol). The mixture was stirred at rt for 4 h. The solvent was then removed under vacuum and the residue was co-evaporated with ethyl ether to yield 192 mg of piperidine HCl salt as a white solid. 1H NMR (400 MHz, CD3OD): δ 2.55–2.62 (m, 2H), 2.66–2.73 (m, 2H), 3.15 (t, J=6.1 Hz, 2H), 3.22–3.26 (t, J=6.1 Hz, 2H), 4.19–4.33 (m, 2H), 4.62–4.78 (m, 2H), 6.52 (s, 1H), 6.93- 6.99 (m, 3H), 7.05 (d, J=8.40 Hz, 1H), 7.37 (t, J=7.91 Hz, 1H) 7.60 (dd, J=9.08, 3.03 Hz, 1H) 7.90 (d, J=2.93 Hz, 1H). LC/MS: 329.2 (M+H). To a mixture of the piperidine HCl salt (200 mg, 0.55 mmol) and Et3N (0.15 mL, 1.1 mmol) in acetonitrile was added 14 [35] and the resulting reaction mixture was stirred at rt for 3.5 h. The reaction mixture was then diluted with saturated NaHCO3 aqueous solution and extracted with EtOAc (3×). The organic layers were combined and washed with water, brine and dried over Na2SO4. Concentration under vacuum afforded the crude residue which was purified by flash chromatography (EtOAc) to yield 111 mg of compound 5 as a white foamy solid upon trituration with pentane (45%). 1H NMR (500 MHz, CDCl3): δ 2.49 (t, J=5.73 Hz, 2H) 2.63 (t, J=5.49 Hz, 2H) 3.57 (t, J=5.85 Hz, 2H) 3.67 (t, J=5.85 Hz, 2H) 4.71–4.78 (m, 2H) 4.72– 4.84 (m, 2H) 6.43 (s, 1H) 6.89–7.04 (m, 4H) 7.33–7.39 (m, 2H) 7.44 (dd, J=9.03, 4.88 Hz, 1H) 7.72 (br. s., 1H) 7.92 (d, J=3.17 Hz, 1H) 8.33 (d, J=9.76 Hz, 1H) 8.86 (d, J=4.15 Hz, 1H); 13 C NMR (500 MHz, CDCl3): δ 157.9, 156.4, 154.9, 151.3, 151.3, 138.7, 133.8, 133.6, 129.4, 129.4, 127.4, 124.7, 120.7, 120.7, 118.5, 112.6, 112.5, 81.7 (d, J=171 Hz), 68.3 (d, J=23 Hz), 35.7, 29.1. LC/MS: 450.2 (M+H).
4.2.5. Synthesis of tert-butyl 4-(3-((5-(2-hydroxyethoxy)pyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate (15)
To a mixture of 12 (0.5 g, 1.3 mmol) and potassium carbonate (0.98 g, 7.1 mmol) in acetone (17 mL) were added (2-bromoethox-y)(tert-butyl)dimethylsilane (0.74 mL, 3.4 mmol) and 18-crown-6 (0.46 g, 1.7 mmol) under nitrogen atmosphere. The reaction mixture was refluxed for 3 h and then cooled and quenched with water. The product was extracted into EtOAc (×3) and the combined organic layers were washed with water and brine and dried over Na2SO4. Concentration under vacuum afforded the crude residue as a colorless liquid. It was dissolved in THF (10 mL) and a solution of tetra-n-butylammonium fluoride (TBAF; 1 M, 2.5 mL, 2 mmol) was added. After stirring for 4 h at room temperature, the reaction was quenched with water and extracted into EtOAc (×3). The combined organic layers were washed with water and brine and dried over Na2SO4. Concentration under vacuum afforded 0.47 g of compound 15 as a yellow gum (83%) with sufficient purity for the next step. 1H NMR (400 MHz, CDCl3-d): δ 1.44 (s, 9 H), 2.28 (t, J=5.4 Hz, 2 H), 2.43 (t, J=6.0 Hz, 2 H), 3.36 (t, J=5.8 Hz, 2 H), 3.46 (t, J=5.7 Hz, 2 H), 3.95 (t, J=4.3 Hz, 2 H), 4.08 (t, J=5.4 Hz, 2 H), 6.30 (s, 1 H), 6.83–6.98 (m, 4 H), 7.25–7.32 (m, 2 H), 7.88 (d, J=3.1 Hz, 1 H). LC/MS: 427.2 (M+H).
4.2.6. Synthesis of tert-butyl 4-(3-((5-(2-(tosyloxy)ethoxy)pyridin-2-yl)oxy)benzylidene)piperidine-1-carboxylate (16)
A mixture of 15 (0.45 g, 1.05 mmol) in DCM (4 mL) was cooled to 0 °C and triethylamine (0.45 mL, 3.15 mmol) was added under nitrogen atmosphere. p-Toluenesulphonyl chloride (0.24 g, 1.26 mmol) was then added portion wise and the reaction stirred at room temperature for 5 h. The reaction mixture was then quenched with water and EtOAc was added. The organic layer was separated and the aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3 solution, water, brine and dried over Na2SO4. Concentration under vacuum afforded the crude residue which was purified by flash chromatography (EtOAc/heptane: 1/1) to yield 0.35 g of compound 16 as a white solid (57%). 1H NMR (400 MHz, CDCl3-d): δ 1.43 (s, 9 H), 2.27 (t, J=,5.6 Hz, 2 H), 2.39–2.45 (m, 5 H), 3.35 (t, J=5.6 Hz, 2 H), 3.46 (t, J=5.6 Hz, 2 H), 4.10–4.14 (m, 2 H), 4.31–4.35 (m, 2 H), 6.29 (s, 1 H), 6.80 (d, J=9.0 Hz, 1 H), 6.86–6.91 (m, 2 H), 6.95 (d, J=7.6 Hz, 1 H), 7.17 (dd, J=9.0, 3.1 Hz, 1 H), 7.25–7.34 (m, 3 H), 7.72 (d, J=3.1 Hz, 1 H), 7.76–7.80 (m, 2 H). LC/MS: 581.7 (M+H).
4.3. Synthesis of labeled compounds
4.3.1. Radiosynthesis of [18F]PF-9811
No-carrier-added [18F]fluoride was prepared by proton irradiation (70 min, 60 μamp) of a 2.4 mL tantalum target [18O(p,n)18F]. The semi-prep HPLC column was equilibrated with the mobile phase 15– 30 min at 3 mL/min before or during the beam.
The activity was unloaded from the target and delivered to the glass V-vial on the General Electric FX-FN synthesis module. The [18F] fluoride was trapped on a Waters QMA Plus Light cartridge (cat# 023531; pre-treated with 5 mL 0.2 M potassium phosphate, then dried under an N2 stream). The [18F]fluoride was eluted with a solution of 1 mL of 0.03 M tetrabutylammonium mesylate in methanol. The methanol was evaporated at 100 °C in vacuo under a stream of helium until dry (about 3.5 min). The reactor was then cooled to 40 °C while still under vacuum.
To the residue was added a solution of 5 mg 16 (5 mg, 8.62 μmol) in 0.6 mL anhydrous acetonitrile and the reaction mixture was heated at 100 °C for 15 min. The reaction mixture was cooled to 50 °C, at which time a solution of 0.5 mL of 1 M trifluoroacetic acid (TFA) in acetonitrile was added. The reaction was heated to 100 °C for 5 min, then cooled to 55 °C. The acetonitrile/TFA mixture was then evaporated in vacuo under a stream of helium for 5 min. The reactor was cooled to 30 °C (under vacuum with no helium stream), followed by the addition of a solution of phenyl pyridazine-3-ylcarbamate (14, ∼3 mg) [35] and diisopropylethylamine (20 μL) in anhydrous aceto-nitrile (0.5 mL). The reaction mixture was heated to 80 °C for 5 min, then cooled to 40 °C. The reaction mixture was diluted with 4 mL of mobile phase (40% acetonitrile/water with 0.1% formic acid) and passed through an Alumina N Sep-Pak Plus Light cartridge (Waters, cat# 23561) into an intermediate vial. The crude reaction mixture was then purified by semi-prep chromatography (vide supra). The fraction containing [18F]PF-9811 was collected between 16.3 and 17.3 min and diluted with 60 mL water, followed by trapping on a Waters 30 mg Oasis HLB vac cartridge (WAT094225). The cartridge was washed with 3 mL water, then eluted with ethanol (0.5 mL) and pH 7.4 phosphate-buffered saline (4.5 mL). The final formulation was filtered (Millipore Durapore PVDF 13 mm, cat# SLGSV013SL) and collected in a sterile 10 mL Hollister-Stier crimp-sealed vial, with a small aliquot (0.4 mL) set aside in a sterile 2 mL QC vial for analytical HPLC analysis on the Agilent system (vide supra).
Synthesis time was 87 min from end of beam. Typically, 80–180 mCi of [18F]PF-9811 was isolated in 10.7±4.4% (n=5) yield decay-corrected starting from 1800 to 2000 mCi [18F]fluoride.
4.4. Quality Control of [18F]PF-9811
The chemical and radiochemical purity of [18F]PF-9811 was determined by injecting an aliquot of the final product into the HPLC system (vide supra). Retention time was 5.6 min, and the identity of the labeled compound was determined by co-injection with the authentic PF-9811 standard. Typically, [18F]PF-9811 was synthesized in 98.6%±0.4% radiochemical purity, 92%±7.7% chemical purity, and >12,000 Ci/mmol specific activity (n=5).
4.5. Log P measurements
Based on a procedure by Wilson et al. [36], log P7.4 values were determined for [18F]PF-9811 by adding 15–30 μCi of the tracer to equal volumes (3.5 mL) of n-octanol and Dulbecco's phosphate buffered saline (0.1 M, pH 7.4; DPBS) in 15 mL polypropylene centrifuge tubes. Each phase was saturated with the other phase before use. The mixture was vortexed (60 s), and then centrifuged (4000 rpm) for 2 min. The aqueous layer was removed and discarded. Fresh DPBS was added, and the process repeated two more times to wash away traces of water-soluble impurities. Next, 0.5 mL of fresh n-octanol, 3.5 mL of fresh DPBS and 3 mL of the washed n-octanol layer containing the radioligand were added to a new centrifuge tube which was vortexed and centrifuged as above. Triplicate samples from the n-octanol (0.05 mL) and DPBS (1.0 mL) phases were obtained, taking care to minimize sample contamination. This procedure was repeated three more times to give a total of four sets of samples from organic layers and aqueous layers (i.e., 3 mL of the remaining 3.35 mL of octanol containing the radiotracer was added to a new centrifuge tube containing 3.5 mL of fresh DPBS, which was then vortexed and centrifuged as before, followed by counting 0.05 mL of octanol and 1.0 mL of the aqueous layer). The samples were counted using an automated gamma counter, and values calculated for each set with the equation:
Log P7.4 = log[(radioactivity in n−octanol×20)/radioactivity in DPBS].
4.6. Evaluation of potency and selectivity by ABPP
4.6.1. In vitro competitive ABPP
Membrane fractions of wild-type C57 Bl/6 J mouse brain (50 μL, 1 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37 °C. After 30 min, FP-Rh (1 μL, 50 μM in DMSO) was added and mixture was incubated for another 30 min at 37 °C. Reactions were quenched with SDS loading buffer (4×), run on SDS-PAGE gel, and visualized with a Hitachi FMBio IIe flatbed fluorescence scanner (MiraiBio). Fluorescence of relevant bands was measured using ImageJ 1.43u to calculate IC50 values.
4.6.2. In vivo competitive ABPP
Wild-type C57 Bl/6 J mice were treated with indicated doses of FAAH inhibitor or vehicle (18:1:1 PBS:Emulphor:ethanol) for 1 h, sacrificed and immediately dissected. Mouse brains were harvested, hemisected and frozen in liquid N2. Each half brain was washed with cold phosphate-buffered saline (PBS) at 0 °C (2×1 mL) to remove excess blood and immediately dounce homogenized in PBS (1 mL). Dounced tissue was sonicated and centrifuged (1000×g, 10 min, 4 °C) to remove cellular debris. The supernatant was centrifuged at high speed (145,000×g, 45 min, 4 °C) to separate membrane and soluble cell components. The supernatant was saved as the soluble fraction. The remaining pellet was gently washed with cold PBS (2×, 500 μL) and sonicated in PBS (300 μL) to resuspend. Total protein concentrations of the soluble and membrane fractions were determined using the Bio-Rad Dc Protein Assay kit. Proteomic mixtures were either diluted to 1.0 mg/mL total protein concentration with PBS for immediate use or aliquoted and stored at − 80 °C. Aliquots (50 μL, 1 mg/mL) of this proteomic mixture were analyzed by ABPP with FP-Rh as describe above.
Acknowledgments
This work was supported in part by the National Institutes of Health, DA017259 (BFC) and DA032541 (MN).
References
- 1.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocanna-binoid signaling in the nervous system. Chem Rev. 2008;108:1687–707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–55. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V, Bisogno T, De Petrocellis L. Endocannabinoids and related compounds: walking back and forth between plant natural products and animal physiology. Chem Biol. 2007;14:741–56. doi: 10.1016/j.chembiol.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 6.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–7. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 7.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Ann Rev Biochem. 2005;74:411–32. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 8.Ezzili C, Otrubova K, Boger DL. Fatty acid amide signaling molecules. Biorg Med Chem Lett. 2010;20:5959–68. doi: 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–74. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 10.Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov. 2009;4:763–84. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–43. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 12.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, et al. Discovery of a potent, selective, and efficacious class of reversible alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem. 2005;48:1849–56. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, et al. Cyclohexylcarbamic acid 3′- or 4′-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 14.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, et al. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–30. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 15.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–20. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, et al. Discovery and molecular basis of potent noncovalent inhibitors of fatty acid amide hydrolase (FAAH) Proc Natl Acad Sci USA. 2011;108:7379–84. doi: 10.1073/pnas.1016167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Sarris K, Kage K, Zhang D, Brown SP, Kolasa T, et al. Synthesis and evaluation of benzothiazole-based analogues as novel, potent, and selective fatty acid amide hydrolase inhibitors. J Med Chem. 2009;52:170–80. doi: 10.1021/jm801042a. [DOI] [PubMed] [Google Scholar]
- 18.Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci USA. 2008;105:12820–4. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol. 2010;400:743–54. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–24. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. Discovery of PF-04457845: a highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med Chem Lett. 2011;2:91–6. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J, Axelrod J, et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–5. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Sanabria-Bohórquez S, Joshi A, Cook J, Holahan M, Posavec D, et al. The discovery and characterization of [11C]MK-3168, a novel PET tracer for imaging fatty acid amide hydrolase (FAAH) J Labelled Comp Radiopharm. 2011;54:S38. [Google Scholar]
- 24.Wyffels L, Muccioli GG, Kapanda CN, Labar G, De Bruyne S, De Vos F, et al. PET imaging of fatty acid amide hydrolase in the brain: synthesis and biological evaluation of an 11C-labelled URB597 analogue. Nucl Med Biol. 2010;37:665–75. doi: 10.1016/j.nucmedbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl Med Biol. 2011;38:247–53. doi: 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Ling GL, Winter H, Arends R, Jay GW, Le V, Young T, et al. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharm. 2011 doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, et al. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci USA. 2010;107:20941–6. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–9. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egertova M, Giang DK, Cravatt BF. A new perspective on cannabinoid signaling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–5. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas EA, Cravatt BF, Danielson PE, Gilula NB, Sutcliffe JG. Fatty acid amide hydrolase, the degenerative enzyme for anandamide and oleamide, has selective distribution within the rat central nervous system. J Neuroscience Res. 1997;50:1047–52. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Ueda N, Puffenbarger RA, Yamamoto S, Deutsch DG. The fatty acid amide hydrolase (FAAH) Chem Phys Lipids. 2000;108:107–21. doi: 10.1016/s0009-3084(00)00190-0. [DOI] [PubMed] [Google Scholar]
- 33.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;3:376–89. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes: identification and partial characterization. J Biol Chem. 1995;270:6030–5. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 35.Fay LK, Johnson DS, Lazerwith SE, Morris MA, Wang LJ, Meyers MJ, et al. Preparation of (hetero)biaryl ether methylidenepiperidinyl ureas as fatty acid amide hydrolase (FAAH) inhibitors. USA: Pfizer Products Inc.; 2008. pp. 57–8. [WO 2008047229] [Google Scholar]
- 36.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl Radiat Isot. 2001;54:203–8. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]