

Abstract
Cold exposure is associated with oxidative stress and cardiac dysfunction. The endothelin (ET) system, which plays a key role in myocardial homeostasis, may participate in cold exposure-induced cardiovascular dysfunction. This study was designed to examine the role of ET-1 in cold stress-induced cardiac geometric and contractile responses. Wild-type (WT) and ETA receptor knockout (ETAKO) mice were assigned to normal or cold exposure (4°C) environment for 2 and 5 weeks prior to evaluation of cardiac geometry, contractile, and intracellular Ca2+ properties. Levels of the temperature sensor transient receptor potential vanilloid (TRPV1), mitochondrial proteins for biogenesis and oxidative phosphorylation, including UCP2, HSP90, and PGC1α were evaluated. Cold stress triggered cardiac hypertrophy, depressed myocardial contractile capacity, including fractional shortening, peak shortening, and maximal velocity of shortening/relengthening, reduced intracellular Ca2+ release, prolonged intracellular Ca2+ decay and relengthening duration, generation of ROS and superoxide, as well as apoptosis, the effects of which were blunted by ETAKO. Western blotting revealed downregulated TRPV1 and PGC1α as well as upregulated UCP2 and activation of GSK3β, GATA4, and CREB in cold-stressed WT mouse hearts, which were obliterated by ETAKO. Levels of HSP90, an essential regulator for thermotolerance, were unchanged. The TRPV1 agonist SA13353 attenuated whereas TRPV1 antagonist capsazepine mimicked cold stress- or ET-1-induced cardiac anomalies. The GSK3β inhibitor SB216763 ablated cold stress-induced cardiac contractile (but not remodeling) changes and ET-1-induced TRPV1 downregulation. These data suggest that ETAKO protects against cold exposure-induced cardiac remodeling and dysfunction mediated through TRPV1 and mitochondrial function.
Keywords: low ambient temperature, myocardial function, ETA receptor, TRPV1, mitochondria
Introduction
Exposure to low ambient temperature represents a major challenge to human health as the seasonal variations in death rates reveal the highest incidences during cold winter months. This phenomenon is referred to as excess winter mortality, which has been described in many countries (Mercer, 2003; Cheng and Su, 2010; Yu et al., 2011). In particular, cold climate stress is associated with adverse cardiovascular events as winter season often represents peaks of adverse cardiac events (Sheth et al., 1999; Kloner, 2006; Okamoto-Mizuno et al., 2009; Cheng and Su, 2010). Although mortality due to ischemic heart disease seems to be better associated with low living-room temperatures and limited bedroom heating in winter as opposed to the low outside temperature (Eurowinter Group, 1997), many clinical and epidemiological studies have confirmed that cold climate is an important variable for higher adverse cardiovascular responses such as increased blood pressure and blood viscosity, compromised myocardial performance and hemodynamics, leading to the onset of stroke, myocardial infarction, and arrhythmias (Sheth et al., 1999; Chen and Sun, 2006; Medina-Ramon et al., 2006; Cheng and Su, 2010; Yu et al., 2011). For example, high prevalence of coronary heart disease and stroke has been found in Alaska Eskimos, despite their low-density lipoprotein (LDL) and high-density lipoprotein (HDL) levels (Howard et al., 2010). Nevertheless, the mechanisms responsible for these adverse myocardial changes under cold climate stress remain essentially unknown. It has been demonstrated that cold exposure increases heart tissue levels of endothelin-1 (ET-1), a key regulator for blood pressure, cardiac growth, myocardial contractility, and hemodynamics (Abassi et al., 2001; Chen and Sun, 2006; Barton and Yanagisawa, 2008; Sun, 2010). ET-1 receptor (ETA and ETA/B) antagonists display proven beneficial effects against hypothermic ischemia-reperfusion disturbances, endothelial dysfunction, hypertension, cardiovascular remodeling, heart failure, and survival (Luscher et al., 2002; Zhang et al., 2002; Barton and Yanagisawa, 2008; Banfor et al., 2009). To further elucidate if ET-1 plays a role in cold stress-induced cardiac geometric and contractile changes, the cardiomyocyte-specific ETA receptor knockout (ETAKO) and wild-type (WT) mice were exposed to low ambient temperature (4°C) chronically prior to assessment of myocardial geometry, contractile, and intracellular Ca2+ properties. Expression of the temperature sensor protein transient receptor potential vanilloid (TRPV1) receptor, a protein with an essential function in the maintenance of myocardial contractile function (Wang et al., 2008; Karashima et al., 2009), was evaluated. At least three heat-activated members of TRPV subfamily (TRPV1, TRPV3, and TRPV4) are involved in temperature sensing (Karashima et al., 2009). Expression of mitochondrial proteins responsible for mitochondrial biogenesis and oxidative phosphorylation (including UCP2, HSP90, and PGC1α) was examined. PGC1 is a transcriptional coactivator associated with regulation of genes related to energy metabolism. PGC1 induces mitochondrial biogenesis and respiration, and regulates adaptive thermogenesis, hepatic gluconeogenesis, and insulin secretion (Finck and Kelly, 2006; Schilling and Kelly, 2011). PGC1 stimulates mitochondrial biogenesis and respiration by inducing UCP2 via regulation of nuclear respiratory factors (Wu et al., 1999). Mitochondrial UCPs possess the potential to generate heat by uncoupling oxidative phosphorylation and play a key role in the regulation of body temperature and energy balance (Rey et al., 2010; Schilling and Kelly, 2011). Furthermore, glycogen synthase kinase-3β (GSK3β), a signaling molecule governing cardiac hypertrophy, cell survival and mitochondrial integrity (Blankesteijn et al., 2008; Cheng et al., 2011), the hypertrophic marker GATA4 and the transcription factor cAMP-response element binding protein (CREB) were evaluated in WT and ETAKO mice following cold exposure. GSK3β, a serine/threonine kinase which is inactivated through phosphorylation at Ser9 residue by Akt, regulates the phosphorylation and subsequent degradation of β-catenin (Blankesteijn et al., 2008). GATA4 and CREB, on the other hand, have been implicated in the GSK3β-regulated cardiac hypertrophy and gene expression (Blankesteijn et al., 2008; Cheng et al., 2011).
Results
General biometric and echocardiographic properties of WT and ETAKO mice
Cold exposure did not significantly affect body, liver and kidney weights or sizes although the heart weight and size were enhanced following 2 or 5 weeks of cold exposure. ETAKO did not affect body or organ weights although it nullified cold exposure-induced cardiac hypertrophy. ETAKO did not affect basal and cold exposure-induced increase in systolic and diastolic blood pressure. Plasma levels of ET-1 were similar among all mouse groups while cold exposure significantly enhanced cardiac ET-1 levels. Heart rate (sedated) and left ventricular end-systolic (LV ESD) were comparable among all groups. While LV wall thickness, calculated LV mass, LV end-diastolic (EDD) and fractional shortening were similar between WT and ETAKO groups in the absence of cold exposure, low temperature significantly increased LV wall thickness, LV EDD, and LV mass while reducing fractional shortening in WT but not ETAKO mice. These deleterious changes indicate concentric ventricular hypertrophy following cold exposure (Supplementary Table S1). In addition, cold exposure exhibited an initial increase in the conscious heart rate within the first 10 min of cold stress before returning to baseline level by 180 min, in a comparable manner, in WT and ETAKO mice. Chronic (2-week) cold exposure did not significantly affect myocardial ETB receptor levels (Supplementary Figure S1).
Cardiomyocyte contractile and intracellular Ca2+ property as well as frequency response
Consistent with the hypertrophied myocardium in response to sustained cold exposure, low temperature exposure but not ETAKO significantly enhanced resting cell length. Moreover, cold exposure for 2 or 5 weeks significantly compromised peak shortening (PS) and maximal velocity of shortening/relengthening (±dL/dt) as well as prolonged time-to-90% relengthening (TR90) without affecting time-to-PS (TPS) in WT mice. Importantly, ETAKO abolished cold exposure-induced mechanical abnormalities without eliciting any notable effects by itself (Figure 1). In addition, cardiomyocytes from mice exposed to cold stress displayed a significantly depressed intracellular Ca2+ rise in response to electrical stimulus (ΔFFI) and reduced intracellular Ca2+ decay rate (single and bi-exponential) associated with an unchanged baseline intracellular Ca2+. ETAKO effectively negated cold exposure-induced changes in ΔFFI and intracellular Ca2+ decay without any effect on intracellular Ca2+ properties by itself (Figure 2A–D).
Figure 1.

Effect of ETAKO on low temperature (LT)-induced cardiomyocyte contractile defects. WT and ETAKO mice were kept at room temperature or 4°C for 2 or 5 weeks. (A) Resting cell length. (B) PS (normalized to cell length). (C) Maximal velocity of shortening (+dL/dt). (D) Maximal of relengthening (−dL/dt). (E) TPS. (F) Time-to-90% relengthening (TR90). Data were represented as mean ± SEM, n = 103 cells from 5 mice per group, *P < 0.05 vs WT, #P < 0.05 vs corresponding WT-LT group.
Figure 2.

Effect of ETAKO on low temperature (LT)-induced intracellular Ca2+ homeostasis and SR Ca2+ store evaluated by frequency (0.1–5.0 Hz)-dependent shortening response in murine cardiomyocytes. WT and ETAKO mice were kept at room temperature or 4°C for 2 or 5 weeks. (A) Resting FFI. (B) Electrically-stimulated rise in FFI (ΔFFI). (C) Single exponential intracellular Ca2+ decay rate. (D) Bi-exponential intracellular Ca2+ decay rate. (E) Frequency response. Each point represents PS normalized to that of baseline value at 0.1 Hz from the same cell. Data were represented as mean ± SEM, n = 82 (A–D) and 23–26 (E) cells from 5 mice per group, *P < 0.05 vs WT, #P < 0.05 vs corresponding WT-LT group.
Mouse hearts beat at high frequencies (>450 beats/min at 37°C) with sarcoplasmic reticulum Ca2+ store being the primary determinant of the frequency-dependent response. We initially stimulated cardiomyocytes to contract at 0.5 Hz for 5 min to ensure steady-state before altering the frequency stepwise from 0.1 to 5 Hz (300 beats/min). All recordings were normalized to the PS obtained at 0.1 Hz of the same cell. Myocytes from cold exposure groups (2 and 5 weeks) exhibited significantly exaggerated depression in PS at 3.0 Hz and higher. Consistent with its effect on cardiac mechanical and intracellular Ca2+ properties, ETAKO mitigated cold exposure-induced accentuation of loss in PS amplitude in response to increased stimulus frequencies (Figure 2E).
Effects of cold exposure on myocardial histology
To assess the impact of ETAKO on myocardial histology following cold exposure, heart gross morphology, cardiomyocyte cross-sectional area, and myocardial interstitial fibrosis were examined. Low-magnification transverse heart sections indicated marked thickening of LV wall following cold exposure. Findings from hematoxylin and eosin (H&E) and fluorescein isothiocyanate (FITC)-conjugated wheat germ staining sections revealed increased cardiomyocyte area following cold exposure, consistent with increased LV mass, wall thickness, EDD, and heart weight in WT mice. The cold temperature-induced cardiac hypertrophy was effectively ablated by ETAKO while ETAKO itself did not affect cardiomyocyte size. Cold exposure prompted overt interstitial fibrosis in the heart, the effect of which was obliterated by ETAKO. ETAKO itself did not affect interstitial fibrosis (Figure 3 and Supplementary Figure S2).
Figure 3.
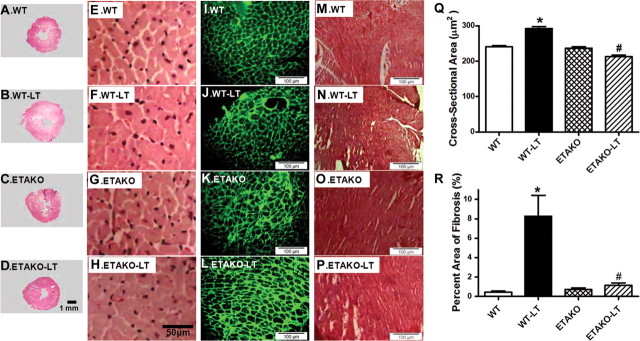
Histological analyses in hearts from WT and ETAKO mice kept at low temperature (LT, 4°C) for 5 weeks. (A–D) Representative photomicrographs from gross morphological view of transverse myocardial sections. Scale bar = 1 mm. (E–H) Representative H&E staining micrographs showing transverse sections of LV myocardium. Scale bar = 50 μm. (I–L) Representative photomicrographs of FITC-conjugated wheat germ agglutinin staining depicting cardiomyocyte size. Scale bar = 100 μm. (M–P) Representative Masson trichrome staining micrographs of LV myocardium. Scale bar = 100 μm. (Q) Quantitative cardiomyocyte cross-sectional (transverse) area from ∼150 cells from 4–5 mice per group. (R) Quantitative analysis of fibrotic area (Masson trichrome stained area in light blue color normalized to the total myocardial area) from ∼ 16 sections from 4 mice per group. Data were represented as mean ± SEM, *P < 0.05 vs WT, #P < 0.05 vs WT-LT group.
Effects of ETAKO on cold exposure-induced generation of  and ROS as well as apoptosis
and ROS as well as apoptosis
To examine the potential mechanism of action behind the ETAKO-elicited protection against cold exposure-induced cardiac geometric and contractile changes, generation of  and ROS as well as apoptosis was examined in cardiomyocytes from WT and ETAKO mice with cold exposure. Results shown in Supplementary Figure S3 indicate that generation of
and ROS as well as apoptosis was examined in cardiomyocytes from WT and ETAKO mice with cold exposure. Results shown in Supplementary Figure S3 indicate that generation of  and ROS as well as caspase 3 activity was significantly elevated in cardiomyocytes from cold-stressed WT mice. Consistent with its mechanical and morphometric responses, ETAKO obliterated or significantly ameliorated cold exposure-induced apoptosis and
and ROS as well as caspase 3 activity was significantly elevated in cardiomyocytes from cold-stressed WT mice. Consistent with its mechanical and morphometric responses, ETAKO obliterated or significantly ameliorated cold exposure-induced apoptosis and  /ROS generation. In line with the caspase-3 data, TUNEL assay also revealed that ETAKO significantly attenuated cold exposure-induced myocardial apoptosis without eliciting any effect by itself (Supplementary Figure S4). ETAKO itself exerted minimal effects on apoptosis and
/ROS generation. In line with the caspase-3 data, TUNEL assay also revealed that ETAKO significantly attenuated cold exposure-induced myocardial apoptosis without eliciting any effect by itself (Supplementary Figure S4). ETAKO itself exerted minimal effects on apoptosis and 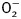 /ROS generation in the absence of cold stress, indicating that the genetic manipulation itself is not innately harmful.
/ROS generation in the absence of cold stress, indicating that the genetic manipulation itself is not innately harmful.
Western blot analysis of TRPV1, mitochondrial proteins, levels of GSK3β, GATA4, and CREB
To elucidate the potential mechanism(s) involved in ETAKO-elicited cardiac protection against cold exposure-induced cardiac hypertrophy and contractile dysfunction, we further examined the expression of the temperature sensor TRPV1, the mitochondrial biogenesis marker PGC1α, mitochondrial proteins HSP90 and UCP2, cardiac hypertrophic regulators GSK3β, GATA4, and CREB. Activity of GSK3β was evaluated using stabilization of β-catenin, a GSK3β-dependent nuclear transcriptional coactivator (Blankesteijn et al., 2008). As shown in Figure 4, cold exposure led to downregulated TRPV1 and PGC1α, upregulated UCP2, as well as enhanced pan and phosphorylated GSK3β, decreased β-catenin (indicating a higher GSK3β activity) as well as phosphorylated GATA4 and CREB without affecting the expression of HSP90 in WT mice. Interestingly, ETAKO reversed cold exposure-induced abnormal upregulation or activation of these proteins without eliciting any effect by itself. Total protein expression of GATA4 and CREB was not affected by either cold exposure or ETAKO (data not shown).
Figure 4.

Effect of ETAKO on low temperature (LT)-induced change in TRPV1 (B), PGC1α (C), HSP90 (D), UCP2 (E), GSK3β (F), total β-catenin (G), GATA4 phosphorylation (normalized to total GATA4; H) and CREB phosphorylation (normalized to total CREB; I). (A) Representative gels using specific antibodies. GAPDH was used as the loading control. Data were represented as mean ± SEM, n = 5–7 mice per group, *P < 0.05 vs WT, #P < 0.05 vs corresponding WT-LT group.
Effect of TRPV1 agonist and antagonist on low temperature-induced echocardiographic and cardiomyocyte contractile properties
To examine if loss of TRPV1 contributes to cold exposure-induced cardiac geometric and contractile changes, WT mice were subjected to cold stress for 2 weeks in the absence or presence of the TRPV1 agonist SA13353 (30 mg/kg, p.o.). A cohort of mice were treated with the TRPV1 antagonist capsazepine (5 mg/kg, i.p.) for 5 days at room temperature. Our data shown in Figure 5 revealed that SA13353 significantly attenuated or nullified cold exposure-induced increase in LV mass, LV wall thickness and LV EDD, decrease in fractional shortening, PS and ±dL/dt as well as prolonged TR90 without affecting LV ESD (data not shown), resting cell length and TPS. SA13353 itself did not elicit any notable effect on echocardiographic and cardiomyocyte contractile properties. Interestingly, cold stress-induced changes in fractional shortening and cardiomyocyte contraction (although not geometric indices) were mimicked by short-term capsazepine treatment. This observation provided compelling evidence for a role of TRPV1 in cold exposure-induced cardiac dysfunction.
Figure 5.

Effect of the TRPV1 agonist SA13353 and antagonist capsazepine on low temperature (LT)-induced changes in echocardiographic and cardiomyocyte contractile indices. WT control mice were treated with or without SA13353 and antagonist capsazepine while being maintained 4°C. (A) LV mass. (B) LV wall thickness. (C) LV EDD. (D) Fractional shortening. (E) PS (normalized to cell length). (F) Maximal velocity of shortening (+dL/dt). (G) Maximal of relengthening (−dL/dt). (H) TPS. (I) Time-to-90% relengthening (TR90). Data were represented as mean ± SEM, n = 6–10 mice (A–D) or 87–98 cells from 5 mice (E–I) per group, *P < 0.05 vs control (cont) group, #P < 0.05 vs LT group.
Effect of GSK3β inhibitor on low temperature-induced cardiomyocyte contractile property
To examine if GSK3β activation plays a role in the cold exposure-induced cardiac contractile defects, WT mice were subjected to low temperature for 2 weeks in the absence or presence of the GSK3β inhibitor SB216763 (0.6 mg/kg, i.p.). Results depicted that SB216763 rescued cold stress-induced loss of mitochondrial membrane potential (MMP) and cardiomyocyte defects including depressed PS and ±dL/dt as well as prolonged TR90 with little effect on TPS. Intriguingly, examination of echocardiographic properties revealed that SB216763 failed to rescue cold exposure-induced cardiac remodeling (increased LV mass, wall thickness, and EDD). Nonetheless, SB216763 treatment prevented cold exposure-induced decrease in fractional shortening. Neither cold exposure nor SB216763 affected heart rate and LV ESD (data not shown). SB216763 itself did not elicit any effect on echocardiographic and cardiomyocyte contractile parameters (Figure 6 and Supplementary Figure S5). These data suggest a possible role of GSK3β in cold exposure-induced cardiac contractile dysfunction but a less likely role in cold exposure-induced cardiac remodeling.
Figure 6.

Effect of the GSK3β inhibitor SB216763 on low temperature (LT)-induced cardiomyocyte contractile defects. WT mice were treated with SB216763 for 2 weeks while being maintained at 4°C. (A) MMP. (B) PS (normalized to cell length). (C) Maximal velocity of shortening (+dL/dt). (D) Maximal of relengthening (−dL/dt). (E) TPS and (F) Time-to-90% relengthening (TR90). Mean ± SEM, n = 116–117 cells from 4 mice per group, *P < 0.05 vs WT, #P < 0.05 vs corresponding WT-LT group.
Effect of pharmacological manipulations on ET-1-induced cardiomyocyte hypertrophy as well as levels of GSK3β (using β-catenin stabilization) and TRPV1
To confirm the role of GSK3β and TRPV1 in cold exposure-induced cardiac hypertrophy, cardiomyocytes from adult WT mice were subjected to ET-1 for 24 h in the absence or presence of the TRPV1 agonist SA13353, the TRPV1 inhibitor capsazepine or SB216763. To consolidate the role of ETA receptor in cold exposure-induced cardiac hypertrophy, cardiomyocytes from adult ETAKO mice were subjected to ET-1 for 24 h prior to assessment of cardiomyocyte surface area. Our data revealed that SA13353 and ETAKO (but not SB216763 and capsazepine) nullified ET-1-induced increase in murine cardiomyocyte size. In addition, neither SA13353 nor capsazepine overtly affected ET-1-induced elevation in GSK3β (shown as reduced β-catenin stabilization) while SB216763 effectively reversed ET-1-induced downregulation of TRPV1 expression. None of the three pharmacological agents affected cardiomyocyte size or levels of β-catenin and TRPV1 by themselves (Figure 7). This observation provided direct evidence for a sequential relationship of GSK3β–TRPV1 in the ET-1-induced cardiac anomalies.
Figure 7.
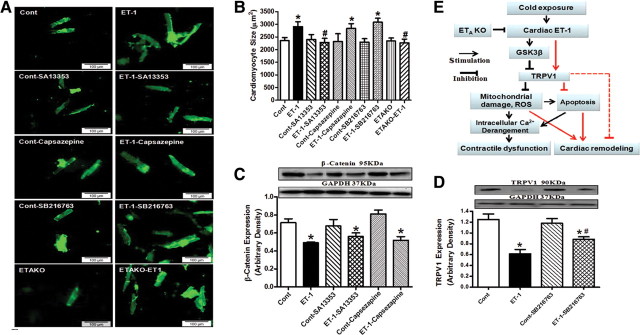
Effect of the TRPV1 agonist SA13353 and antagonist capsazepine as well as the GSK3β inhibitor SB216763 and ETAKO on ET-1-induced myocyte hypertrophy, GSK3β activity (measured by β-catenin levels) and TRPV1 expression. WT mouse cardiomyocytes were exposed to ET-1 (10 nM) for 24 h in the absence or presence of SA13353 (1 µM), capsazepine (1 μM), or SB216763 (10 µM). Cardiomyocytes from ETAKO mice was also incubated with ET-1 (10 nM) for 24 h prior to evaluation. (A) Representative images of murine cardiomyocytes exposed to ET-1 with or without pharmacological agents (or ETAKO). (B) Cardiomyocyte size. (C) Total β-catenin. (D) TRPV1 expression. (E) Schematic diagram depicting possible mechanisms involved in cold exposure-induced changes in cardiac contractile and geometry. Red color denotes pathway unique for cardiac remodeling (GSK3β-independent). It is likely that changes in ROS generation and mitochondrial integrity may also regulate cardiac remodeling. Dashed line indicates the known cardiac remodeling action of TRPV1. Data were represented as mean ± SEM, n = 15–17 fields (B) and 5 isolations (C and D), *P < 0.05 vs control (cont) group, #P < 0.05 vs ET-1 group.
Role of Akt phosphorylation in cold stress- or ET-1-induced GSK3β dephosphorylation
Akt phosphorylates GSK3β to promote stabilization of the GSK3β target protein β-catenin (Blankesteijn et al., 2008; Cheng et al., 2011). To examine the possible role of Akt in cold exposure- or ET-1-induced dephosphorylation of GSK3β, Akt phosphorylation was examined in myocardium from cold-stressed WT and ETAKO mice. Our data revealed suppressed Akt phosphorylation (absolute or normalized) associated with unchanged Akt in cold-stressed WT mouse hearts, the effect of which was prevented by ETAKO. ETAKO itself did not affect pan or phosphorylated Akt levels. To further examine the role of Akt in ET-1-induced GSK3β dephosphorylation, murine cardiomyocytes from cardiomyocyte-specific constitutively active Akt mice were subjected to ET-1 (10nM) for 24 h prior to determination of GSK3β phosphorylation and β-catenin stabilization. Our data revealed significantly elevated phosphorylation of Akt and GSK3β accompanied with β-catenin stabilization in cardiomyocytes from Akt transgenic mice. ET-1 overtly decreased phosphorylation of Akt and GSK3β as well as β-catenin expression, the effect of which was masked by constitutive Akt activation in cardiomyocytes (Supplementary Figure S6). These data suggest a possible role of dampened Akt phosphorylation in cold exposure- and ET-1-induced GSK3β dephosphorylation.
Discussion
The myocardial morphometric and functional observation from our study demonstrated that ETAKO significantly attenuated or ablated cold exposure-induced cardiac geometric and contractile dysfunction. Our data further revealed that ETAKO significantly ameliorated cold exposure-induced generation of  and ROS, apoptosis, loss of the temperature sensor protein TRPV1, mitochondrial dysfunction (PGC1α and UCP2) as well as activation of GSK3β, GATA4, and CREB, indicating a role of these signaling molecules in cold stress-induced cardiac contractile and geometric changes. Our observation also depicted that cold exposure-induced cardiac contractile and geometric defects may be rescued by the TRPV1 agonist SA13353 while the TRPV1 antagonist capsazepine mimicked cold exposure-induced cardiomyocyte dysfunction. The GSK3β inhibitor SB216763 mitigated cold stress-induced cardiac contractile but not geometric alterations, indicating a disparate role of GSK3β in cold stress-induced geometric and functional alterations in the heart. Given that cardiac ET-1 levels are elevated following cold exposure, the effect of pharmacological regulators of GSK3β and TRPV1 as well as ETAKO was tested in ET-1-treated murine cardiomyocytes. Our findings supported a pivotal role of TRPV1 but not GSK3β in ET-1-triggered cardiomyocyte hypertrophic response and a possible sequential role of GSK3β–TRPV1 in the ET-1-induced cardiac responses. These data support the notion that GSK3β and TRPV1 signaling cascades may be essential players in the pathogenesis of cold stress-induced myocardial anomalies and suggest the likely therapeutic potential of ETA receptor antagonism in the management of cold exposure-induced cardiovascular complications.
and ROS, apoptosis, loss of the temperature sensor protein TRPV1, mitochondrial dysfunction (PGC1α and UCP2) as well as activation of GSK3β, GATA4, and CREB, indicating a role of these signaling molecules in cold stress-induced cardiac contractile and geometric changes. Our observation also depicted that cold exposure-induced cardiac contractile and geometric defects may be rescued by the TRPV1 agonist SA13353 while the TRPV1 antagonist capsazepine mimicked cold exposure-induced cardiomyocyte dysfunction. The GSK3β inhibitor SB216763 mitigated cold stress-induced cardiac contractile but not geometric alterations, indicating a disparate role of GSK3β in cold stress-induced geometric and functional alterations in the heart. Given that cardiac ET-1 levels are elevated following cold exposure, the effect of pharmacological regulators of GSK3β and TRPV1 as well as ETAKO was tested in ET-1-treated murine cardiomyocytes. Our findings supported a pivotal role of TRPV1 but not GSK3β in ET-1-triggered cardiomyocyte hypertrophic response and a possible sequential role of GSK3β–TRPV1 in the ET-1-induced cardiac responses. These data support the notion that GSK3β and TRPV1 signaling cascades may be essential players in the pathogenesis of cold stress-induced myocardial anomalies and suggest the likely therapeutic potential of ETA receptor antagonism in the management of cold exposure-induced cardiovascular complications.
Cold stress may rapidly exceed the body's capacity for self-regulation, and present a threat to the overall human health (Mercer, 2003; Swoap et al., 2004; Cheng and Su, 2010). Our findings revealed that chronic cold exposure for 2 or 5 weeks elicited cardiac hypertrophy and contractile dysfunction. Although cold exposure-induced cardiac geometric and functional changes may be associated with cold-induced increase in blood pressure and hemodynamic effects (Swoap et al., 2004; Cheng and Su, 2010), data from our study did not favor a major role of hypertension or increased afterload in ETAKO-induced beneficial responses on the heart as ETAKO rescued cold stress-induced cardiac anomalies in the absence of blood pressure regulation. Our study did not reveal a major role of heart rate although contribution from the early-on tachycardia (possibly due to sympathetic activation) cannot be completely ruled out. Cold exposure has been reported to lead to both tachycardia and bradycardia, depending on route, duration and species subjected to cold stress, through regulation of autonomic nervous tone (Swoap et al., 2004; Sun, 2010). Nonetheless, our result indicated that heart rate fully returned to baseline following 3 h, 2- and 5-week of cold exposure.
Long-term cold exposure may promote ROS generation accompanied with a compensatory increase in antioxidant defense enzymes (Hong et al., 2008). This is in line with our observation of enhanced production of  and ROS, overt apoptosis and mitochondrial damage in myocardium following cold challenge. The ability of
and ROS, overt apoptosis and mitochondrial damage in myocardium following cold challenge. The ability of  and ROS generation to facilitate myocardial hypertrophy and apoptosis is consistent with our findings of enhanced cardiomyocyte size (H&E and WTA staining), interstitial fibrosis, enlarged heart (gross section and LV chamber), and apoptosis. Cardiomyocyte-specific ETAKO rescued cold stress-induced oxidative stress and apoptosis, indicating a role of cardiomyocytes in cold stress-induced cardiac injury although contribution from non-cardiomyocyte components (such as fibroblasts) cannot be completely ruled out. Results from immunoblotting analysis indicated that cold exposure-induced cardiac hypertrophic and contractile changes were associated with preserved GSK3β, GATA4, and CREB signaling. Our data showed that ETAKO prevented cold stress-induced cardiac remodeling as evidenced by heart weight, heart size, LV wall thickness, LV mass and histological changes despite persistent elevation in cardiac (but not plasma) ET-1 levels. ET-1 has a predominant paracrine action. Plasma ET-1 levels usually denote ET-1 spillover to the circulation which poorly reflects the local ET-1 action under pathological conditions (Barton and Yanagisawa, 2008). In our hands, ETAKO ameliorated cold stress (ET-1)-induced dephosphorylation of GSK3β (evidenced by pan GSK3β and β-catenin stabilization finding), as well as hyper-phosphorylation of GATA4 and CREB, suggesting a role of these signaling molecules in the beneficial effects of ETAKO. However, GSK3β inhibition mitigated cold-induced cardiac contractile but not geometric changes, indicating a GSK3β-dependent and -independent mechanism responsible for the functional and geometric changes, respectively, under cold stress. Such discrepant regulatory mechanisms of GSK3β under cold stress are illustrated in Figure 7E. The GSK3β-independent regulatory machinery of cardiac remodeling for cold stress and ETAKO appears to be reasonable as cold stress (and ET-1) promotes GSK3β activity (dephosphorylation) involved in negative regulation of protein synthesis and cardiac hypertrophy (Cheng et al., 2011). Our observation that ETAKO reversed cold stress-induced phosphorylation of GATA4 and CREB suggested a role of these hypertrophic signaling molecules in cold stress- and ETAKO-induced regulation of cardiac hypertrophy.
and ROS generation to facilitate myocardial hypertrophy and apoptosis is consistent with our findings of enhanced cardiomyocyte size (H&E and WTA staining), interstitial fibrosis, enlarged heart (gross section and LV chamber), and apoptosis. Cardiomyocyte-specific ETAKO rescued cold stress-induced oxidative stress and apoptosis, indicating a role of cardiomyocytes in cold stress-induced cardiac injury although contribution from non-cardiomyocyte components (such as fibroblasts) cannot be completely ruled out. Results from immunoblotting analysis indicated that cold exposure-induced cardiac hypertrophic and contractile changes were associated with preserved GSK3β, GATA4, and CREB signaling. Our data showed that ETAKO prevented cold stress-induced cardiac remodeling as evidenced by heart weight, heart size, LV wall thickness, LV mass and histological changes despite persistent elevation in cardiac (but not plasma) ET-1 levels. ET-1 has a predominant paracrine action. Plasma ET-1 levels usually denote ET-1 spillover to the circulation which poorly reflects the local ET-1 action under pathological conditions (Barton and Yanagisawa, 2008). In our hands, ETAKO ameliorated cold stress (ET-1)-induced dephosphorylation of GSK3β (evidenced by pan GSK3β and β-catenin stabilization finding), as well as hyper-phosphorylation of GATA4 and CREB, suggesting a role of these signaling molecules in the beneficial effects of ETAKO. However, GSK3β inhibition mitigated cold-induced cardiac contractile but not geometric changes, indicating a GSK3β-dependent and -independent mechanism responsible for the functional and geometric changes, respectively, under cold stress. Such discrepant regulatory mechanisms of GSK3β under cold stress are illustrated in Figure 7E. The GSK3β-independent regulatory machinery of cardiac remodeling for cold stress and ETAKO appears to be reasonable as cold stress (and ET-1) promotes GSK3β activity (dephosphorylation) involved in negative regulation of protein synthesis and cardiac hypertrophy (Cheng et al., 2011). Our observation that ETAKO reversed cold stress-induced phosphorylation of GATA4 and CREB suggested a role of these hypertrophic signaling molecules in cold stress- and ETAKO-induced regulation of cardiac hypertrophy.
Perhaps the most interesting findings from our study were that ETAKO reversed cold exposure-induced downregulation of TRPV1 expression. This is in concert with the findings that TRPV1 agonist rescued cold exposure-induced cardiac geometric and functional defects. It is likely that TRPV1 governs the GSK3β-independent cardiac remodeling response under cold stress and/or ETAKO. This is supported by the in vitro data that TRPV1 agonist SA13353 but not GSK3β inhibitor SB216763 ablated ET-1-induced cardiomyocyte hypertrophy. TRPV1 antagonist mimicked cold stress-induced myocardial contractile dysfunction (lack of geometric changes may be due to the short treatment nature). TRP channels serve as receptors for a variety of biological events, including regulation of temperature, mechanical force, and noxious sensation (Wang et al., 2008). TRPV1, a ligand-gated cation channel found mainly in sensory nerves in the cardiovascular system, plays a key role in cardiovascular and neurological diseases, asthma, cancer, and renal diseases (Nilius and Owsianik, 2010). Deletion of TRPV1 has been shown to impair postischemic recovery in isolated perfused heart, exacerbate inflammation, and cardiac remodeling after myocardial infarction (Wang and Wang, 2005; Wang et al., 2008). Recent evidence has consolidated the beneficial cardiovascular effects of TRPV1 agonists such as capsaicinoids and capsinoids through ischemic preconditioning, release of neurotransmitters and inhibition of platelet aggregation (Luo et al., 2011). Our in vitro data shown in Figure 7 suggested an upstream regulatory role of GSK3β for TRPV1. We noted that GSK3β inhibition ablated ET-1-induced changes in TRPV1, but not vice versa. Given that both GSK3β inhibitor and TRPV1 agonist effectively rescued cold stress-induced contractile defects, our data support a GSK3β–TRPV1-mediated mechanism in the regulation of cardiac contractile function and possibly intracellular Ca2+ homeostasis under cold stress and ETAKO. As shown in our study, cold exposure triggers ET-1 release (Chen and Sun, 2006), en route to induction of oxidative stress, apoptosis, cellular damage, and cardiac dysfunction through the NAPDH oxidase-mediated production of 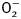 (Xu et al., 2004). Interestingly, TRPV1 participates in the regulation of NADPH oxidase-mediated ROS production (Schilling and Eder, 2009). Our data revealed that cold exposure promotes mitochondrial damage as evidenced by reduced MMP, an indicator for mitochondrial function (Juhaszova et al., 2004). GSK3β is a double-edged sword retarding pathological cardiac remodeling while promoting mitochondrial permeability transition pore opening (Juhaszova et al., 2004; Blankesteijn et al., 2008). GSK3β-induced mitochondrial permeation pore opening promotes mitochondrial damage in pathological conditions such as aging, diabetes and ischemia-reperfusion (Cheng et al., 2011; Zhang et al., 2011). Our present data revealed that ETAKO nullified cold stress-induced GSK3β activity, favoring preserved mitochondrial integrity. Furthermore, TRPV1 agonist nullified cold exposure-induced MMP depolarization, favoring a role of TRPV1 downstream of GSK3β in ETAKO-offered protection of mitochondria in cold stress. Mitochondrial dysfunction triggers a cascade of unfavorable cellular events leading to intracellular Ca2+ mishandling and contractile dysfunction (Ren et al., 2010; Zhang et al., 2011). Further study is warranted to better elucidate the mechanism of action behind the GSK3β–TRPV1-engaged regulation on mitochondrial integrity, intracellular Ca2+ homeostasis and contractile function in the heart under cold stress.
(Xu et al., 2004). Interestingly, TRPV1 participates in the regulation of NADPH oxidase-mediated ROS production (Schilling and Eder, 2009). Our data revealed that cold exposure promotes mitochondrial damage as evidenced by reduced MMP, an indicator for mitochondrial function (Juhaszova et al., 2004). GSK3β is a double-edged sword retarding pathological cardiac remodeling while promoting mitochondrial permeability transition pore opening (Juhaszova et al., 2004; Blankesteijn et al., 2008). GSK3β-induced mitochondrial permeation pore opening promotes mitochondrial damage in pathological conditions such as aging, diabetes and ischemia-reperfusion (Cheng et al., 2011; Zhang et al., 2011). Our present data revealed that ETAKO nullified cold stress-induced GSK3β activity, favoring preserved mitochondrial integrity. Furthermore, TRPV1 agonist nullified cold exposure-induced MMP depolarization, favoring a role of TRPV1 downstream of GSK3β in ETAKO-offered protection of mitochondria in cold stress. Mitochondrial dysfunction triggers a cascade of unfavorable cellular events leading to intracellular Ca2+ mishandling and contractile dysfunction (Ren et al., 2010; Zhang et al., 2011). Further study is warranted to better elucidate the mechanism of action behind the GSK3β–TRPV1-engaged regulation on mitochondrial integrity, intracellular Ca2+ homeostasis and contractile function in the heart under cold stress.
Our current study suggested that the cold stress-induced dephosphorylation of GSK3β may be underscored by overtly dampened Akt phosphorylation under cold stress (or in response to ET-1). Akt phosphorylation inactivates GSK3β through phosphorylation at Ser9 position to promote stabilization of the GSK3β target protein β-catenin (Blankesteijn et al., 2008; Cheng et al., 2011). ET-1 has been shown to inhibit Akt phosphorylation and compromises insulin signaling (Shemyakin et al., 2011), consistent with our finding. However, other mechanisms such as ROS accumulation may also contribute to cold stress- and ET-1-induced regulation of GSK3β signaling cascade and should not be discounted. It is worth mentioning that the natural TRPV1 ligand capsaicin has been reported to activate Akt (Malagarie-Cazenave et al., 2009). Thus reduced TRPV1 expression under cold stress may initiate a vicious cycle in further dampening Akt signaling and subsequently dephosphorylation of GSK3β, a downstream signaling molecule of Akt (Zhao et al., 2011).
Cold stress is associated with a higher cardiovascular mortality rate (Sheth et al., 1999; Cheng and Su, 2010; Sun, 2010) where accumulation of ET-1 and decrease in the temperature sensor TRPV1 are present. Both ETA receptor blockers and TRPV1 analogues have shown beneficial clinical outcome (Luscher et al., 2002; Barton and Yanagisawa, 2008; Banfor et al., 2009; Luo et al., 2011) although if and how ETA receptor and TRPV1 contribute to cold climate-induced comorbidities still remain elusive. Although several TRPV1 agonists have received FDA approval in pain control, the therapeutic potential of TRPV1 analogues in cardiovascular diseases is just emerging.
In summary, our present study provides evidence, for the first time, that ETAKO rescues cold exposure-induced cardiac hypertrophy and contractile dysfunction. Our data indicated that GSK3β dephosphorylation, loss of TRPV1, oxidative stress and mitochondrial damage are involved in cold stress- and ETAKO-elicited changes in cardiac anomalies. Given that TRPV1 is capable of mediating vascular protection against hypertension (Yang et al., 2010), our study using the ETAKO model further suggest the potential of ETA receptor and TRPV1 as therapeutic targets in cold stress-associated comorbidities.
Materials and methods
ETAKO transgenic mice, cold exposure and drug treatment
All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee at the University of Wyoming (Laramie, WY, USA). Generation of cardiomyocyte-specific ETAKO mice was described previously in detail (Kedzierski et al., 2003). Eight to 10-month-old male ETAKO mice and their WT littermates were housed at room temperature or low temperature in a cold room (4°C) within the School of Pharmacy Animal Facility with free access to food and water for 2 or 5 weeks before myocardial morphology and function were evaluated. Systolic and diastolic blood pressures were examined with a semi-automated, amplified tail cuff device. A cohort of WT mice were subjected to cold exposure for 2 weeks while receiving the TRPV1 agonist SA13353 (30 mg/kg, p.o.) (Tsuji et al., 2010), the TRPV1 antagonist capsazepine (5 mg/kg, i.p.) (Nguyen et al., 2010), or the GSK3β inhibitor SB216763 (0.6 mg/kg, i.p.) (Obame et al., 2008). Information of cardiomyocyte-specific constitutively active Akt model was described in Supplementary material.
Plasma and cardiac levels of ET-1
Plasma and cardiac ET-1 levels were determined using an ET-1 enzyme immunometric assay kit (Assay Designs, Inc.) based on a double-antibody sandwich technique, as described in our earlier report (Xu et al., 2008). The detection threshold for ET-1 was 0.14 pg/ml. The inter-assay and intra-assay coefficients of variation were below 3.3%.
Echocardiographic assessment
Cardiac geometry and function were evaluated in anesthetized (avertin 2.5%, 10 μl/g body weight, i.p.) mice using 2-D guided M-mode echocardiography (Sonos 5500) equipped with a 15–6 MHz linear transducer. LV anterior and posterior wall dimensions during diastole and systole were recorded from three consecutive cycles in M-mode using methods adopted by the American Society of Echocardiography. Fractional shortening was calculated from LV EDD and ESD diameters using the equation (EDD − ESD)/EDD. Heart rates were averaged over 10 cardiac cycles (Doser et al., 2009).
Isolation of cardiomyocytes
After ketamine/xylazine sedation, hearts were removed and perfused with Krebs-Henseleit bicarbonate (KHB) buffer containing (in mM): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES, and 11.1 glucose. Hearts were digested with collagenase D for 20 min. Left ventricles were removed and minced before being filtered. Myocyte yield was ∼ 75%, which was not affected by cold exposure or ETAKO. Only rod-shaped myocytes with clear edges were selected for mechanical and intracellular Ca2+ study (Doser et al., 2009).
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using an IonOptix™ soft-edge system (IonOptix). Myocytes were placed in a chamber mounted on the stage of an Olympus IX-70 microscope and superfused (∼2 ml/min at 25°C) with a KHB buffer containing 1 mM CaCl2. Myocytes were field stimulated at 0.5 Hz unless otherwise stated. Cell shortening and relengthening were assessed, including PS, TPS, TR90, and ±dL/dt (Doser et al., 2009). In the case of altering stimulus frequency from 0.1 to 5.0 Hz, the steady-state contraction of myocyte was achieved (usually after the first 5–6 beats) before PS was recorded.
Intracellular Ca2+ transients
A cohort of myocytes were loaded with fura-2/AM (0.5 μM) for 10 min and fluorescence intensity was recorded with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed onto an Olympus IX-70 inverted microscope and imaged through a Fluor ×40 oil objective. Cells were exposed to light emitted by a 75 W lamp and passed through either a 360 or a 380 nm filter, while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480–520 nm and qualitative change in fura-2 fluorescence intensity (FFI) was inferred from FFI ratio at the two wavelengths (360/380). Fluorescence decay time was calculated as an indicator of intracellular Ca2+ clearing (Doser et al., 2009).
Histological examination
Following anesthesia, hearts were excised and immediately placed in 10% neutral-buffered formalin at room temperature for 24 h after a brief rinse with phosphate buffered saline (PBS). The specimens were embedded in paraffin, cut in 5 µm sections and stained with H&E as well as FITC-conjugated wheat germ agglutinin. Heart sections were stained with H&E for gross morphology analysis. Cardiomyocyte cross-sectional areas were calculated on a digital microscope (400×) using the Image J (version1.34S) software. The Masson's trichrome staining was used to detect fibrosis in heart sections. The percentage of fibrosis was calculated using the histogram function of the photoshop software. Briefly, 5 random fields (6 mm2) at 200× magnification from each section were assessed for fibrosis. The fraction of the light blue stained area normalized to the total area was used as an indicator of myocardial fibrosis while omitting fibrosis of the perivascular, epicardial, and endocardial areas from the study (Doser et al., 2009; Turdi et al., 2011).
Intracellular fluorescence measurement of 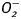 and ROS
and ROS
Intracellular 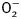 and ROS were measured by changes in fluorescence intensity resulting from intracellular probe oxidation (Privratsky et al., 2003; Dong et al., 2006). In brief, cardiomyocytes were loaded with dihydroethidium (DHE, 5 μM) and 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, 1 μM) (Molecular Probes) for 30 min at 37°C for detection of
and ROS were measured by changes in fluorescence intensity resulting from intracellular probe oxidation (Privratsky et al., 2003; Dong et al., 2006). In brief, cardiomyocytes were loaded with dihydroethidium (DHE, 5 μM) and 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, 1 μM) (Molecular Probes) for 30 min at 37°C for detection of  and ROS, respectively. Cells were sampled randomly using an Olympus BX-51 microscope equipped with Olympus MagnaFire™ SP digital camera and ImagePro image analysis software (Media Cybernetics). Fluorescence was calibrated with InSpeck microspheres (Molecular Probes). An average of 100 cells were evaluated using the grid crossing method in 15 visual fields per isolation.
and ROS, respectively. Cells were sampled randomly using an Olympus BX-51 microscope equipped with Olympus MagnaFire™ SP digital camera and ImagePro image analysis software (Media Cybernetics). Fluorescence was calibrated with InSpeck microspheres (Molecular Probes). An average of 100 cells were evaluated using the grid crossing method in 15 visual fields per isolation.
TUNEL assay
TUNEL staining of myonuclei positive for DNA strand breaks were determined using a fluorescence detection kit (Roche). Paraffin-embedded sections (5 μm) were deparaffinized and rehydrated before incubation with proteinase K solution at room temperature for 30 min. TUNEL reaction mixture containing terminal deoxynucleotidyl transferase, fluorescein-dUTP was added to the sections in 50-μl drops and incubated for 60 min at 37°C in a humidified chamber in the dark. Following embedding, sections were visualized with an Olympus BX-51 microscope equipped with an Olympus MaguaFire SP digital camera. DNase I and label solution were used as positive and negative controls. To determine the percentage of apoptotic cells, micrographs of TUNEL-positive and DAPI-stained nuclei were captured using an Olympus fluorescence microscope and counted using the ImageJ software (ImageJ version 1.43r; NIH) from 15 random fields at 400× magnification (Ren et al., 2009).
Western blot analysis
The protein was prepared as described (Doser et al., 2009). Samples containing equal amount of proteins were separated on 10% SDS–polyacrylamide gels in a minigel apparatus (Mini-PROTEAN II; Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk in TBS-T, and were incubated overnight at 4°C with anti-TRPV1, anti-HSP90, anti-PGC1α, anti-UCP2, anti-GSK3β, anti-pGSK3β (Ser9), anti-GATA4, anti-pGATA4 (Ser105), anti-CREB, and anti-pCREB (Ser133) antibodies. After immunoblotting, the film was scanned and the intensity of immunoblot bands was detected with a Bio-Rad Calibrated Densitometer. GAPDH was used as the loading control. To evaluate the sequential role of GSK3β and TRPV1 in ET-1-induced responses, H9C2 myoblasts were incubated for 24 h with ET-1 (10 nM) in the absence or presence of the TRPV1 agonist SA13353 (1 µM), the TRPV1 inhibitor capsazepine (1 µM) or the GSK3β inhibitor SB216763 (10 µM) for 24 h before levels of GSK3β, phosphorylated GSK3β and TRPV1 were examined.
Measurement of MMP
Cardiomyocytes were suspended in HEPES-saline buffer and MMP (ΔΨm) was detected as described (Ma et al., 2009). Briefly, after pre-incubation with 5 µM JC-1 for 10 min at 37°C, cells were washed two times by sedimentation using HS buffer free of JC-1 and resuspended in modified Eagle's medium (MEM) supplemented with MG-AGE or bovine serum albumin (BSA) (2.5 µM) at 37°C for 4 h. During the incubation period, cardiomyocytes were examined periodically under confocal laser scanning microscope (Leica TCS SP2) at excitation wavelength of 490 nm and emission fluorescence was recorded at 530 nm (monomer form of JC-1, green) and at 590 nm (aggregate form of JC-1, red). Alternatively, fluorescence of each sample was read at excitation wavelength of 490 nm and emission wavelength of 530 and 590 nm using a spectrofluorimeter (Spectra MaxGeminiXS) at an interval of 10 sec. Results in fluorescence intensity were expressed as 590/530 nm emission ratio.
Evaluation of murine cardiomyocyte surface area
Adult mouse cardiomyocytes were isolated as described and maintained in MEM. Following treatment of ET-1 (10 nM, 24 h) with or without the preincubation of SA13353 (1 µM), capsazepine (1 µM), and SB216763 (10 µM) for 30 min and then throughout the ET-1 exposure duration, cardiomyocytes on chamber slides were rinsed with PBS and fixed for 15 min in 4% paraformaldehyde at room temperature. Cells were then blocked with 5% BSA for 30 min prior to incubation with an antibody against muscle-specific α-actin at 37°C for 1 h. The cells were then incubated with a goat anti-rabbit IgG-FITC antibody at 37°C for 60 min. Following four rinses with PBS, cells were stained with propidium iodide (PI, 1 mg/ml) for 30 min at 37°C and were viewed using a Nikon Eclipse TE300 microscope with a Cascade-cooled CCD digital camera (Roper Scientific, Inc.). Areas of cardiomyocytes were measured with the NIH ImageJ software using images of myocytes stained with the anti-α-actin antibody (Hua et al., 2011).
Data analysis
Data were mean ± SEM. Statistical significance (P < 0.05) for each variable was estimated by analysis of variance followed by a Tukey's post hoc analysis.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by National Center for Research Resource (NCRR) P20RR016474.
Conflict of interest: none declared.
Acknowledgements
The authors wish to thank Santen Pharmaceutical Co., Ltd, Japan for providing SA13353. This work was presented at the 2011 American Heart Association Keystone Meeting (Keystone, CO, USA).
References
- Abassi Z.A., Ellahham S., Winaver J., et al. The intrarenal endothelin system and hypertension. News Physiol. Sci. 2001;16:152–156. doi: 10.1152/physiologyonline.2001.16.4.152. [DOI] [PubMed] [Google Scholar]
- Banfor P.N., Franklin P.A., Segreti J.A., et al. ETA receptor blockade with atrasentan prevents hypertension with the multitargeted tyrosine kinase inhibitor ABT-869 in telemetry-instrumented rats. J. Cardiovasc. Pharmacol. 2009;53:173–178. doi: 10.1097/FJC.0b013e3181993493. [DOI] [PubMed] [Google Scholar]
- Barton M., Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can. J. Physiol. Pharmacol. 2008;86:485–498. doi: 10.1139/Y08-059. [DOI] [PubMed] [Google Scholar]
- Blankesteijn W.M., van de Schans V.A., ter Horst P., et al. The Wnt/frizzled/GSK-3 beta pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharmacol. Sci. 2008;29:175–180. doi: 10.1016/j.tips.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Chen G.F., Sun Z. Effects of chronic cold exposure on the endothelin system. J. Appl. Physiol. 2006;100:1719–1726. doi: 10.1152/japplphysiol.01407.2005. [DOI] [PubMed] [Google Scholar]
- Cheng X., Su H. Effects of climatic temperature stress on cardiovascular diseases. Eur. J. Intern. Med. 2010;21:164–167. doi: 10.1016/j.ejim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Cheng H., Woodgett J., Maamari M., et al. Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J. Mol. Cell. Cardiol. 2011;51:607–613. doi: 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F., Zhang X., Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor-NADPH oxidase pathway. Hypertension. 2006;47:222–229. doi: 10.1161/01.HYP.0000198555.51645.f1. [DOI] [PubMed] [Google Scholar]
- Doser T.A., Turdi S., Thomas D.P., et al. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Eurowinter Group. Cold exposure and winter mortality from ischaemic heart disease, cerebrovascular disease, respiratory disease, and all causes in warm and cold regions of Europe. The Eurowinter Group. Lancet. 1997;349:1341–1346. [PubMed] [Google Scholar]
- Finck B.N., Kelly D.P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J.H., Kim K.J., Suzuki K., et al. Effect of cold acclimation on antioxidant status in cold acclimated skaters. J. Physiol. Anthropol. 2008;27:255–262. doi: 10.2114/jpa2.27.255. [DOI] [PubMed] [Google Scholar]
- Howard B.V., Comuzzie A., Devereux R.B., et al. Cardiovascular disease prevalence and its relation to risk factors in Alaska Eskimos. Nutr. Metab. Cardiovasc. Dis. 2010;20:350–358. doi: 10.1016/j.numecd.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y., Zhang Y., Ren J. IGF-1 deficiency resists cardiac hypertrophy and myocardial contractile dysfunction: role of microRNA-1 and microRNA-133a. J. Cell. Mol. Med. 2012;16:83–95. doi: 10.1111/j.1582-4934.2011.01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M., Zorov D.B., Kim S.H., et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karashima Y., Talavera K., Everaerts W., et al. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl Acad. Sci. USA. 2009;106:1273–1278. doi: 10.1073/pnas.0808487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski R.M., Grayburn P.A., Kisanuki Y.Y., et al. Cardiomyocyte-specific endothelin A receptor knockout mice have normal cardiac function and an unaltered hypertrophic response to angiotensin II and isoproterenol. Mol. Cell. Biol. 2003;23:8226–8232. doi: 10.1128/MCB.23.22.8226-8232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloner R.A. Natural and unnatural triggers of myocardial infarction. Prog. Cardiovasc. Dis. 2006;48:285–300. doi: 10.1016/j.pcad.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Luo X.J., Peng J., Li Y.J. Recent advances in the study on capsaicinoids and capsinoids. Eur. J. Pharmacol. 2011;650:1–7. doi: 10.1016/j.ejphar.2010.09.074. [DOI] [PubMed] [Google Scholar]
- Luscher T.F., Enseleit F., Pacher R., et al. Hemodynamic and neurohumoral effects of selective endothelin A (ET(A)) receptor blockade in chronic heart failure: the Heart Failure ET(A) Receptor Blockade Trial (HEAT) Circulation. 2002;106:2666–2672. doi: 10.1161/01.cir.0000038497.80095.e1. [DOI] [PubMed] [Google Scholar]
- Ma H., Li S.Y., Xu P., et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J. Cell. Mol. Med. 2009;13:1751–1764. doi: 10.1111/j.1582-4934.2008.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Malagarie-Cazenave S., Olea-Herrero N., Vara D., et al. Capsaicin, a component of red peppers, induces expression of androgen receptor via PI3K and MAPK pathways in prostate LNCaP cells. FEBS Lett. 2009;583:141–147. doi: 10.1016/j.febslet.2008.11.038. [DOI] [PubMed] [Google Scholar]
- Medina-Ramon M., Zanobetti A., Cavanagh D.P., et al. Extreme temperatures and mortality: assessing effect modification by personal characteristics and specific cause of death in a multi-city case-only analysis. Environ. Health Perspect. 2006;114:1331–1336. doi: 10.1289/ehp.9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J.B. Cold—an underrated risk factor for health. Environ. Res. 2003;92:8–13. doi: 10.1016/s0013-9351(02)00009-9. [DOI] [PubMed] [Google Scholar]
- Nguyen T.L., Nam Y.S., Lee S.Y., et al. Effects of capsazepine, a transient receptor potential vanilloid type 1 antagonist, on morphine-induced antinociception, tolerance, and dependence in mice. Br. J. Anaesth. 2010;105:668–674. doi: 10.1093/bja/aeq212. [DOI] [PubMed] [Google Scholar]
- Nilius B., Owsianik G. Transient receptor potential channelopathies. Pflugers. Arch. 2010;460:437–450. doi: 10.1007/s00424-010-0788-2. [DOI] [PubMed] [Google Scholar]
- Obame F.N., Plin-Mercier C., Assaly R., et al. Cardioprotective effect of morphine and a blocker of glycogen synthase kinase 3 beta, SB216763 [3-(2,4-dichlorophenyl)-4(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione], via inhibition of the mitochondrial permeability transition pore. J. Pharmacol. Exp. Ther. 2008;326:252–258. doi: 10.1124/jpet.108.138008. [DOI] [PubMed] [Google Scholar]
- Okamoto-Mizuno K., Tsuzuki K., Mizuno K., et al. Effects of low ambient temperature on heart rate variability during sleep in humans. Eur. J. Appl. Physiol. 2009;105:191–197. doi: 10.1007/s00421-008-0889-1. [DOI] [PubMed] [Google Scholar]
- Privratsky J.R., Wold L.E., Sowers J.R., et al. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003;42:206–212. doi: 10.1161/01.HYP.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- Ren J., Babcock S.A., Li Q., et al. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol. Lett. 2009;187:149–156. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Pulakat L., Whaley-Connell A., et al. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. (Berl) 2010;88:993–1001. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey B., Roussel D., Romestaing C., et al. Up-regulation of avian uncoupling protein in cold-acclimated and hyperthyroid ducklings prevents reactive oxygen species production by skeletal muscle mitochondria. BMC Physiol. 2010;10:5. doi: 10.1186/1472-6793-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T., Eder C. Importance of the non-selective cation channel TRPV1 for microglial reactive oxygen species generation. J. Neuroimmunol. 2009;216:118–121. doi: 10.1016/j.jneuroim.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Schilling J., Kelly D.P. The PGC-1 cascade as a therapeutic target for heart failure. J. Mol. Cell. Cardiol. 2011;51:578–583. doi: 10.1016/j.yjmcc.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemyakin A., Salehzadeh F., Esteves Duque-Guimaraes D., et al. Endothelin-1 reduces glucose uptake in human skeletal muscle in vivo and in vitro. Diabetes. 2011;60:2061–2067. doi: 10.2337/db10-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth T., Nair C., Muller J., et al. Increased winter mortality from acute myocardial infarction and stroke: the effect of age. J. Am. Coll. Cardiol. 1999;33:1916–1919. doi: 10.1016/s0735-1097(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Sun Z. Cardiovascular responses to cold exposure. Front Biosci. (Elite Ed) 2010;2:495–503. doi: 10.2741/e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoap S.J., Overton J.M., Garber G. Effect of ambient temperature on cardiovascular parameters in rats and mice: a comparative approach. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R391–R396. doi: 10.1152/ajpregu.00731.2003. [DOI] [PubMed] [Google Scholar]
- Tsuji F., Murai M., Oki K., et al. Transient receptor potential vanilloid 1 agonists as candidates for anti-inflammatory and immunomodulatory agents. Eur. J. Pharmacol. 2010;627:332–339. doi: 10.1016/j.ejphar.2009.10.044. [DOI] [PubMed] [Google Scholar]
- Turdi S., Kandadi M.R., Zhao J., et al. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J. Mol. Cell. Cardiol. 2011;50:712–722. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang D.H. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation. 2005;112:3617–3623. doi: 10.1161/CIRCULATIONAHA.105.556274. [DOI] [PubMed] [Google Scholar]
- Wang Y., Babankova D., Huang J., et al. Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension. 2008;52:264–270. doi: 10.1161/HYPERTENSIONAHA.108.110197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Puigserver P., Andersson U., et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Xu F.P., Chen M.S., Wang Y.Z., et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 2004;110:1269–1275. doi: 10.1161/01.CIR.0000140766.52771.6D. [DOI] [PubMed] [Google Scholar]
- Xu H., Duan J., Dai S., et al. alpha-zearalanol attenuates oxLDL-induced ET-1 gene expression, ET-1 secretion and redox-sensitive intracellular signaling activation in human umbilical vein endothelial cells. Toxicol. Lett. 2008;179:163–168. doi: 10.1016/j.toxlet.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Yang D., Luo Z., Ma S., et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010;12:130–141. doi: 10.1016/j.cmet.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Mengersen K., Wang X., et al. Daily average temperature and mortality among the elderly: a meta-analysis and systematic review of epidemiological evidence. Int. J. Biometeorol. 2011 doi: 10.1007/s00484-011-0497-3. 10.1007/s00484-011-0497-3. [DOI] [PubMed] [Google Scholar]
- Zhang X.Y., Francis R.J., Sun C.K., et al. Endothelin receptor A blockade ameliorates hypothermic ischemia-reperfusion-related microhemodynamic disturbances during liver transplantation in the rat. J. Surg. Res. 2002;102:63–70. doi: 10.1006/jsre.2001.6246. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xia Z., La Cour K.H., et al. Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeation pore opening. Antioxid. Redox Signal. 2011;15:2407–2424. doi: 10.1089/ars.2010.3751. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao Y., Wang Y., Zhu W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 2011;3:276–282. doi: 10.1093/jmcb/mjr013. [DOI] [PubMed] [Google Scholar]