

Abstract
Mitochondrial DNA (mtDNA) deletion mutations are frequently observed in aged postmitotic tissues and are the cause of a wide range of human disorders. Presently, the molecular bases underlying mtDNA deletion formation remain a matter of intense debate, and it is commonly accepted that several mechanisms contribute to the spectra of mutations in the mitochondrial genome. In this work we performed an extensive screening of human mtDNA deletions and evaluated the association between breakpoint density and presence of non-canonical DNA elements and over-represented sequence motifs. Our observations support the involvement of helix-distorting intrinsically curved regions and long G-tetrads in eliciting instability events. In addition, higher breakpoint densities were consistently observed within GC-skewed regions and in the close vicinity of the degenerate sequence motif YMMYMNNMMHM. A parallelism is also established with hot spot motifs previously identified in the nuclear genome, as well as with the minimal binding site for the mitochondrial transcription termination factor mTERF. This study extends the current knowledge on the mechanisms driving mitochondrial rearrangements and opens up exciting avenues for further research.
Introduction
Rearrangement of mitochondrial DNA (mtDNA) and consequent loss of mitochondrial function has been implicated in the aging process and in a broad range of clinical phenotypes (reviewed in [1]). Short direct and inverted repeats are found to flank the majority of such rearrangements (∼85%), a fact that has led to the assumption that deletion formation arises from slipped mispairing during replication or repair of damaged DNA [2]. It appears, however, that no significant correlation exists between the density of repeat pairs and distribution of deletion breakpoints [3], suggesting that additional factors are likely to contribute to the mutational spectra. In human mtDNA rearrangements, the 5′ breakpoints have a typical unimodal distribution with a maximum around 8 to 9 kb, whereas the 3′ breakpoints are preferentially clustered above 13 kb [3]. This distribution bias has led Samuels and co-workers [3] to present evidence strongly favoring the existence of a unique and similar mechanism involved in the formation of all mtDNA deletions, irrespective of their immediate deletion breakpoints or presence of repeated sequences. The authors show that the 13 bp direct repeats responsible for the 4,977 bp “common deletion”, lie in the center of the distribution of other deletions, and are therefore at least partially responsible for governing the latter. Later on, an elegant computational analysis by Guo and colleagues [4] has refuted the bimodal hypothesis proposed in Samuels’s work, by essentially showing that the distribution of deletions in individuals lacking the 13 bp repeat pair is roughly the same as those found in control samples containing it. In alternative, they suggest that deletions arise preferentially through the formation of distant segments of mtDNA capable of forming stable imperfect duplexes. According to this view, after the duplex is formed, the deletion will occur at one of the many perfect repeats available nearby. Further evidence point towards an evolutionary selection pressure against long and stable repeats in long living mammals such as the human, but not in short-lived ones [5], [6].
More recently, Damas and co-workers [7] have found that the most frequent deletion breakpoints occur within or near regions showing non-canonical (non-B) conformations, namely hairpins, cruciforms and cloverleaf-like elements. These important findings are in line with two earlier studies that support non-B-mediated instability: the first, published more than a decade ago, found that bent-inducing sequences render certain regions of the mitochondrial genome more labile to attack by reactive oxygen species or more prone to undergo deletions or duplications [8], [9]. The second observed that several direct repeats flanking mtDNA rearrangements have a skewed base composition rich in pyrimidines at the level of the light strand, thus suggesting the formation of a triple-helix structure between repeats [10].
When considering all of the above, it becomes clearer that none of the theories proposed so far is broad enough to explain all variants of the mutational spectra. Instead, they collectively point for a multifactorial view of human mtDNA instability, as previously suggested [4]. In this work we started by evaluating the importance of alternative non-canonical structures (e.g. intrinsically curved DNA, G-quadruplexes, triplex DNA and Z-DNA), whose impact on mtDNA instability is unknown or has so far been poorly explored in the literature. Most interestingly, our analysis revealed that the biased distribution of breakpoints along the mitochondrial genome correlates significantly with local compositional skews and with the presence of a degenerate sequence motif whose biological significance is discussed. In combination with past studies, the data shown here may help to understand and redefine the multiple mechanisms by which deletion formation occurs in the human mitochondrial genome.
Methods
Wild-type Human Mitochondrial DNA Sequence
The light strand of the mitochondrial revised Cambridge reference sequence (rCRS, accession number: NC_012920) was used throughout this study.
Meta-analysis of Deletion Breakpoints in Human mtDNA
We compiled 754 different mitochondrial deletions available at the Mitomap database (http://www.mitomap.org) and in published literature describing pathological and non-pathological clinical situations (see Table S1). These breakpoints were defined as the positions upstream of the 5′ break and downstream of the 3′ break and numbered according to the L-strand positions of the rCRS mtDNA sequence. During the breakpoint selection process, we have excluded breakpoint pairs in which both extremities were repeated. However, those deletions sharing one breakpoint and differing in another were considered as distinct, and were therefore included. Due to the presence of flanking repeats, intervals of values are often provided in the literature instead of the exact breakpoint positions (e.g. 7,508–7,515; 15,939–15,946). In these cases, we have maintained the smallest value for each breakpoint in the interval (7,508–15,939 in the latter example). This allowed for an easier comparison between our data and that provided by earlier reports [7]. The mutational spectra encompassing all of the 5′ and 3′ deletion breakpoints were plotted using R (http://cran.r-project.org).
Curvature/Bendability Profiles and Three-dimensional Representation of DNA Sequences
Curvature propensity plots were obtained using the BEND algorithm [11] by submission of DNA sequences to the bend.it server (http://hydra.icgeb.trieste.it/dna/bend_it.html) [12] using the DNAse I-based parameters of [13]. This server calculates DNA curvature as a vector sum of dinucleotide geometries (roll, tilt and twist angles) and expresses it as degrees per helical turn (10.5°/helical turn = 1°/bp). DNA sequences were submitted in raw format and the predicted curvature and bendability were collected by E-mail in ASCII format. Three-dimensional representation of the curvature profiles was performed with the model.it server (http://hydra.icgeb.trieste.it/dna/model_it.html) [12] and the output was displayed and visualized with MOLEGRO Molecular Viewer (http://www.molegro.com/mmv-product.php).
Sliding Window Analysis of GC-content and GC-skew
We have used the DNA base composition analysis tool (http://molbiol-tools.ca/Jie_Zheng/dna.html) to evaluate GC-content and GC-skew along the human mitochondrial genome using non-overlapping 20 bp sliding windows. GC-skew was calculated as (G-C)/(G+C).
Randomization of Breakpoint Positions
For breakpoint randomization we have generated 200 datasets using two approaches. In the so-called random approach, 1,508 arbitrary deletion breakpoints (twice the number of deletions) were randomly distributed throughout the mitochondrial genome with no restrictions. In the partially random approach the same number of breakpoints was generated while maintaining their original abundance within the mitochondrial arcs and origins of replication. In both cases, repeated events were allowed to occur. In order to assess the significance of a given variable P we computed a z-score as:
where 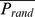 is the average of the randomized variable P and
is the average of the randomized variable P and  represents its standard deviation. The corresponding p values were obtained from
represents its standard deviation. The corresponding p values were obtained from 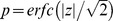 , where erfc is the complement error function.
, where erfc is the complement error function.
De novo Motif Finding, Selection and Validation
Identification of G-quadruplex structures was performed with the Quadparser algorithm [14] by searching for sequences complying with the canonical folding rule G≥3N1–7G≥3N1–7G≥3N1–7G≥3 and C≥3N1–7C≥3N1–7C≥3N1–7C≥3. It should be noted that Quadparser outputs only distinctive and non-overlapping sequences, irrespectively of the number of G or C runs present in the motif. Also, if runs of different length coexist in the same motif, more than one topological rearrangement could occur, and in this case, Quadparser will again output it as a single site. Search for triplex DNA and Z-DNA motifs was performed with the non-B DNA motif search tool (nBMST) [15]. For de novo search of over-represented motifs in the vicinity of the breakpoint dataset, we started by extracting all the ±15 bp flanking regions using the window extractor DNA feature of the Sequence Manipulation Suite (http://www.bioinformatics.org/sms2/window_extract_dna.html). Two observations should be made on this: first, the length chosen for the flanking windows results from a compromise between the more common range of motif lengths (6–12 nt) and the need to minimize the chance of getting false positives. Even so, similar results were observed when conducting a motif search considering ±25 bp flanking regions and motif lengths between 6–20 nt (data not shown). Second, data extraction was performed using the non-repeated breakpoint dataset to avoid biasing the results. Motif search was performed with the Multiple Expectation Maximization for Motif Elicitation tool (MEME) [16] and the Gibbs sampling algorithm AlignACE [17]. MEME search was carried out to detect motifs of length 6–20 nt, using both 'zoops' (zero or one occurrence per sequence) and 'anr' (any number of repetitions) options. In AlignACE, the background GC-content parameter ('gcback') was set to 0.471, which corresponds to the fractional GC-content of the breakpoint regions. Moreover, the number of columns to align ('numcols' parameter) was set to 6–20 nt and the expected number of motifs to find ('expect' parameter) was arbitrarily set to 3. From the output file, only motifs with maximum a priori log likelihood (MAP) scores higher than 200 were accepted, since this value is above the typical ranges considered as biologically significant (usually above 10). Each motif set obtained was then visually compared, and only those consistently and simultaneously predicted by both algorithms were considered as strong. Closely related motif groups identified by the same program were discarded. Motifs consisting of single nucleotide repeats of the type Pn were manually parsed out, irrespectively of their positions or number of occurrences. For consensus analysis, the position-specific probability matrix (PSPM)-derived motifs were plotted with Weblogo [18]. To confirm that the similar motif sets were properly grouped, we used the web-tool STAMP [19], which allowed motif edge trimming whenever the information content was below 0.4. To statistically validate the significance of the motifs found, we have calculated its background occurrence by randomly shuffling the mitochondrial genome 130 times preserving k-tuples of length 1 and 3, respectively using the shuffleseq tool from the EMBOSS suite [20] and the gshuf program (kindly provided by Eduardo Rocha from Institut Pasteur). Moreover, the physical distances between each breakpoint and the closest motif were computed in both the mitochondrial genome and shuffled data sets. Statistical significance (p-values) was calculated from corresponding z-scores.
Results
Deletion Breakpoints are Non-randomly Distributed in the Human Mitochondrial Genome
The set of 754 deletions gathered in this work, has only minor differences to that recently published in [7] (see Table S1). Among the 1,508 deletion breakpoints (twice the number of deletions), 1,115 were found to be different. There is a clear preference for 5′ breakpoints to map in the vicinity of position 7.7 kb and 3′ breakpoints to map in the vicinity of positions 14.5 and 16.1 kb (Fig. 1A). The latter positions fall within or around the CO2 gene (5′ breakpoints) and ND6 and CYTB genes (3′ breakpoints). Well-known examples of deletion hot spots located in the close vicinity of these positions are the “common deletion” (nucleotide positions 5′ 8,470–8,482; 3′ 13,447–13,459) or the displacement loop (D-loop) 16,070 regions. The large majority of human mtDNA deletions (86%) affect solely the major arc (nucleotide positions 5,799–16,569 and 1–109), 2% affect the minor arc (nucleotide positions 442–5,720) and 12% affect the origins of replication (nucleotide positions 110–441 and 5,721–5,798 respectively for OH and OL) (Fig. 1B). The average global density of breakpoints (per 0.1 kb) is 9.1, whereas partial densities in the minor arc and major arc are respectively 1.8 and 12.7. Minor arc deletions are typically smaller, harder to detect and not as widely associated with disease phenotypes as those found in the major arc. This fact may contribute to the disparity of available data between arcs. Moreover, there is a general consensus that the strand-asynchronous asymmetric replication mode of the mtDNA favors the occurrence of aberrations within the major arc. As pointed out before [7], this non-stochastic distribution of deletion breakpoints departs significantly from that obtained in a non-restricted simulated random model, which reinforces the idea that certain drivers of instability might be over-represented in the abovementioned regions.
Figure 1. Human mitochondrial deletion spectra.

(A) Distribution of the 5′ and 3′ positions corresponding to 1,508 breakpoints, as well as corresponding histograms and positions along the mitochondrial genome. CR-control region; RNR-Ribosomal RNA; ND-NADH dehydrogenase; CO-cytochrome oxidase; CYTB-Cytochrome B. Black arrows correspond to the 22 tRNA genes. (B) Pie chart indicating the proportion of deletions occurring exclusively in the major arc (nucleotide positions 1–109 and 5,799–16,569), minor arc (nucleotide positions 442–5,720) or involving the origins of replication OH (nucleotide positions 110–441 bp) and OL (nucleotide positions 5,721–5,798).
Breakpoints are Preferentially Clustered in the Close Vicinity of Intrinsically Curved Regions
It was recently shown that intra-strand DNA hairpins and cloverleaf-like elements are enriched in common breakpoint sites of the human and mouse mitochondrial genomes [7]. These observations prompted us to investigate if breakpoints were preferentially located within or in the close vicinity of other classes of non-B DNA elements. The intrinsic flexibility of a DNA molecule (bendability) and its tendency to form a bent structure in the absence of external forces (curvature propensity) are parameters commonly used to describe secondary structure. A highly bendable molecule is less rigid, and does not necessarily retain intrinsic curvature as it allows a mixture of many different conformational states [21]. Thus, regions having high curvature/bendability ratios are more prone to adopt curved and rigid conformations with elevated topological stress. In this sense, we decided to evaluate if human mitochondrial deletion breakpoints were preferentially clustered in regions under high torsional stress, and if known hotspots such as the “common deletion” are located in regions with particularly high ratios. While analyzing the mtDNA curvature/bendability profile (Figure 2A), we observed that the locations of the highest peaks (nucleotide positions 7,444; 8,510; 14,512; 15,951) fall within regions of high breakpoint density (compare with Fig. 1A). In particular, the 8,510 peak closely matches the 5′ breakpoint of the “common deletion”, and the 15,951 peak locates in the vicinity of the 16,070 hotspot. Given the fact that the curvature/bendability ratio can change abruptly in just a few base pairs, we hypothesized that a considerable number of breakpoint positions may have a low ratio but still locate in the vicinity (±50 bp) of a curvature maximum. To evaluate the possibility for such distribution bias, we considered all breakpoints mapping in 0.1 kb bins centered in each local maximum of the mitochondrial genome and computed their corresponding densities (Fig. 2B). The highest breakpoint densities were found in those regions with the highest curvature/bendability ratios, and departed significantly from density values estimated to occur randomly (Fig. 2B). These high breakpoint density values were found within the CO1, tRNA Ser, ATPase8 and ND6 genes. Despite the generalized decrease in breakpoint density observed at ratios below 3, 90.5% of all breakpoints locate in the close vicinity of regions with ratios above the average value for the human mitochondrial genome (0.85). The increased topological stress of regions harboring high breakpoint density becomes more obvious after inspection of their three-dimensional structure. Fig. 1C depicts the three regions with the highest ratios in the genome, whereas the remaining regions are shown as supplementary material (Fig. S1). The adoption of S- or elbow-like structures with low flexibility might contribute to an increased frequency of genetic instability events, likely due to replication fork stalling or increased susceptibility to reactive oxygen species.
Figure 2. Impact of intrinsically bent DNA in the distribution of deletion breakpoints.

(A) Curvature/bendability profile of the entire mtDNA genome as computed by the bend.it algorithm (see Methods section). The exact positions corresponding to the highest curvature/bendability ratios are indicated above the corresponding peaks. (B) Density of deletion breakpoints (∑ number of breakpoints/∑ fragment sizes) computed in 0.1 kb bins flanking each curvature maximum (black bars). Also shown are the density values computed after randomization of breakpoint positions (shown in blue) or after partially randomization of breakpoint positions (shown in green) (see also Methods section). (C) Three-dimensional representations of three 0.2 kb regions harboring highly curved sequences. The exact position corresponding to each curvature maximum is highlighted in red. Additional three-dimensional representations of the remaining peaks highlighted in (A) can be found as supplementary material (Fig. S1). Error bars represent standard deviations. * p<0.05; *** p<0.001.
Large G-quadruplexes are Enriched in Deletion Breakpoints
Our previous observations prompted us to pinpoint additional non-B structures within the human mtDNA, particularly sequences capable of forming G-quadruplexes, triplex DNA and Z-DNA, and evaluate their enrichment in deletion breakpoints. A burgeoning body of evidence supports the involvement of such structures in genomic instability events [22]–[24], but their role in the human mitochondrial genome has not been thoroughly explored. Both the Quadparser and nBMST tools were used to search for sequences that can potentially fold into such structures (see Methods for more details). We have found five G-tetrads, three sequences prone to generate triplex DNA and one sequence prone to generate Z-DNA (Fig. 3A). The local average density of breakpoints found in these nine sequences was 14.0 per 0.1 kb, which corresponds to a fold increase of 1.5 when compared to the average for the mitochondrial genome. When we compared the real breakpoint densities within these structures with those predicted from the random and partially random models, we verified that only the G2 and G5 elements were significantly enriched in breakpoints (Fig. 3B). The average breakpoint density found for these two elements was 41.7 per 0.1 kb, which corresponds to a fold increase of 4.6 and 3.3 when respectively compared to the genome average and major arc densities. Interestingly, G2 and G5 show the largest sizes among their class (Fig. 3A). In view of these observations, it is plausible to speculate that the presence of G-quadruplexes above a certain threshold size may generate more stable and bulkier structures capable of causing replication fork stalling. Apart from carrying a local over-representation of deletion breakpoints, G2 and G5 are also located in the close vicinity of regions displaying the highest frequency of instability events (compare Fig. 3A and Fig. 1A). G2 maps within the CO2 and tRNA Lys genes whereas G5 maps within the CYTB gene. Concerning triplex DNA, it does not seem to play an influential role in promoting mitochondrial instability, since the number of breakpoints found was generally under-represented when compared to the random and partially-random models, even in large elements such as T1 (Fig. 3B). No deletion breakpoints were found within the Z-DNA element (Fig. 3B), which was expected since mutations occurring in the D-loop region tend to be strongly selected against due to their potential effect on replication and copy number.
Figure 3. Impact of the presence of non-canonical (non-B) DNA structures on the distribution of deletion breakpoints.

(A) Sequences and locations of the DNA strings predicted to fold into G-quadruplexes, triplexes and Z-DNA. (B) Density of deletion breakpoints per 0.1 kb bins of each non-B element (black bars). Also shown are the density values computed after randomization of breakpoint positions (shown in blue) or after partially randomization of breakpoint positions (shown in green). Error bars represent standard deviations. * p<0.05; ** p<0.01; *** p<0.001.
Deletion Breakpoints are Over-represented in GC-skewed Regions and in the Close Vicinity of a Degenerate Sequence Motif
During our analysis, we frequently observed a GC- and, to a lesser extent, an AT-rich DNA context next to deletion breakpoints, a fact that is consistent with the formation of several non-B structures (reviewed in [23]). The particularly high density of breakpoints observed in such a small and compositionally biased portion of the genome (G-quadruplexes) prompted us to further investigate if mtDNA instability events could be concurrent to regions showing variations in GC-skew or GC-content. Both GC-content and GC-skew were measured in non-overlapping 20 bp sliding windows along the human mtDNA. The average % GC-content was found to be 44.4% while the average GC-skew was −0.41, due to the predominance of cytosine (and adenine) residues in the light strand [25]. In line with our finding that breakpoints were over-represented in G2 and G5, we also found a significant percentage of breakpoints (64%) mapping in regions in which GC-skew is below the genome average (Fig. 4A). In particular, regions having a GC-skew below −0.6, show the highest density of deletion breakpoints (above 12 per 0.1 kb), significantly departing from the values expected to occur by chance (Fig. 4A). On the other hand, roughly 60% of all breakpoints are located in regions with a % GC-content above the genome average (Fig. 4B). Concomitantly, breakpoints located in regions having a % GC-content between [55–75[ were found to be over-represented when comparing to the random and partially random models (Fig. 4B).
Figure 4. Impact of GC-skew and % GC-content in the distribution of deletion breakpoints.

Variation in the density of deletion breakpoints per 0.1 kb with GC-skew (A) and % GC-content (B). Black bars represent the density values obtained for the human mtDNA, whereas blue and green bars respectively represent the values computed after randomization and partial randomization of breakpoint positions. Error bars represent standard deviations. * p<0.05; ** p<0.01; *** p<0.001.
These observations on the presence of compositional asymmetries near unstable regions are not only in line with our previous findings of non-B elements, but together with literature evidence (see Discussion section below), raise the possibility for the presence of other over-represented motifs. To evaluate this scenario, we carried out a search for conserved motifs in the close vicinity (±15 bp) of our non-repeated breakpoint dataset (n = 1,115). For this purpose, as well as to attain more reliable conclusions on over-represented motifs, we have used two different motif discovery tools, MEME and AlignAce, followed by motif edge trimming using STAMP (see Methods section for further details). An 11–mer degenerate consensus [C/T][C/A][C/A][C/T][C/A]NN[C/A][C/A][C/A/T][C/A] (or alternatively YMMYMNNMMHM) was found to be over-represented in our dataset (Fig. 5A and Fig. S2). This motif occurs 469 times in the human mtDNA, and was found to be over-represented when compared to shuffled mitochondrial genomes (Fig. 5B). 50.3% of all mtDNA breakpoints were observed at a distance of less than 5 bp from one of such motifs (Fig. 5C). This percentage increased to 73.7% when we considered a maximum breakpoint-motif distance of 20 bp (Fig. 5C). Also, the distribution of this motif was globally well correlated with the distribution of deletion breakpoints in both arcs (Spearman ρ = 0.38; p<0.001) (Fig. 5D). Still, some regions depart from this tendency and show extremely high counts of breakpoints, despite a weak increase in the number of YMMYMNNMMHM motifs (e.g. nucleotide positions position 5.5 in the minor arc and 7.5 and 16 kb in the major arc) (Fig. 5D). Bearing in mind our findings on intrinsic curvature, these local discrepancies correlate with the nearby presence of highly bent regions at positions 5,517, 7,444 and 15,951, which as we mentioned previously, likely play a destabilizing role in these regions.
Figure 5. Search for over-represented motifs in the close vicinity of deletion breakpoints.

(A) Sequence logo of the degenerate 11-mer motif over-represented in the close vicinity (±15 bp) of the non-repeated breakpoint dataset. Representative logos were obtained from MEME and AlignACE (see Fig. S2), and compared both manually and using the STAMP tool. Degenerate nucleotides are as follows: Y = (C or T); M = (A or C); H = (A or T or C); N = (A or T or G or C). (B) The number of occurrences of the YMMYMNNMMHM motif in the human mtDNA (black bar) is compared with those obtained from randomly shuffled genomes preserving k-tuples of 1 and 3 (respectively blue and green bars). (C) Percentage of breakpoints in terms of distance (bp) to the nearest YMMYMNNMMHM motif. Black bars represent the percentage values obtained for the human mtDNA, whereas blue and green bars respectively represent the values computed after randomization and partial randomization of deletion breakpoints. Error bars represent standard deviations. * p<0.05; ** p<0.01; *** p<0.001. (D) Distribution profiles of breakpoints (red) and YMMYMNNMMHM motif (blue) along the minor arc (left) and major arc (right). Stippled lines indicate the positions of the previously identified highly bent regions as well as of the G2 and G5 motifs.
Discussion
It is now commonly accepted that the generation of large-scale mtDNA deletions can be attributed either to the formation of slipped structures during DNA replication, or alternatively, to the repair of strand breaks originated by fork stalling or ionizing radiation (reviewed in [26]). Although the causes behind fork arrest in the mitochondrial genome can be numerous, those that can be attributed to the formation of higher-order DNA structures, have only recently been given a predominant role in mitochondrial deletion formation [7]. The authors of this study point out the importance of non-B elements such as hairpins, cruciforms and cloverleaf-like elements in eliciting human mitochondrial DNA rearrangements either by facilitating fork arrest or nucleolytic attack. Building on this information, we examined whether additional DNA architectures could similarly impact the stability of the human mitochondrial genome. Our analysis revealed that intrinsically curved regions as well as large G-quadruplexes are enriched in deletion breakpoints. Bent DNA typically arises from the presence of short runs of regularly phased adenine:thymine tracts (helical periodicity of 10–11 bp), and its presence has been implicated in functionally relevant cellular processes including transcription [27], replication [28], and recombination [29], [30]. It has also been suggested that bent DNA might serve as recognition motif to the binding of topoisomerases and nucleases, thus facilitating breakage and subsequent illegitimate recombination or attack by reactive oxygen species [8], [9]. In a former study, the mobility of PCR-amplified and digested fragments of human mtDNA was evaluated by two-dimensional gel electrophoresis [9]. The authors found evidence for the presence of bent-like DNA, locating near or within deletion-prone regions (nucleotide positions 5,221–5,988, 6,928–7,493, 7,901–8,732 and 15,327–16,228). In our analysis we were able to narrow down these large regions to four peaks, respectively mapping at positions 5,517, 7,444, 8,510 and 15,951 (Fig. 2A). Together with the peak predicted at nucleotide position 14,512, these locations were found to concentrate in their ±50 bp vicinity, some of the highest breakpoint densities seen in the human mitochondrial genome (see Fig. 2A and 2B). We considered that deletion breakpoints might arise in poorly bent regions, but still as a consequence (or under partial influence) of the topological distortion induced by the presence of nearby curvature/bendability maxima. And despite the fact that this “proximity” effect becomes more obvious at highly distorted regions such as those mentioned above, we found that 90.6% of all breakpoints actually locate in the close vicinity of regions with curvature/bendability ratios above the average value found for the human mitochondrial genome (0.85).
In this work we also found an over-representation of deletion breakpoints in two large G-quadruplexes located at nucleotide positions 8,252–8,295 and 15,516–15,545. Such findings are consistent with the fact that quadruplexes are fork pausing sites capable of promoting recombination in vitro [31] and also over-represented in human recombination hotspots [22]. Although the detection of G-quadruplexes was made using the rule commonly associated with its canonical form (see Methods section), we do not discard the possibility that progenitor or degenerate forms eventually present (e.g. having different G runs or loop sizes) can also impact the stability of the human mtDNA.
The observation that mitochondrial deletion breakpoints were over-represented in negatively GC-skewed regions (Fig. 4A), prompted us to investigate the possibility of the presence of nearby hot motifs. This decision was further supported on the basis of three lines of evidence, according to which, distinct compositionally skewed motifs have been implicated in mitochondrial instability. The first line of evidence, refers to an intriguing finding brought to light in a recent study, in which was found that 12 out of the 13 bp direct repeats of the “common deletion” perfectly match a degenerate consensus motif (CCNCCNTNNCCNC) strongly over-represented in human nuclear recombination hot spots [32]. The latter, as well as the 9-mer CCCCACCCC were found to be implicated in allelic crossover activity during meiosis, nonallelic homologous recombination, and instability at hypervariable human minisatellites [32]. By also finding the presence of such motifs associated with the mitochondrial “common deletion”, the authors suggest in the same study, their implication in repeat-associated rearrangements, for example, by stimulating the formation of double-stranded breaks. A second line of evidence comes from a previous study on the mitochondrial transcription terminator factor mTERF, where it was shown that its binding sites (minimal consensus CCN8CC) in the human mitochondrial DNA are also replication pausing sites, which match frequent breakpoints in rearranged mtDNA “sublimons” [33]. The third line of evidence comes from the observation that poly(C) motifs such as CCTC and ACCC found in the D-loop hypervariable segment I and NADH dehydrogenase genes, are associated with a higher rate of point substitutions, small deletions and duplications [34], [35]. Also, local recombination rates were found to be positively correlated with GC-content across several human chromosomes [36], presumably resulting from non-adaptive processes such as GC-biased gene conversion [37]. Our search returned a unique highly degenerate motif, YMMYMNNMMHM, over-represented in the close vicinity (±15 bp) of our breakpoint dataset. Occurrences of this 11-mer motif match the positions of CCNCCNTNNCCNC flanking the common deletion, as well as one out of two occurrences of the CCCCACCCC motif. Its frequency distribution along the human mtDNA also correlates with that of the minimal mTERF binding site (Spearman ρ = 0.58; p<0.001). We therefore anticipate a biological role for the YMMYMNNMMHM motif in eliciting instability events. One hypothesis is that its homopolymeric runs are capable of leading to an intracellular local depletion of a particular nucleotide, ultimately resulting in replication fork stalling followed by double strand break (DSB) formation [38]. In fact, replication stalling at homopolymeric runs has been previously pointed out as primary cause of mtDNA deletion formation in patients with autosomal dominant progressive external opthalmoplegia (adPEO) [38]. Another possibility is that the presence of the YMMYMNNMMHM motif somehow stimulates the formation of DNA•RNA hybrids (R-Loops) during replication, leading to genomic instability. Such structures have been shown to form preferentially at regions populated by short iterated repeats with a high GC-content, and to block the progression of the replication fork (reviewed in [39]). R-loops are known to form during the initiation of mammalian mtDNA replication [40], but in the last few years have been recurrently linked to DNA instability phenomena [41].
In conclusion, this study provides evidence supporting the idea that mtDNA instability arises from the concerted action of a potpourri of mechanisms, likely to provide adverse sequence contexts that favor genetic rearrangements (Fig. 6). Notably, our findings support the idea that local abrupt shifts in DNA composition provided by certain non-B structures or compositionally skewed motifs may represent important markers for genetic instability. Further research, will provide us with additional information on the YMMYMNNMMHM motif, and eventually extend it to a more general family of motifs.
Figure 6. Diagram illustrating the sequence of events (I, II) capable of driving functional mitochondria to shift to a partially functional or non-functional state.

The mutational events (I) may arise as a consequence of unusual DNA conformations, fragile motifs, exogenous factors, among others. These mutations will co-exist with the wild-type mtDNA in a heteroplasmic state, or eventually be selected (II) until a homoplasmic state is reached.
Supporting Information
Three-dimensional reconstruction of the remaining 0.2 kb highly curved sequences highlighted in Fig. 2A . The exact position corresponding to each curvature maximum is highlighted in red.
(TIF)
Sequence logos for the most significant motifs found in regions flanking (±15 bp) deletion breakpoints using MEME and AlignACE. MEME E values correspond to the expected number of motifs with equal or higher likelihood, with same width and number of occurrences in a set of random sequences of similar size and composition than the input sequence. The logos obtained were then trimmed using the STAMP tool, and the result is shown in Fig. 5A.
(TIF)
List of the 5′ and 3′ breakpoints from the 754 deletions analyzed in this study. With the exception of two entries (marked with an asterisk), the list is similar to that recently published in [7]. Breakpoints were obtained from the references listed.
(DOCX)
Acknowledgments
The authors gratefully acknowledge Eduardo P.C. Rocha (Institut Pasteur, Paris) for critical reading and for providing the C program gshuf, which was used to simulate random nucleic acid sequences.
Funding Statement
This work was supported by Fundação para a Ciência e a Tecnologia (FCT) through the MIT-Portugal Program, Bioengineering Focus Area and Project PTDC/EQU-EQU/114231/2009. PHO acknowledges FCT for the Post-Doctoral Grant BPD/64652/2009. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Tuppen HA, Blakely EL, Turnbull DM, Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797: 113–128. [DOI] [PubMed] [Google Scholar]
- 2. Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, et al. (2008) What causes mitochondrial DNA deletions in human cells? Nat Genet 40: 275–279. [DOI] [PubMed] [Google Scholar]
- 3. Samuels DC, Schon EA, Chinnery PF (2004) Two direct repeats cause most human mtDNA deletions. Trends Genet 20: 393–398. [DOI] [PubMed] [Google Scholar]
- 4. Guo X, Popadin KY, Markuzon N, Orlov YL, Kraytsberg Y, et al. (2010) Repeats, longevity and the sources of mtDNA deletions: evidence from 'deletional spectra'. Trends Genet 26: 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lakshmanan LN, Gruber J, Halliwell B, Gunawan R (2012) Role of direct repeat and stem-loop motifs in mtDNA deletions: cause or coincidence? PLoS One 7: e35271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Samuels DC (2004) Mitochondrial DNA repeats constrain the life span of mammals. Trends Genet 20: 226–229. [DOI] [PubMed] [Google Scholar]
- 7. Damas J, Carneiro J, Goncalves J, Stewart JB, Samuels DC, et al. (2012) Mitochondrial DNA deletions are associated with non-B DNA conformations. Nucleic Acids Res 40: 7606–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hou JH, Wei YH (1998) AT-rich sequences flanking the 5'-end breakpoint of the 4977-bp deletion of human mitochondrial DNA are located between two bent-inducing DNA sequences that assume distorted structure in organello. Mutat Res 403: 75–84. [DOI] [PubMed] [Google Scholar]
- 9. Hou JH, Wei YH (1996) The unusual structures of the hot-regions flanking large-scale deletions in human mitochondrial DNA. Biochem J 318 (Pt 3): 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rocher C, Letellier T, Copeland WC, Lestienne P (2002) Base composition at mtDNA boundaries suggests a DNA triple helix model for human mitochondrial DNA large-scale rearrangements. Mol Genet Metab 76: 123–132. [DOI] [PubMed] [Google Scholar]
- 11. Goodsell DS, Dickerson RE (1994) Bending and curvature calculations in B-DNA. Nucleic Acids Res 22: 5497–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vlahovicek K, Kajan L, Pongor S (2003) DNA analysis servers: plot.it, bend.it, model.it and IS. Nucleic Acids Res 31: 3686–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brukner I, Sanchez R, Suck D, Pongor S (1995) Sequence-dependent bending propensity of DNA as revealed by DNase I: parameters for trinucleotides. EMBO J 14: 1812–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huppert JL, Balasubramanian S (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33: 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cer RZ, Bruce KH, Mudunuri US, Yi M, Volfovsky N, et al. (2011) Non-B DB: a database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res 39: D383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- 17. Roth FP, Hughes JD, Estep PW, Church GM (1998) Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol 16: 939–945. [DOI] [PubMed] [Google Scholar]
- 18. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahony S, Benos PV (2007) STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35: W253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- 21. Perez-Martin J, de Lorenzo V (1997) Clues and consequences of DNA bending in transcription. Annu Rev Microbiol 51: 593–628. [DOI] [PubMed] [Google Scholar]
- 22. Mani P, Yadav VK, Das SK, Chowdhury S (2009) Genome-wide analyses of recombination prone regions predict role of DNA structural motif in recombination. PLoS One 4: e4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J, Bacolla A, Wang G, Vasquez KM (2010) Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci 67: 43–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, et al. (2012) Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 8: 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanaka M, Ozawa T (1994) Strand asymmetry in human mitochondrial DNA mutations. Genomics 22: 327–335. [DOI] [PubMed] [Google Scholar]
- 26. Chen T, He J, Huang Y, Zhao W (2011) The generation of mitochondrial DNA large-scale deletions in human cells. J Hum Genet 56: 689–694. [DOI] [PubMed] [Google Scholar]
- 27. Cress WD, Nevins JR (1996) A role for a bent DNA structure in E2F-mediated transcription activation. Mol Cell Biol 16: 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gimenes F, Takeda KI, Fiorini A, Gouveia FS, Fernandez MA (2008) Intrinsically bent DNA in replication origins and gene promoters. Genet Mol Res 7: 549–558. [DOI] [PubMed] [Google Scholar]
- 29. Kusakabe T, Sugimoto Y, Maeda T, Nakajima Y, Miyano M, et al. (2001) Linearization and integration of DNA into cells preferentially occurs at intrinsically curved regions from human LINE-1 repetitive element. Gene 274: 271–281. [DOI] [PubMed] [Google Scholar]
- 30. Milot E, Belmaaza A, Wallenburg JC, Gusew N, Bradley WE, et al. (1992) Chromosomal illegitimate recombination in mammalian cells is associated with intrinsically bent DNA elements. EMBO J 11: 5063–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boan F, Gomez-Marquez J (2010) In vitro recombination mediated by G-quadruplexes. Chembiochem 11: 331–334. [DOI] [PubMed] [Google Scholar]
- 32. Myers S, Freeman C, Auton A, Donnelly P, McVean G (2008) A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129. [DOI] [PubMed] [Google Scholar]
- 33. Hyvarinen AK, Pohjoismaki JL, Reyes A, Wanrooij S, Yasukawa T, et al. (2007) The mitochondrial transcription termination factor mTERF modulates replication pausing in human mitochondrial DNA. Nucleic Acids Res 35: 6458–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maliarchuk BA (2002) [The effect of the nucleotide context on induction of mutations in hypervariable segment 1 of the human mitochondrial DNA]. Mol Biol (Mosk) 36: 418–423. [PubMed] [Google Scholar]
- 35. Bianchi NO, Bianchi MS, Richard SM (2001) Mitochondrial genome instability in human cancers. Mutat Res 488: 9–23. [DOI] [PubMed] [Google Scholar]
- 36. Fullerton SM, Bernardo Carvalho A, Clark AG (2001) Local rates of recombination are positively correlated with GC content in the human genome. Mol Biol Evol 18: 1139–1142. [DOI] [PubMed] [Google Scholar]
- 37. Katzman S, Capra JA, Haussler D, Pollard KS (2011) Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol Evol 3: 614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wanrooij S, Luoma P, van Goethem G, van Broeckhoven C, Suomalainen A, et al. (2004) Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res 32: 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McIvor EI, Polak U, Napierala M (2010) New insights into repeat instability: role of RNA*DNA hybrids. RNA Biol 7: 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee DY, Clayton DA (1998) Initiation of mitochondrial DNA replication by transcription and R-loop processing. J Biol Chem 273: 30614–30621. [DOI] [PubMed] [Google Scholar]
- 41. Aguilera A, Garcia-Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Three-dimensional reconstruction of the remaining 0.2 kb highly curved sequences highlighted in Fig. 2A . The exact position corresponding to each curvature maximum is highlighted in red.
(TIF)
Sequence logos for the most significant motifs found in regions flanking (±15 bp) deletion breakpoints using MEME and AlignACE. MEME E values correspond to the expected number of motifs with equal or higher likelihood, with same width and number of occurrences in a set of random sequences of similar size and composition than the input sequence. The logos obtained were then trimmed using the STAMP tool, and the result is shown in Fig. 5A.
(TIF)
List of the 5′ and 3′ breakpoints from the 754 deletions analyzed in this study. With the exception of two entries (marked with an asterisk), the list is similar to that recently published in [7]. Breakpoints were obtained from the references listed.
(DOCX)