

Abstract
We sought to create a population pharmacokinetic model for total mycophenolic acid (MPA), to study the effects of different covariates on MPA pharmacokinetics, to create a limited sampling schedule (LSS) to characterize MPA exposure (i.e., area under the curve or AUC) with maximum a posteriori Bayesian estimation, and to simulate an optimized dosing scheme for allogeneic hematopoietic cell transplantation (HCT) recipients. 4,496 MPA concentration-time points from 408 HCT recipients were analyzed retrospectively using a nonlinear mixed effects modeling approach. MPA pharmacokinetics was characterized with a two-compartment model with first-order elimination and a time-lagged first-order absorption process. Concomitant cyclosporine and serum albumin were significant covariates. The median MPA clearance and volume of the central compartment were 24.2 L/hr and 36.4 L, respectively, for a 70 kg patient receiving tacrolimus with a serum albumin of 3.4 g/dL. Dosing simulations indicated that higher oral MMF doses are needed with concomitant cyclosporine, which increases MPA clearance by 33.8%. The optimal LSS was immediately before and at 0.25, 1.25, 2, and 4hr after oral MMF administration. MPA AUC in an individual HCT recipient can be accurately estimated using a five-sample LSS and maximum a posteriori Bayesian estimation.
Keywords: Mycophenolic acid, population pharmacokinetics, limited sampling schedule, hematopoietic cell transplantation, dosing simulation
INTRODUCTION
The therapeutic potential and availability of allogeneic hematopoietic cell transplantation (HCT) have expanded with the development of less toxic nonmyeloablative conditioning regimens.1 In these regimens, postgrafting immunosuppression is often achieved with a calcineurin inhibitor (CNI) and mycophenolate mofetil (MMF). MMF is a prodrug hydrolyzed by esterases to form its primary metabolite, mycophenolic acid (MPA).2 MPA is subsequently metabolized to an inactive hydroxyl-β-glucuronide (MPAG), which can undergo enterohepatic recirculation and produce a secondary peak in plasma MPA concentration-time profiles. MPA selectively and reversibly blocks the activity of inosine monophosphate dehydrogenase (IMPDH), the inhibition of which blocks the de novo pathway of purine synthesis in T and B lymphocytes.2
Nonmyeloablative HCT relies on a delicate balance between recipient and donor cells, ideally ensuring adequate immunosuppression of the recipient, optimal anti-tumor effect, and minimal toxicity. In nonmyeloablative HCT recipients of an HLA–matched related donor graft, oral MMF is administered Q12hr, similar to treatment for solid organ transplant patients. While these patients rarely experience graft rejection, graft versus host disease (GVHD) continues to be a therapeutic challenge.3–4 Pharmacokinetic studies5–7 suggested that HCT recipients exhibit a shorter MPA half-life than solid organ transplant recipients.2 In patients undergoing nonmyeloablative conditioning with an HLA-matched unrelated donor graft, Q12hr MMF administration and low total MPA area under the plasma concentration-time curve (AUC) are related to a higher risk of graft rejection.3, 8 Increasing the frequency of MMF administration from Q12hr to Q8hr reduced graft rejection from 15% to 5%; acute GVHD, non-relapse mortality, and progression free and overall survival were similar between Q8hr and Q12hr administration.8 In HCT patients, low total MPA AUC is also related to low (<50%) T-cell donor chimerism, making it the only modifiable risk factor for donor chimerism.3 Optimal donor chimerism is 50–90% in nonmyeloablative HCT recipients. Donor chimerism greater than 40% is associated with lower rejection risk,9 while donor chimerism greater than 50% is associated with higher complete remission rates through the graft-versus-tumor (GVT) effect, which involves the immunoreactivity of donor cells against recipient cells. Donor chimerism <90% is associated with lower rates of grade 2–4 acute GVHD.9–10 Thus, optimization of donor T-cell chimerism could lower rates of graft rejection and GVHD while maximizing the GVT effect.1
Pharmacodynamic studies are needed to improve our understanding of the relationship between MPA exposure and clinical outcomes. As nonmyeloablative HCT recipients are treated in an ambulatory setting, we sought to maximize compliance with pharmacokinetic sampling by developing a population pharmacokinetic-based minimally intrusive limited sampling schedule (LSS). We also sought to identify alternative starting doses of oral MMF to maximize the proportion of patients reaching a target exposure based on a previous pharmacodynamic study.3
METHODS
Study and treatment plan
Between March 1998 to July 2006, 408 HCT recipients underwent blood sampling to determine MPA pharmacokinetics (371 after oral MMF only, 37 after IV MMF only, and 40 after IV and then oral MMF). Postgrafting immunosuppression for these patients also included a CNI. According to HCT treatment protocols, MMF doses were based on body weight (BW) and rounded to the nearest 250 mg dose. MMF doses were calculated using actual body weight, if less than ideal body weight, or adjusted ideal body weight: AIBW = 0.25 × (actual weight − ideal weight) + ideal weight. The ideal body weight was calculated as follows: for males = 50 kg + (2.3 kg/inch over 5 feet); for females = 45.5 kg + (2.3 kg/inch over 5 feet). There was no adjustment of MMF dose based on MPA plasma concentrations. MMF administration frequency was specified by the HCT treatment protocol. Written informed consent was obtained from all patients, and all study procedures were approved by the Institutional Review Board at the Fred Hutchinson Cancer Research Center and monitored by independent Data Safety Monitoring Boards. Approval was obtained for this retrospective analysis.
Pharmacokinetic sample collection and analytical assay
Of the 4,496 concentration-time points, 3,620 samples were obtained after oral MMF administration and 876 samples after IV MMF administration. Steady-state blood samples were collected on various days after allogeneic graft infusion based on treatment protocols. The blood sampling scheme for patients receiving IV MMF has been previously described.6, 11 For the oral MMF regimen, samples were scheduled to be drawn before and at 1, 2, 4, 6, 8, and 10 hr after the dose; the 10 hr sample was not collected in patients receiving Q8hr MMF. The median number of AUCs determined per patient was 2 (range of 1 to 4; 61% of patients with two AUCs). Blood samples were obtained at the following times post-transplant: one AUC on day 2, 260 AUCs on day 7 ± 2, 34 AUCs on days 10 to 17, 288 AUCs on day 21 ± 2, 30 AUCs on days 24 through 31, 15 AUCs one to three months post-transplant, and one AUC nine months post-transplant. Total MPA plasma concentrations were quantified as previously described.3 The dynamic range was 0.2 to 30 μg/mL and the inter-day coefficient of variation was less than 10%.
Population pharmacokinetic analysis
Total MPA plasma concentration-time data were analyzed using nonlinear mixed effects modeling (NONMEM® VI 1.2; Icon Development Solutions, LLC, Ellicott City, MD, USA). The first-order (FO) method was used during model exploration, and the first-order conditional estimation (FOCE) method was used to determine the final model and for validation. The hybrid method was applied when certain parameters (e.g., lag time) were difficult to estimate by FOCE. MMF doses were converted to the equivalent MPA content by multiplying values by 0.739.2, 11 Clearances and volumes of distribution were allometrically scaled to body weight, which was normalized to 70 kg and linearly incorporated into clearance (CL), volume of distribution of the central compartment (Vc), volume of distribution of the peripheral compartment (Vp), and the intercompartmental clearance (Q). Allometric exponents were fixed to 0.75 for CL and Q and to 1 for Vc and Vp.12 To deconvolve the absorption and disposition components, data after IV and oral MMF administration were fit simultaneously.
A two-compartment model with first-order elimination was applied to characterize MPA pharmacokinetics. Various models were tested to describe the formation and absorption processes for MPA following oral MMF administration, including first-order absorption with or without a lag time, a transit compartment model, and the Weibull function. A one-compartment model with first-order elimination was used to describe MMF pharmacokinetics following the 2 hr IV infusion of MMF. Inter-individual and inter-occasion variability (IIV and IOV) in pharmacokinetic parameters were modeled using an exponential error model based on the assumption of log-normal parameter distribution. The estimation of the IOV occurred over three occasions: occasion 1 was 7 ± 3 days after HCT, occasion 2 was days 11 to 24 after HCT, and occasion 3 was 25 days or later after HCT. Residual unknown variability was estimated using an additive error model as data were natural log-transformed for model fitting. A partial variance-covariance matrix was used where correlation between CL and Vc was estimated. Model selection criteria included the objective function value (OFV), precision of parameter estimates, goodness-of-fit (GOF) plots, and visual predictive check (VPC) plots. GOF plots included observed versus predicted concentrations as well as weighted residuals (WRES) versus model predicted values and time post dose. VPC plots were used to internally evaluate the predictive performance of the final model. One hundred data sets were simulated from parameter estimates of the final model, and 5th, 50th, and 95th percentiles of simulated data were compared with observed data. Data were binned according to nominal sampling times: predose and 1, 2, 4, 6, 8, and 10 hr post dose. Diagnostic graphics were generated in S-PLUS 8.0 (Insightful Corp., Seattle, WA, USA).
Relationships between patient characteristics or clinical laboratory tests (Table 1) and the IIV and IOV in pharmacokinetic parameters were investigated. For continuous covariates, values were centered to their median and modeled on pharmacokinetic parameters in a linear or nonlinear manner. For categorical covariates, the difference in pharmacokinetic parameters between the categories was estimated. A two-step approach of stepwise forward selection and backward elimination was applied for covariate model building. For forward selection, all covariates were introduced into the structural model individually. Inclusion of a covariate was determined by the likelihood ratio test and reduction in unexplained IIV or IOV. The difference in OFV between a model with and without a covariate is approximately χ2 distributed with degrees of freedom equal to 1. A decrease of at least 6.6 units in the OFV (p<0.01) was considered to be a statistically significant improvement over the base structural model. Such a covariate was considered significant and included in an intermediate model. For backward elimination, all covariates selected in the first step were excluded one by one from the intermediate model. A covariate was retained in the final model if an increase of at least 10.8 units in the OFV (p<0.001) occurred after excluding the covariate.
Table 1.
Patient characteristicsa
| Number of participants | 408 |
|---|---|
| Demographic characteristics | |
| Gender (male/female) | 257/151 |
| Age (year) | 53 [1–74] |
| N ≤ 18 years | 9 (2.2%) |
| Adjusted ideal body weight (kg) | 72 [11–99] |
| N BMI > 30 kg/m2 | 106 (25%) |
| Height (cm) | 172 [79–199] |
| Body surface area (m2) | 1.98 [0.47–2.67] |
| HCT characteristics | |
| Type of conditioning | |
| Nonmyeloablativeb | 347 (85%) |
| Myeloablativec | 61 (15%) |
| Received fludarabine administration (yes/no) | 294/114 |
| Donor type (HLA-matched related/unrelated) | 202/206 |
| Concomitant calcineurin inhibitor | |
| Cyclosporine | 327 (80%) |
| Tacrolimus | 81 (20%) |
| Biochemistryd | |
| Serum creatinine (mg/dL) | 1.1 [0.3–10.4] |
| Creatinine clearance (mL/min) | 86 [22–159] |
| Serum alanine aminotransferase (U/L) | 17 [4–856] |
| Serum aspartate aminotransferase (U/L) | 21 [7–466] |
| Serum alkaline phosphatase (U/L) | 68 [18–637] |
| Serum lactate dehydrogenase (U/L) | 213 [55–1708] |
| Serum total bilirubin (μmol/L) | 1.0 [0.2–37.5] |
| Serum direct bilirubin (μmol/L) | 0.3 [0.1–23.7] |
| Serum albumin (g/dL) | 3.4 [1.6–4.8] |
Categorical data presented as number of participants meeting stated criteria; continuous data presented as median [min-max];
fludarabine with low-dose total body irradiation, with or without alemtuzumab;
cyclophosphamide with either busulfan or total body irradiation;
biochemistry values obtained within 7 days before MPA AUC and thus, those patients with multiple AUCs can have multiple biochemistry values
LSS for AUC Estimation
Two optimal sampling strategies were determined for prediction of AUC0–8hr or AUC0–12hr after oral MMF administration. Optimal sampling times were selected from a proposed set of time points: 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, and 6 hr post dose, as well as one pre-dose time point (time = −0.1 hr). The optimal LSS was determined by a simulation approach that was described previously.13 Briefly, two datasets were simulated for 1000 subjects whose pharmacokinetic parameters were based on the final model and estimated population mean parameters and IIV. In the first dataset, serial MPA concentrations were simulated without residual error for calculating the true AUC for each individual. In the second dataset, each subject was replicated ten times using different simulated residual errors to account for unknown residual variability (i.e., assay error or sampling error). The MPA concentration-time profile was then simulated for each residual error within each subject. Five samples were randomly selected from the simulated MPA profile and used to obtain maximum a posteriori (MAP) Bayesian estimates of the pharmacokinetic parameters for each individual at each replicate. Bayesian analysis was performed in NONMEM using options of POSTHOC = 1 and MAXEVAL = 0. Predicted AUCs were calculated based on MAP Bayesian estimates (integration approach in NONMEM) and were compared with true AUC values.
The agreement between predicted AUC values calculated from the LSS and the true AUC values was assessed by scaled mean square error (sMSE):
| (Eq.1) |
| (Eq.2) |
| (Eq.3) |
with MSEj and sMSEj calculated for the jth simulated subject; N is the ten error replicates for the jth subject, is the predicted AUC from the LSS for each error replicate i of jth subject, and is the true AUC of the jth subject. The LSS with the minimum root mean sMSE (across 1000 subjects) was considered optimal. Root mean sMSE was calculated across 1000 simulated subjects (M=1000). In addition, the Pearson correlation coefficient (r) and mean relative bias, one component of MSE, were also computed as an assessment of the performance of the LSS:
| (Eq.4) |
The comparison between the optimal LSS and the standard sampling protocol was conducted for 1000 simulated subjects with ten residual errors. All simulations and statistical analyses were performed in NONMEM VI and the open-source statistical software R (v.2.10.0).
MMF Dosing Regimen Simulations
Using the final population PK model for oral MMF, different dosing regimens were evaluated to find a regimen that maximized the proportion of patients who achieved the target total MPA steady-state plasma concentration (Css) of 3 mg/L with Q8hr MMF oral administration. The Css was calculated as AUC0-τ divided by τ (with τ as the dosing interval).3 The Css term was used because of the varied administration schedules for oral MMF, with Q8hr and Q12hr being the most common. The target MPA Css was chosen because nonmyeloablative HCT recipients of an HLA-matched unrelated graft are at increased risk of graft rejection or low T-cell chimerism with total MPA Css < 2.5 and 3 mg/L, respectively.3 Simulations with more frequent administration schedules were not considered, as a phase I study in HCT recipients suggested that MMF administration at Q6hr increased toxicity without improved efficacy.6 Fixed-dosing regimens of 1250 and 1750 mg oral MMF Q8hr for seven days were simulated and compared with body weight based regimens of 15 and 20 mg/kg oral MMF Q8hr for seven days. For each dosing scenario, 1000 hypothetical subjects were simulated from the final population model with mean and intersubject variability parameters. Body weight was normally distributed with mean of 81.7 kg and standard deviation of 20.2 kg. Co-medication regimens with cyclosporine or tacrolimus were also compared. Steady-state AUC0-τ was determined and Css was calculated as AUC0-τ/τ (with τ as the dosing interval).3 All simulations were performed in NONMEM VI and R (v.2.10.0).
RESULTS
In this retrospective analysis, 4,496 total MPA plasma concentration time points were obtained from 408 HCT recipients who participated in various clinical transplant protocols at the Fred Hutchinson Cancer Research Center (FHCRC). Patient characteristics are summarized in Table 1. The dose, administration route, and administration frequency of MMF, along with the timing of MPA pharmacokinetic sampling, were determined by the clinical transplant protocols.
Population Pharmacokinetic Model
Preliminary graphical analysis revealed large inter- and intra-individual pharmacokinetic variability and complex MPA profiles, with some showing delayed absorption and enterohepatic recirculation (Supplemental Figure 1). All pharmacokinetic data were fit simultaneously to evaluate absorption and disposition processes controlling MPA pharmacokinetics. The final model consists of a single compartment for MMF pharmacokinetics featuring first-order rate constants for oral absorption and metabolism, leading to a linear two-compartment model with first-order elimination for the disposition of MPA (Figure 1). The first-order absorption rate constant (Ka) is a hybrid parameter encompassing the transformation of MMF to MPA, as well as pre-formed MPA absorption. An estimated time lag was required to account for the delayed increase in MPA plasma concentrations following oral MMF administration. Alternative absorption models, such as transit compartments14 or the Weibull function,15 failed to improve model fitting criteria (see Methods). In addition, a model incorporating enterohepatic cycling was not supported; fewer than 4% of MPA AUC profiles exhibited a secondary peak. Bioavailability (F) was initially estimated close to 1 and was fixed to 1 in the final parameter estimation to ensure computational stability. Population parameter estimates from this base model are summarized in Table 2; both IIV and IOV of model parameters were estimated. IOV was included to account for the potential influence of various factors on MPA pharmacokinetics, including conditioning regimen, presence of acute or chronic GVHD, concomitant antibiotics, co-administration of corticosteroids to treat GVHD and/or time after graft infusion. The IIV was relatively large for the volumes of distribution (> 70%) and the IOV for Ka was about 50% (Table 2).
Figure 1.
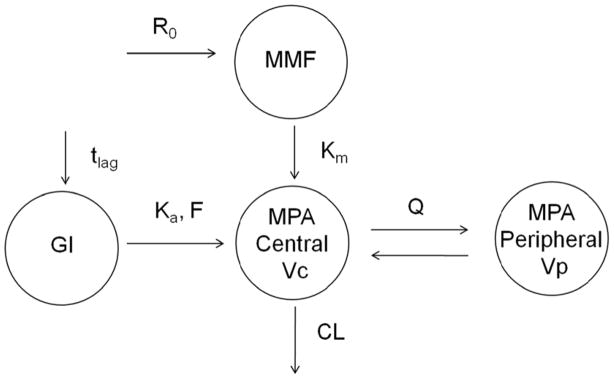
Final MPA pharmacokinetic model following oral and 2 hr intravenous infusion of MMF. R0 = zero-order IV infusion rate constant; Km = first-order metabolizing rate constant; Ka = first-order rate constant representing both formation and absorption process; tlag = lag time; F=bioavailability; CL = clearance; Vc = volume of central compartment of MPA; Q = inter-compartment clearance; Vp = volume of peripheral compartment of MPA.
Table 2.
Population pharmacokinetic parameters for MPA after oral MMF administration in HCT recipients
| Parameter | Explanation | Estimate [RSE%a] from
|
|
|---|---|---|---|
| base structural model | final covariate model | ||
| Ka (1/hr) | First-order rate constant representing both formation and absorption process | 0.582 [6.3] | 0.602 [6.7] |
| Tlag (hr) | Lag time of oral absorption | 0.228 [3.8] | 0.228 [3.7] |
| Km (1/hr) | First-order metabolizing rate constant | 6.24 [35.4] | 5.99 [35.4] |
| CL (L/hr/70kg) | Clearance | 31.4 [2.3] | 24.2 [3.2] |
| Vc (L/70kg) | Volume of central compartment | 34.7 [10.4] | 36.4 [10.8] |
| Q (L/hr/70kg) | Inter-compartment clearance | 18.5 [5.0] | 19.0 [4.8] |
| Vp (L/70kg) | Volume of peripheral compartment | 243 [14.7] | 247 [14.0] |
|
| |||
| Inter-individual variability
| |||
| Ka (CV%a) | 34.2 [54.1] | 32.1 [63.0] | |
| Tlag (CV%) | Not estimated (NE) | ||
| Km (CV%) | NE | ||
| CL (CV%) | 36.1 [7.9] | 28.1 [9.8] | |
| Vc (CV%) | 82.7 [19.3] | 79.9 [20.1] | |
| Q (CV%) | 32.7 [34.9] | 29.2 [40.3] | |
| Vp (CV%) | 72.3 [56.6] | 66.6 [60.5] | |
| corr (CL, Vc)a | Correlation between CL and Vc | 0.584 | 0.50 |
|
| |||
| Inter-occasion variability
| |||
| Ka (CV%) | 48.6 [21.0] | 49.3 [22.1] | |
| CL (CV%) | 13.0 [20.8] | 14.1 [19.1] | |
|
| |||
| Covariate effects
| |||
| θALBUMIN | NE | −0.686 [11.5] | |
| θCYCLOSPORINE | NE | 0.338 [14.1] | |
| Residual variability (mg/L)b | 0.487 [0.9] | 0.489 [1.0] | |
CV = coefficient of variation; RSE = relative standard error. Bioavailability was fixed to 1.
Additive residual error was on a natural logarithmic-scale. Typical CL values were calculated as followed: CL = 24.2 ×(body weight/70)0.75 × (albumin/3.4)−0.686× (1+0.338×CSP); CSP = 1 for cyclosporine, 0 for tacrolimus. Body weight calculations described in Methods, “Study and treatment plan.”
A comprehensive covariate analysis was conducted to identify objective factors influencing MPA pharmacokinetics; changes in model OFV during covariate modeling are listed in Supplemental Table 1. The covariate analysis identified serum albumin concentration and concomitant CNI as significant factors influencing MPA clearance. MPA clearance was negatively correlated with albumin concentration (Supplemental Figure 2). The model OFV decreased more than 6.6 units (p <0.01) and IIV in total clearance decreased from 36.1 to 31.1% with the inclusion of albumin concentration in the model. Concomitant cyclosporine administration was associated with a significant increase in MPA clearance, as compared to tacrolimus (Supplemental Figure 2), and this explained 3.8% of the IIV in MPA clearance. The model OFV decreased 122 units and the IIV for clearance decreased from 36.1 to 28.1% after serum albumin concentration and concomitant CNI were both included in the model. Together, these two covariates explain 22% of the IIV in MPA clearance. Backward elimination of either covariate resulted in an increase in OFV of more than 10.8 units. Including serum creatinine concentration as a covariate on MPA clearance significantly decreased the model OFV; the decrease in IIV of clearance, however, was negligible (0.28%). After allometric scaling of CL and Vc, creatinine clearance was no longer correlated with MPA clearance. No covariates were associated with IOV terms.
Parameters of the final model were precisely estimated, with relative standard error (RSE) less than 15% for most fixed-effect parameters and less than 40% for the majority of random effect parameters (Table 2). The RSE was low for MPA clearance (2.3%), demonstrating precise estimation of this parameter. MPA clearance was eventually defined by the following equation:
| (Eq.5) |
with CSP = 1 for cyclosporine and 0 for tacrolimus. The RSE was greater (35.4%) for the metabolism of MMF to MPA. Furthermore, the IOV was less than the IIV for clearance (CV% = 14.1 vs. 28.1%). The absorption rate constant, Ka, showed moderate IIV (CV% = 32.1%) and high IOV (CV% = 49.3%). GOF plots, including comparisons of i) individual and population model predicted and observed values and ii) weighted residuals, confirm satisfactory model precision with negligible bias (Supplemental Figure 3). The predictive performance of the final model was also assessed using VPC plots (Figure 2), wherein MPA concentration profiles were simulated for 40,800 subjects. The overlap of observed and model predicted 5th, 50th, and 95th percentiles of plasma drug concentrations suggest accurate predictive capabilities of the final model (Figure 2).
Figure 2.

Visual predictive check of MPA concentrations on a semi-log scale (left panel) and a linear scale (right panel). Dashed lines represent the 5th and 95th percentile of the observed data. Solid lines represent the 5th and 95th percentile of simulated data. Open circles and crosses represent 50th percentile of observed and simulated data.
LSS for Estimating AUC Values
We sought to develop a five-sample LSS over the shortest possible sampling duration to optimize patient compliance. Sampling durations of 2, 4, and 6 hr were compared, and the 4 hr sampling duration provided a good compromise between satisfactory predictive performance (i.e., AUC0–12 hr) and convenient clinical visits. The ten best sampling designs within a 4 hr time-frame post oral MMF administration are listed in Supplemental Table 2. The sampling schedule of −0.1hr (pre-dose), 0.25, 1.25, 2, and 4 hr post dose produced the minimum sMSE and was thus selected as the optimal LSS for Q12hr MMF administration. These five time points reflect the trough and peak concentrations (−0.1 and 1.25 hrs) and are distributed within the drug distribution (2 hr) and elimination (4 hr) phases. The early sampling time-point (0.25 hr) is important for estimating the lag time of delayed MPA absorption. The optimal LSS for Q8 hr MMF administration is: −0.1hr (pre-dose), 0.25, 1.25, 2, and 3 hr post dose.
The predictive performances of the reference sampling design (seven samples over a 10 hr sampling duration) and the optimal LSS are compared in Table 3 and Supplemental Figure 4. The LSS using five samples collected over 4 hr post dose performed favorably against the reference design for estimating net MPA exposure, AUC0–12hr, based on sMSE, Pearson correlation coefficient (r), mean relative bias (%), and the percentage of patients with unsatisfactory estimation (relative bias >10%). Thus, collecting fewer blood samples over a shorter sampling duration does not appear to sacrifice the precision achieved by a standard empirical design.
Table 3.
Comparison between optimal LSS and reference design for estimating AUCa0-τ after oral MMF administration
| Pharmacokinetic Sampling Schedule | Sampling times (hr) | Root mean sMSEb % (AUC0-τ) | Mean relative bias (rbias)% (AUC0-τ) | Pearson r | Percent individuals with rbias% > 10% |
|---|---|---|---|---|---|
| Q12 hr oral MMF administration
| |||||
| Reference design | Pre-dose, 1, 2, 4, 6, 8, 10 | 14.82 | 7.31 | 0.970 | 0.3% |
| 6 hr duration LSS | Pre-dose, 0.25, 1, 2, 5 | 16.23 | 8.57 | 0.961 | 0.7% |
| 4 hr duration LSS | Pre-dose, 0.25, 1.25, 2, 4 | 16.29 | 8.55 | 0.961 | 0.5% |
| 2 hr duration LSS | Pre-dose, 0.25, 1.25, 1.75,2 | 16.82 | 9.5 | 0.944 | 1.5% |
|
| |||||
| Q8 hr oral MMF administration
| |||||
| Reference design | Pre-dose, 1, 2, 4, 6, 8 | 16.06 | 8.31 | 0.964 | 0.4% |
| 6 hr duration LSS | Pre-dose, 0.25, 1, 2, 5 | 16.69 | 8.71 | 0.963 | 0.7% |
| 4 hr duration LSS | Pre-dose, 0.25, 1, 2, 3 | 16.86 | 8.69 | 0.962 | 0.7% |
| 2 hr duration LSS | Pre-dose, 0.25, 1.25, 1.75,2 | 16.90 | 8.92 | 0.957 | 1.5% |
AUC0-τ where τ is dosing frequency, so AUC0–12hr for PO MMF administered Q12 hrs and AUC0–8hr for PO MMF administered Q8hrs;
sMSE: scaled mean square error.
Simulation study for MMF dosing regimen
Compared to a body weight-based dosing regimen, a fixed-dosing regimen results in large variation in Css values, which is more pronounced for subjects weighing less than 50 kg (Supplemental Figure 6). For subjects weighing 75–100 kg and taking concomitant tacrolimus, the fixed-dosing regimen (1250 mg) and weight-based regimen (15 mg/kg) result in similar Css values, with 13.4 and 9.6% of subjects, respectively, with an MPA Css < 3 mg/L (Figure 3). Co-medication with cyclosporine increases MPA clearance and results in lower MPA Css values. Increasing the dose of MMF to 1750 mg or 20 mg/kg should compensate for the effect of cyclosporine on MPA clearance and optimize the proportion of subjects who achieve the target MPA Css of 3 mg/L (Figure 3).
Figure 3.

Simulated total MPA Css (mg/L) values in subjects weighing between 75 to 100 kg and serum albumin concentration of 3.4 g/dL for oral administration of MMF every 8 hr at 1250 mg, 1750 mg, 15 mg/kg, and 20 mg/kg with a calcineurin inhibitor, either cyclosporine and tacrolimus. Dashed line represents the lower therapeutic Css of 3 mg/L.
DISCUSSION
This population pharmacokinetic analysis of MPA, in the largest population of allogeneic HCT recipients to date, successfully characterized the disposition of total MPA in HCT recipients and identified large IIV in total MPA AUC after a weight-based dose of 15 mg/kg of oral MMF (Supplemental Figure 3). This IIV is similar to that observed for CNIs, for which kinetics-based dosing is accepted in HCT.16–17 The IIV in MPA AUC persists even after considering covariates associated with MPA clearance (Supplemental Figure 5). The current model suggests nonspecific plasma protein binding (albumin concentration) and concomitant cyclosporine administration influence MPA clearance in HCT patients (Supplemental Figures 2 and 5). These covariates were previously reported in renal transplant recipients.18 Rapid total MPA clearance occurred in patients with decreased serum albumin concentrations, most likely due to increased unbound MPA fraction, as MPA is highly bound to serum albumin (~ 97–99%). Furthermore, in nonmyeloablative HCT recipients, a ≥ 0.5 g/dL decrease in serum albumin concentration predicted the development of grade III/IV acute GVHD.19 Such a decrease in albumin concentrations would lead to a more rapid MPA clearance, potentially placing the patient at higher risk for inadequate postgrafting immunosuppression and subsequent GVHD. The impact of hypoalbuminea upon free MPA AUC, however, is not known, especially because lower total MPA AUC may not equate to a low free MPA AUC.20 Cyclosporine increases MPA clearance by 33.8% compared to concomitant tacrolimus. The inhibition effect of cyclosporine on ATP binding cassette protein C2 likely results in decreased enterohepatic cycling and thus, more rapid MPA clearance.21–22
In contrast to MMF pharmacokinetic and pharmacodynamic research in solid organ transplantation, similar data in homogenous populations of HCT recipients is scarce. A mechanistic understanding is needed for the observation that HCT recipients5–7 have a shorter MPA half-life compared to solid organ transplant recipients.2 Identifying concomitant cyclosporine as a covariate for MPA clearance is one potential explanation. A separate analysis suggested that predose cyclosporine and plasma albumin concentrations account for the differences in MPA clearance between HCT recipients, renal transplant recipients, and patients with autoimmune disorders.23 After oral MMF administration, HCT recipients had a 50% higher median clearance of MPA (45.6 L/hr) compared to renal transplant patients (30.2 L/hr).23
Population pharmacokinetic models of various renal transplant patient populations receiving oral MMF have varying results, with the apparent oral clearance (CL/F) of MPA ranging from 11.9 to 34.9 L/hr.2 The apparent MPA clearance in the present study is 24.2 L/hr/70 kg with 3.4 g/dL serum albumin concentration and concomitant tacrolimus. In general, MPA pharmacokinetic parameter estimates for HCT recipients (Table 2) are similar to those for renal transplant recipients, with the exception of Ka.24–30 Currently, it is unclear whether population pharmacokinetic models created for solid organ transplant recipients can be used in HCT recipients. HCT recipients demonstrate a slower first-order absorption rate process (0.602/hr) compared to renal transplant recipients (0.64 to 4.1/hr).24, 26–30 The large IOV of Ka (49.3% CV) could be a consequence of inconsistent eating at the time of MMF self-administration, antibiotics, gastrointestinal GVHD, or the gastrointestinal epithelium recovering from administration of the conditioning regimen. Gastrointestinal disturbances caused by the conditioning regimen (e.g., total body irradiation) could be responsible for the decreased Ka value in HCT recipients. The conditioning regimen was not, however, a significant covariate, potentially because only 61 (15%) patients received myeloablative conditioning. We considered evaluating the presence of gastrointestinal GVHD and antibiotics as covariates in this population pharmacokinetic model. Gastrointestinal GVHD was not considered because assessing the severity of GVHD involves some subjective judgment both in the interpretation of medical records and at the bedside.31 Antibiotics may affect the enterohepatic recirculation of MPA by impairing conversion of MPA glucuronides to MPA by gastrointestinal bacterial β-glucuronidase. Antibiotics were not a covariate in a population pharmacokinetic analysis from 468 renal transplant patients, the largest such study to date.18 Three contradictory studies from smaller populations (ranging from two32 to 64 participants33) have been reported, including a prospective pharmacokinetic study in 64 renal transplant recipients receiving concomitant tacrolimus.33 Oral ciprofloxacin or amoxicillin with clavulanic acid reduced the 12 hour predose MPA concentration to 46% of pre-antibiotic levels within 3 days of antibiotic commencement; MPA concentrations recovered after 14 days of antibiotics. A case series of two patients suggests amoxicillin and clavulanic acid decreased MPA AUC.32 The impact of antibiotics on MPA pharmacokinetics may differ based on the bacterial spectrum. A healthy volunteer (N=11) cross-over study reported that MPA AUC was reduced by an average of 10%, 19%, and 33% when MMF was given with norfloxacin, metronidazole, and norfloxacin plus metronidazole, respectively.34 Antibiotic use in HCT patients can influence the low (<4%) number of concentration-time profiles demonstrating a secondary peak. Unfortunately, concomitant antibiotic use was not evaluated when comparing MPA clearance between HCT recipients, renal transplant recipients, and patients with autoimmune disorders.23 We did not evaluate antibiotic use as a covariate because of inconsistent documentation regarding the precise initiation and discontinuation of antibiotic therapy relative to the days of MPA pharmacokinetic sampling. This is a limitation and future research is needed to address this issue.
One critical barrier to pharmacokinetic/dynamic studies in nonmyeloablative HCT recipients is the time commitment of blood sampling in an ambulatory clinic. Many HCT recipients may not feel well enough to remain in the clinic for pharmacokinetic sampling, which is supported by the 50% participation rate of patients in the various clinical trials from which this retrospective analysis was conducted (data not shown). Two sampling strategies for oral MMF pharmacokinetics were created – one each for Q8 and Q12hr MMF dosing regimens (Table 3). To enhance patient compliance with pharmacokinetic sampling, varying numbers of samples (n = 4–5) and time durations (2, 4, and 6 hr; Table 3) were evaluated during the LSS development. The sMSE in a four-sample LSS was large; sMSE was only satisfactorily minimized in a LSS with five samples (data not shown). As shown in Table 3, the five time points are allocated around the trough and peak concentrations (−0.1 and 1.25 hrs), within the distribution phase (2 hr), and within the elimination phase (4 hr). The early sampling time-point (0.25 hr) is important for estimating the lag time of delayed MPA absorption. Notably, the LSS over 2 hr does have a higher sMSE and relative bias than longer schedules (Table 3). The shorter LSS could, however, be advantageous because it minimizes the time patients must wait in the clinic for phlebotomy. The 4 hr LSS requires an additional 2 hr stay, but adds a sample in the MPA elimination phase, which might allow for assessing the role of MPA enterohepatic recirculation in AUC0–τ. Enterohepatic recirculation results in a prolonged half-life, as represented by a relatively flat terminal phase. In a cohort of 14 HCT patients receiving reduced intensity conditioning and oral MMF with cyclosporine, a LSS with draws at 0.33, 1.5, and 4 hr had the highest precision on Bayesian estimates of the population pharmacokinetic model parameters using a D-optimality criterion.35 The large IOV in Ka might influence the efficiency of the LSS. Food intake has been shown to alter MPA absorption, delaying Tmax and decreasing Cmax.2 Information on food consumption was not available in this retrospective study. None of the covariates could explain the IOV on absorption; to minimize the variability in MPA absorption between occasions, we suggest that MPA should be taken on an empty stomach. In addition, greater initial MMF doses appear necessary for patients receiving concomitant cyclosporine. Simulations suggest that oral MMF doses should be increased to 20 mg/kg for patients also receiving cyclosporine as part of their immunosuppressive regimen (Figure 3).
Monitoring MPA trough concentrations (i.e., predose) is appealing in terms of patient convenience, but total MPA trough concentrations correlate poorly with AUC0-τ in HCT recipients.3–4 CNI doses are personalized to trough concentrations because of their pharmacodynamic association with GVHD, nonrelapse mortality, and overall survival.36 Currently, there is no firmly established target MPA exposure for HCT recipients. Low trough concentrations of either unbound or total MPA have been associated with graft failure4, 37 and more frequent acute GVHD4, 37 in select HCT populations. A previous pharmacodynamic analysis conducted at our center, however, indicated that MPA trough concentrations were not associated with T-cell donor chimerism or other clinical outcomes in nonmyeloablative HCT recipients with an unrelated donor graft.3 Graft rejection and low donor T-cell chimerism were associated with low total MPA exposure. Specifically, patients receiving oral MMF are at increased risk of graft rejection or low T-cell chimerism with total MPA Css < 2.5 or 3 mg/L, respectively.3 Thus, in settings where total MPA exposure is associated with clinical outcomes, the developed population pharmacokinetic-based LSS provides a useful tool for conducting future pharmacodynamic studies.
Supplementary Material
Acknowledgments
We are grateful to the study participants, nurses, and laboratory staff for their assistance with collection and analysis of the MPA pharmacokinetic data. We also thank Dr. David H. Salinger for providing initial R-code for LSS calculations. This study was funded by NIH Grants HL091744, HL36444, CA78902, and CA18029.
Footnotes
CONFLICT OF INTEREST/DISCLOSURE
None of the authors declare conflicts of interest that could be perceived as influencing this research.
References
- 1.Deeg HJ, Maris MB, Scott BL, Warren EH. Optimization of allogeneic transplant conditioning: not the time for dogma. Leukemia. 2006 Oct;20(10):1701–1705. doi: 10.1038/sj.leu.2404327. [DOI] [PubMed] [Google Scholar]
- 2.Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin Pharmacokinet. 2007;46(1):13–58. doi: 10.2165/00003088-200746010-00002. [DOI] [PubMed] [Google Scholar]
- 3.Giaccone L, McCune JS, Maris MB, et al. Pharmacodynamics of mycophenolate mofetil after nonmyeloablative conditioning and unrelated donor hematopoietic cell transplantation. Blood. 2005 Dec 15;106(13):4381–4388. doi: 10.1182/blood-2005-06-2217. Epub 2005 Sep 4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson P, Rogosheske J, Barker JN, et al. Relationship of mycophenolic acid exposure to clinical outcome after hematopoietic cell transplantation. Clin Pharmacol Ther. 2005 Nov;78(5):486–500. doi: 10.1016/j.clpt.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Maris MB, Niederwieser D, Sandmaier BM, et al. HLA-matched unrelated donor hematopoietic cell transplantation after nonmyeloablative conditioning for patients with hematologic malignancies. Blood. 2003 Sep 15;102(6):2021–2030. doi: 10.1182/blood-2003-02-0482. [DOI] [PubMed] [Google Scholar]
- 6.Nash RA, Johnston L, Parker P, et al. A phase I/II study of mycophenolate mofetil in combination with cyclosporine for prophylaxis of acute graft-versus-host disease after myeloablative conditioning and allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2005 Jul;11(7):495–505. doi: 10.1016/j.bbmt.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 7.van Hest RM, Doorduijn JK, de Winter BC, et al. Pharmacokinetics of mycophenolate mofetil in hematopoietic stem cell transplant recipients. Ther Drug Monit. 2007 Jun;29(3):353–360. doi: 10.1097/FTD.0b013e31805d8816. [DOI] [PubMed] [Google Scholar]
- 8.Maris MB, Sandmaier BM, Storer BE, et al. Unrelated donor granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cell transplantation after nonmyeloablative conditioning: the effect of postgrafting mycophenolate mofetil dosing. Biol Blood Marrow Transplant. 2006 Apr;12(4):454–465. doi: 10.1016/j.bbmt.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 9.Baron F, Little MT, Storb R. Kinetics of engraftment following allogeneic hematopoietic cell transplantation with reduced-intensity or nonmyeloablative conditioning. Blood Rev. 2005 May;19(3):153–164. doi: 10.1016/j.blre.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Baron F, Baker JE, Storb R, et al. Kinetics of engraftment in patients with hematologic malignancies given allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. Blood. 2004 Oct 15;104(8):2254–2262. doi: 10.1182/blood-2004-04-1506. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Mager DE, Bemer MJ, et al. A Limited Sampling Schedule to Estimate Mycophenolic Acid Area Under the Concentration-Time Curve in Hematopoietic Cell Transplantation Recipients. J Clin Pharmacol. 2011 Dec 14; doi: 10.1177/0091270011429567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 13.Salinger DH, Blough DK, Vicini P, et al. A limited sampling schedule to estimate individual pharmacokinetic parameters of fludarabine in hematopoietic cell transplant patients. Clin Cancer Res. 2009 Aug 15;15(16):5280–5287. doi: 10.1158/1078-0432.CCR-09-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkins JJ, Savic RM, Karlsson MO, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008 Jun;52(6):2138–2148. doi: 10.1128/AAC.00461-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ene I, Ette PJW. Pharmacometrics: The Science of Quantitative Pharmacology. In: Olivier Petricoul VC, Fuseau Eliane, Marchano Mathilde, editors. Population Models for Drug Absorption and Enterohepatic Recycling. 2007. [Google Scholar]
- 16.Venkataramanan R, Jain A, Cadoff E, et al. Pharmacokinetics of FK 506: preclinical and clinical studies. Transplant Proc. 1990 Feb;22(1):52–56. [PMC free article] [PubMed] [Google Scholar]
- 17.Przepiorka D, Nash RA, Wingard JR, et al. Relationship of tacrolimus whole blood levels to efficacy and safety outcomes after unrelated donor marrow transplantation. Biol Blood Marrow Transplant. 1999;5(2):94–97. doi: 10.1053/bbmt.1999.v5.pm10371361. [DOI] [PubMed] [Google Scholar]
- 18.van Hest RM, Mathot RA, Pescovitz MD, Gordon R, Mamelok RD, van Gelder T. Explaining variability in mycophenolic acid exposure to optimize mycophenolate mofetil dosing: a population pharmacokinetic meta-analysis of mycophenolic acid in renal transplant recipients. J Am Soc Nephrol. 2006 Mar;17(3):871–880. doi: 10.1681/ASN.2005101070. [DOI] [PubMed] [Google Scholar]
- 19.Rezvani AR, Storer BE, Storb RF, et al. Decreased serum albumin as a biomarker for severe acute graft-versus-host disease after reduced-intensity allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011 Nov;17(11):1594–1601. doi: 10.1016/j.bbmt.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tett SE, Saint-Marcoux F, Staatz CE, et al. Mycophenolate, clinical pharmacokinetics, formulations, and methods for assessing drug exposure. Transplant Rev (Orlando) 2011 Apr;25(2):47–57. doi: 10.1016/j.trre.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 21.van Gelder T, Klupp J, Barten MJ, Christians U, Morris RE. Comparison of the effects of tacrolimus and cyclosporine on the pharmacokinetics of mycophenolic acid. Ther Drug Monit. 2001 Apr;23(2):119–128. doi: 10.1097/00007691-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Meiser BM, Groetzner J, Kaczmarek I, et al. Tacrolimus or cyclosporine: which is the better partner for mycophenolate mofetil in heart transplant recipients? Transplantation. 2004 Aug 27;78(4):591–598. doi: 10.1097/01.tp.0000129814.52456.25. [DOI] [PubMed] [Google Scholar]
- 23.de Winter BC, Mathot RA, Sombogaard F, et al. Differences in clearance of mycophenolic acid among renal transplant recipients, hematopoietic stem cell transplant recipients, and patients with autoimmune disease. Ther Drug Monit. 2010 Oct;32(5):606–614. doi: 10.1097/FTD.0b013e3181efd715. [DOI] [PubMed] [Google Scholar]
- 24.van Hest RM, van Gelder T, Vulto AG, Mathot RA. Population pharmacokinetics of mycophenolic acid in renal transplant recipients. Clin Pharmacokinet. 2005;44(10):1083–1096. doi: 10.2165/00003088-200544100-00006. [DOI] [PubMed] [Google Scholar]
- 25.Le Guellec C, Bourgoin H, Buchler M, et al. Population pharmacokinetics and Bayesian estimation of mycophenolic acid concentrations in stable renal transplant patients. Clin Pharmacokinet. 2004;43(4):253–266. doi: 10.2165/00003088-200443040-00004. [DOI] [PubMed] [Google Scholar]
- 26.de Winter BC, van Gelder T, Glander P, et al. Population pharmacokinetics of mycophenolic acid: a comparison between enteric-coated mycophenolate sodium and mycophenolate mofetil in renal transplant recipients. Clin Pharmacokinet. 2008;47(12):827–838. doi: 10.2165/0003088-200847120-00007. [DOI] [PubMed] [Google Scholar]
- 27.van Hest RM, van Gelder T, Bouw R, et al. Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol. 2007 Jun;63(6):741–752. doi: 10.1111/j.1365-2125.2006.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shum B, Duffull SB, Taylor PJ, Tett SE. Population pharmacokinetic analysis of mycophenolic acid in renal transplant recipients following oral administration of mycophenolate mofetil. Br J Clin Pharmacol. 2003 Aug;56(2):188–197. doi: 10.1046/j.1365-2125.2003.01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staatz CE, Duffull SB, Kiberd B, Fraser AD, Tett SE. Population pharmacokinetics of mycophenolic acid during the first week after renal transplantation. Eur J Clin Pharmacol. 2005 Aug;61(7):507–516. doi: 10.1007/s00228-005-0927-4. [DOI] [PubMed] [Google Scholar]
- 30.Musuamba FT, Rousseau A, Bosmans JL, et al. Limited sampling models and Bayesian estimation for mycophenolic acid area under the curve prediction in stable renal transplant patients co-medicated with ciclosporin or sirolimus. Clin Pharmacokinet. 2009;48(11):745–758. doi: 10.2165/11318060-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 31.Martin PJ, McDonald GB, Sanders JE, et al. Increasingly frequent diagnosis of acute gastrointestinal graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2004 May;10(5):320–327. doi: 10.1016/j.bbmt.2003.12.304. [DOI] [PubMed] [Google Scholar]
- 32.Ratna P, Mathew BS, Annapandian VM, et al. Pharmacokinetic drug interaction of mycophenolate with co-amoxiclav in renal transplant patients. Transplantation. 2011 Mar 27;91(6):e36–38. doi: 10.1097/TP.0b013e31820a6a79. [DOI] [PubMed] [Google Scholar]
- 33.Borrows R, Chusney G, Loucaidou M, et al. The magnitude and time course of changes in mycophenolic acid 12-hour predose levels during antibiotic therapy in mycophenolate mofetil-based renal transplantation. Ther Drug Monit. 2007 Feb;29(1):122–126. doi: 10.1097/FTD.0b013e31803111d5. [DOI] [PubMed] [Google Scholar]
- 34.Naderer OJ, Dupuis RE, Heinzen EL, Wiwattanawongsa K, Johnson MW, Smith PC. The influence of norfloxacin and metronidazole on the disposition of mycophenolate mofetil. J Clin Pharmacol. 2005 Feb;45(2):219–226. doi: 10.1177/0091270004271555. [DOI] [PubMed] [Google Scholar]
- 35.Saint-Marcoux F, Royer B, Debord J, et al. Pharmacokinetic modelling and development of Bayesian estimators for therapeutic drug monitoring of mycophenolate mofetil in reduced-intensity haematopoietic stem cell transplantation. Clin Pharmacokinet. 2009;48(10):667–675. doi: 10.2165/11317140-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ram R, Storer B, Mielcarek M, et al. Association between calcineurin inhibitor blood concentrations and outcomes after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012 Mar;18(3):414–422. doi: 10.1016/j.bbmt.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osunkwo I, Bessmertny O, Harrison L, et al. A pilot study of tacrolimus and mycophenolate mofetil graft-versus-host disease prophylaxis in childhood and adolescent allogeneic stem cell transplant recipients. Biol Blood Marrow Transplant. 2004 Apr;10(4):246–258. doi: 10.1016/j.bbmt.2003.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.