

Abstract
Kappa opioid receptors (KORs) belong to the G-protein coupled class of receptors (GPCRs). They are activated by the endogenous opioid peptide dynorphin (DYN) and expressed at particularly high levels within brain areas implicated in modulation of motivation, emotion, and cognitive function. Chronic activation of KORs in animal models has maladaptive effects including increases in behaviors that reflect depression, the propensity to engage in drug-seeking behavior, and drug craving. The fact that KOR activation has such a profound influence on behaviors often triggered by stress has led to interest in selective KOR antagonists as potential therapeutic agents. This perspective provides a description of preclinical research conducted in the development of several different classes of selective KOR antagonists, a summary of the clinical studies conducted thus far, and recommendations for the type of work needed in the future to determine if these agents would be useful as pharmacotherapies for neuropsychiatric illness.
INTRODUCTION
Opioid receptors were discovered in 1973 using opioid radioligand binding assays in brain homogenates.1–3 Subsequent cloning studies in the early 1990s differentiated three receptors: μ, δ, and κ.4–7 The cDNA clones of κ opioid receptors (KORs) have been isolated and characterized from several species, including humans. The KOR belongs to the G-protein-coupled class of receptors (GPCRs) that are widely expressed throughout the brain and are located in brain areas that are implicated in the modulation of reward, mood state, and cognitive function such as the ventral tegmental area (VTA), nucleus accumbens (NAc), prefrontal cortex (PFC), hippocampus (HPC), striatum (ST), amygdala (AMYG), locus coeruleus (LC), substantia nigra (SN), dorsal raphe nucleus (DRN), and hypothalamus (HL) of both the rat and human brains.8–11 KORs are activated by the endogenous opioid peptides derived from prodynorphin.12–14 Even though these peptides bind to the μ and δ receptors (MOR and DOR, respectively) as well as the KOR, dynorphin A (1–17) (Figure 1) shows preference for the KOR.15 Upon activation, the KOR couples to the pertussis toxin-sensitive heterotrimeric Gαi/o protein resulting in inhibition of adenylate cyclase, increase in potassium conductance, decrease in calcium conductance, and mobilization of intracellular calcium.16 In addition, KOR activates the extracellular signal-regulated kinase (ERK 1/2) and p38 mitogen-activated protein kinase (MAPK)17–19 and can activate c-Jun amino-terminal kinase (JNK).20 Interestingly, prototypical KOR antagonists can also activate JNK via a pharmacological process called biased agonism or ligand-directed signaling, whereby ligands can inhibit one intracellular signaling pathway while simultaneously activating another.21,22
Figure 1.

Structure of dynorphin A (1–17) amino acid abbreviation YGGFLRRIRPKLKWDNQ
Relatively early studies showed that activation of the MOR elevates mood, whereas KOR activation produces dysphoria and psychotomimetic effects in humans23 and anhedonia-, dysphoria-, and anxiety-like effects in rodents24 (see ref. 25 for a review). These results led to the hypothesis that opposing endogenous opioid systems regulate emotional and perceptual experiences.23 Figure 2 shows that stimulation of MORs by β-endorphins or a μ agonist in the ventral tegmental area increases dopamine (DA) release. In contrast, KOR activation in the nucleus accumbens region by dynorphin (DYN) and κ agonists decreases DA transmission, whereas blockade of the KOR with a κ antagonist increases basal DA release.26,27 More recent studies have expanded on these early reports. For example, Nestler and Carlezon have proposed that neuroplastic processes that result in enhanced DYN within the NAc promote depressive-like effects in rodents, whereas KOR antagonists have antidepressant-like effects.28
Figure 2.

Model for modulating the endogenous opioid system26
While KOR activation resulting from acute stress can facilitate motivation to escape stimuli that represent threats to homeostasis, chronic stress can have adverse effects such as increased risk of depression, increased propensity to participate in drug-seeking behavior, and increased drug-craving.25 Several studies suggest that DYN activation of the KOR is a key element of these responses. Increased levels of DYN expression are thought to contribute to the negative mood states precipitated by cocaine (1) (Figure 3) withdrawal, a state resembling depression.29 Rewarding as well as stressful stimuli increase cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) function in the NAc29,30 (see ref. 31 for a review). The increased levels of dynorphin resulting from activation of CREB seen after stress or drug exposure could contribute to symptoms of emotional numbing.32 Several studies have established that elevation of CREB function in the NAc elicits the rodent equivalent of signs of major depression.29,33,34 In addition, the studies showed that stress, a common trigger for addictive as well as depressive disorders, also activates CREB in the NAc.29,35 In contrast, disruption of CREB function in the NAc had antidepressant-like effects indistinguishable from those of standard antidepressants.29 The results from these studies suggested that activation of CREB in the NAc is a molecular mediator for aversive or depressive-like symptoms.29,36,37 Dynorphin released during chronic stress exposure likely produces a variety of depressive-signs in rodents, including immobility in the Porsolt forced-swim test (FST),38–40 social defeat behavior in the Miczek assay,41 and potentiation of the rewarding effects of cocaine in the conditioned place preference (CPP) assay.40 Microinjections of KOR agonists directly into the NAc demonstrate that KOR activation within the region is sufficient to produce anhedonia, a hallmark sign of depressive illness.42,43
Figure 3.
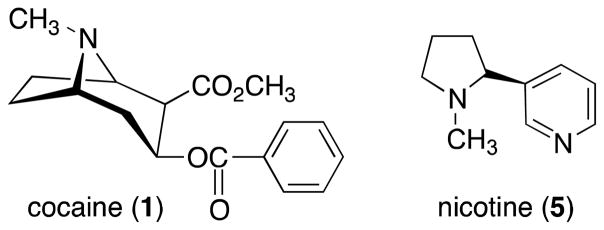
Structures of cocaine and nicotine
The fact that KOR function appears to have a profound influence on behaviors that are thought to reflect motivational and emotional states in animal models of depression and anxiety led to interest in the use of selective κ opioid antagonists as potential pharmacotherapies for treating mood disorders.30,38,44 Initial reports that the prototypical KOR antagonist nor-binaltorphimine (nor-BNI) (2) (Figure 4) produced antidepressant-like effects in the FST in rats29 were quickly followed up with similar findings in other tests33 and with (3R)-1,2,3,4-tetrahydro-7-hydroxy-N-[(1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl]-3-isoquinolinecarboxamide (JDTic) (3) (Figure 4), a novel, chemically dissimilar KOR antagonist.38 Additional work showed that KOR antagonists have anxiolytic-like effects and prevent the development of stress-related behavioral adaptations.45 It has also been shown that KOR antagonists inhibit stress-induced but not cocaine-primed reinstatement of cocaine-associated CPP in mice46,47 and similarly inhibit stress-induced but not cocaine-primed reinstatement for responding for cocaine in rats.38 In addition, genetic deletion of the KOR or prodynorphin also abolishes stress-induced reinstatement of CPP in mice.47 These data clearly suggest that the κ opioid system plays a role in stress-induced cocaine-seeking. Since the relationship between stress and drug addiction in humans is strong, 48,49 κ opioid antagonists, which enhance stress resilience, also have potential as pharmacotherapies to treat stress-induced relapse to drug seeking. It is important to note that stress is a common factor in these mood- and addiction-related studies, which has led to more broad theories that KOR antagonists may have protective effects against stress.25,50 Indeed, KOR antagonists can block the aversive and cognitive-disrupting effects of corticotropin-releasing factor,51–53 a key regulator of stress effects.54–56 A broad ability to reduce the impact of stress may explain how KOR antagonists can have efficacy in such a wide variety of animal models that would appear to represent different disease states.
Figure 4.

Structures of nor-BNI and JDTic
Development of KOR Antagonists
Even though KOR-selective antagonists were initially developed as tools for studying the in vitro and in vivo properties of the KOR agonists, more recent studies have led to the development of KOR antagonists as potential drugs to treat several CNS disorders. In the following sections, the naltrexone-related compounds such as nor-BNI and irreversible binding KOR antagonists which were developed as tools to study the KOR agonists will be briefly summarized, followed by more detailed summaries of several classes of KOR antagonists developed as potential pharmacotherapies to treat mood disorders and drug addiction, as well as to protect against the effects of stress. The status of clinical development of κ opioid antagonist is presented in the Clinical Studies.
Naltrexone-related Compounds
Even though potent and selective naltrexone (4) (Figure 5) related KOR antagonists were discovered in the mid-to late-1980s, they were developed and only used as pharmacological tools to characterize in vitro and in vivo properties of KOR antagonists. An increased understanding of the KOR system as it related to cocaine and opiate abuse as well as mood disorders in the late 1990s and early 2000s led to increased interest in KOR antagonists and their potential to treat human diseases. Since several excellent reviews have covered these early studies, they will not be detailed in this perspective.57,58 However, a brief summary as it relates to the development of potential pharmacotherapies for treatment of mood disorders, such as depression and anxiety, and addictions, including cocaine, nicotine (5), alcohol, and opiates, will be presented.21,30,31,56,59–65
Figure 5.
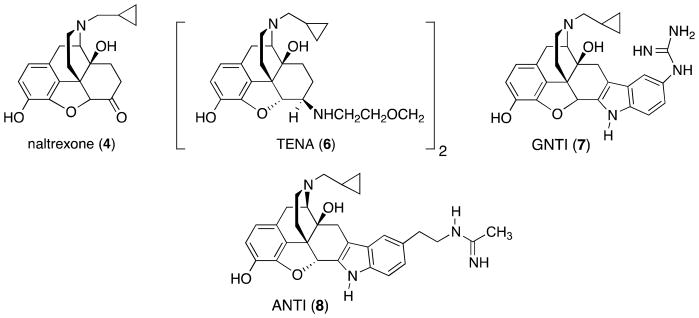
Structures of naltrexone, TENA, GNTI, and ANTI
The first selective KOR antagonist was the β-naltrexamine derivative 1,8-bis(β-naltrexamino)-3,6-dioxaoctane (TENA) (6), which was reported by Portoghese and co-workers in 1982.66 Since its κ selectivity over μ and δ was only 4- and 2.5-fold, respectively, TENA did not prove to be a very useful compound for studying the KOR. In 1987 Portoghese and co-workers reported the design and development of the naltrexone-derived nor-BNI as the first truly potent and selective KOR antagonist useful for animal studies.67–70 For example, nor-BNI has selective, centrally mediated κ antagonist effects in mice if the time separating the administration of nor-BNI and the κ agonist is at least 24 h.70 The resulting κ opioid antagonist activity and selectivity can last up to 28 days.71,72 Thus, under these conditions nor-BNI is a KOR-selective antagonist.
Portoghese and co-workers also developed 5′-guanidinonaltrindole (GNTI) (7) as a more potent and more selective κ opioid antagonist.73–76 nor-BNI and GNTI have similar features in that they are structural derivatives of the opioid antagonist naltrexone. Like nor-BNI, GNTI also was found to have a slow onset and long duration of action in studies with rhesus monkeys;76 in fact, GNTI appeared inactive in rats when given by systemic administration, although it had strong behavioral effects in this species when administered by intracerebroventricular (ICV) infusion.39 The poor bioavailability to GNTI, in particular, has been attributed to specific elements of the molecule (i.e., the high pKa of the guanidinium group).39 More recent work with nor-BNI in rat demonstrates that the KOR antagonist effects remain detectable for at least 86 days, the time point at which the studies were terminated.77 Although these types of molecular and pharmacological issues are not understood completely, they have limited enthusiasm for the development of the naltrexone/nor-BNI derivatives as pharmacotherapies. Based on the molecular properties of these compounds, they were not studied as possible pharmacotherapies. Even though a number of nor-BNI analogues have been developed,57,58,68,75 nor-BNI remains the most widely used KOR antagonist as a pharmacological tool. Some of the seminal studies in which nor-BNI and GNTI were used as pharmacological tools are described below.
Pliakas and co-workers29 first reported that intracerebroventricularly (i.c.v.) administration of the KOR antagonist nor-BNI decreased immobility in the FST. More thorough analyses that included more doses, drugs (e.g., GNTI, 5′-acetamidinoethylnaltrindole (ANTI) (8)), and tests (e.g., intracranial self-stimulation (ICSS)) suggested that these KOR antagonists possess antidepressant-like effects.39,78 As pointed out in the Introduction, these authors also suggested that their findings are consistent with the hypothesis that CREB-mediated induction of DYN, an endogenous κ agonist15 in the nucleus accumbens (NAc), triggers immobility behavior in the FST.
A major problem in treating drug addiction is the vulnerability to relapse during abstinence.79 To gain a better understanding of the mechanisms of relapse, animal models of drug reinstatement were developed to help identify triggers for reinstatement of drug self-administration.80–83 Drug reinstatement studies in rodents have shown that presentation of drug-associated cues, drug priming, and stress (acute foot shock) each increased drug self-administration.38,47,84–86 In addition, activation of the κ opioid system by foot shock, forced swim, or KOR agonist 2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide (U50,488)87 (Figure 6) administration reinstated cocaine-seeking behavior in mice.47 Pretreatment with the selective κ antagonist nor-BNI or by KOR or prodynorphin gene disruption abolishes cocaine reinstatement.47 Since the relationship between stress and drug addiction is strong,48 these studies showed that KOR antagonists, which enhance stress resilience, also have potential as pharmacotherapies to treat stress-induced relapse to drug seeking.
Figure 6.

Structures of U50,488, THC, WIN 55,212-2, and CP 55,940
There are conflicting results on the use of KOR antagonists for ethanol consumption. Williams and Woods88 reported no alteration in ethanol consumption following nor-BNI administration in nonhuman primates. nor-BNI also did not alter the alcohol deprivation effects in rats.89 Mitchell and co-workers90 reported that treatment with nor-BNI resulted in a significant increase in ethanol consumption in animals with a history of stable ethanol self-administration. In contrast KOR knockout mice showed decreased ethanol drinking.91 Walker and Koob92 reported that the effects of nor-BNI on ethanol self-administration vary depending upon whether an animal is naïve or physically dependent. For example, recent work by Walker and co-workers93 shows that nor-BNI prevents the normal ability of cues that are associated with ethanol withdrawal to trigger ethanol consumption, again suggesting that KOR antagonists can alleviate the impact of aversive or stressful stimuli that contribute to maladaptive (excessive drinking) behaviors.
Forced swim stressed (FSS)-induced potentiation of ethanol reward and self-administration of ethanol is blocked by nor-BNI in mice.94 Even though Dyn−/− mice consumed a similar volume of ethanol as wild-type littermates, they did not demonstrate significant stress-induced increases in consumption. These studies suggest that KOR antagonists may prove beneficial in preventing stress-induced increases in ethanol consumption. nor-BNI (i.c.v. or subcutaneous (s.c.) administration) was also shown to decrease ethanol self-administration in ethanol-dependent animals but had no effect in non-dependent animals.95 Dependence was induced using an intermittent alcohol vapor exposure schedule.95 As shown in the elevated plus maze (EPM) test, male Wistar rats fed on an ethanol liquid diet showed a decrease in the percentage of time spent exploring the open arm and had fewer open arm entries.96 Pretreatment with nor-BNI attenuated both effects relative to controls. These results are consistent with the hypothesis that the KOR/DYN system is involved in stress-related chronic exposure to ethanol. Even though the results with ethanol are variable, the evidence suggests that KOR antagonist treatment may be effective in the treatment of drug and ethanol addiction.
Smith and co-workers97 demonstrated that during stress exposure, KOR activation is necessary, and KOR activation in the amygdala alone is sufficient to increase nicotine-seeking behavior as measured by CPP. This increase in nicotine CPP was blocked by the KOR antagonist nor-BNI either by intraperitoneal (i.p.) administration or by local injection in the amygdala without affecting nicotine reward in the absence of stress. Pretreatment with nor-BNI also significantly attenuated stress-induced (forced swim) reinstatement of nicotine-CPP in mice but had no effect on nicotine-primed reinstatement.98 These results suggest that KOR antagonists will be useful pharmacological tools for studying nicotine addiction and may be useful as pharmacotherapies to prevent relapse to nicotine craving.
It is also interesting to note that cannabinoids and κ opioids act on common elements of the circuitry in the brain and the spinal cord that produce analgesia. For example, antinociception produced by intrathecal administration of the cannabinoid receptor agonist Δ-9-tetrahydrocannabinol (THC) (9) (Figure 6) and (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55,212-2) (10) (Figure 6) was attenuated by prior administration of nor-BNI. In contrast, antinociception produced by the non-classical cannabinoid agonist 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol (CP 55,940) (11) (Figure 6) remained unaffected by prior administration of nor-BNI.99 Interestingly, nor-BNI also blocked the antinociception produced by spinal administration of THC.100
Even though the degree of selectivity and potency following systemic administration of these naltrexone-related ligands was limited, the discovery of opioid-selective antagonists for KORs such as nor-BNI and GNTI by Portoghese and collaborators was of major significance in studies of the relationship to function of opioid receptors. Moreover, they provided valuable information for the design of new antagonists and highlighted the need to develop other selective antagonists.
Irreversible Binding Compounds
(1S,2S)-trans-2-Isothiocyanato-4,5-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide) (−)-UPHIT101 (12) and 2-(3,4-dichlorophenyl)-N-methyl-N-[(1S)-1-(3-isothiocyanatophenyl)-2-(1-pyrrolidinyl)ethyl]acetamide102,103 (DIPPA) (13) (Figure 7) are two irreversible binding ligands that behave like KOR antagonists (see ref. 57 for review). (−)-UPHIT was evaluated for its ability to antagonize the KOR-selective agonist [5R-(5α,7α,8β)]-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetamide (U69,593),87 the κ agonist/μ antagonist bremazocine (14), the μ agonist [D-Ala2, NMe-Phe4, Gly-ol5]enkephalin (DAMGO) (15), and the δ antagonist [D-Pen2,D-Pen5]enkephalin (DPDPE) (16)-induced (Figure 8) antinociception in the warm-water tail-flick test after i.c.v. administration.104 Pretreatment with (−)-UPHIT antagonized U69,593-induced antinociception for up to 48 hours but did not affect bremazocine, the μ-selective DAMGO, or the δ-selective DPDPE-induced antinociception. These results strongly suggested that (−)-UPHIT is a long-lasting κ-selective opioid antagonist. Similarly, DIPPA produced a long-lasting antagonism of κ-selective agonist (±)-U50,488-induced antinociception in the mouse tail-flick test.103 DIPPA also behaved as a mixed agonist/antagonist in the mouse abdominal stretch assay.103 More recently, DIPPA has been shown to produce anxiolytic-like effects in the novelty-induced hypophagia and defensive burying test in Wistar Kyoto (WKY) and Sprague Dawley (SD) rats.105 While it is unlikely that these irreversibly binding compounds will become drug candidates, they can serve as useful pharmacologic or biochemical tools to investigate KORs.
Figure 7.
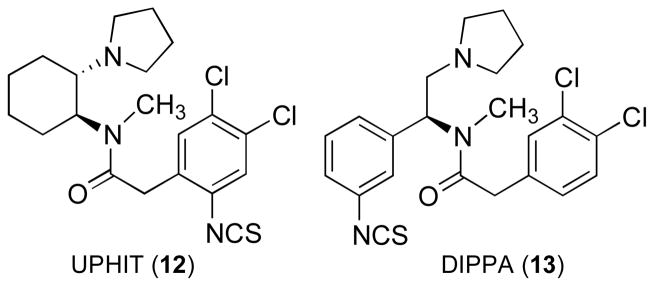
Structures of UPHIT and DIPPA
Figure 8.

Structures of U69,593, bremazocine, DAMGO, and DPDPE
N-Substituted trans-3,4-Dimethy-4-(3-hydroxyphenyl)piperidines
The N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidines (17) (Figure 9) are a novel class of opioid antagonists where the intrinsic antagonist activity is mediated by the trans-3,4-dimethyl orientation of the methyl group on the piperidine ring with the (3R,4R)-enantiomer being the most potent antagonist.106–112 All N-substituted analogues, including the N-methyl analogue (17, R = CH3), were pure antagonists.110,111,113,114 As a strategy for obtaining a potent and selective κ opioid selective N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine analogue, a library of compounds based on the general structure 18 was synthesized.115 Evaluation of this library of compounds in inhibition of opioid receptor binding assays identified N-{(2′S)-[3-(4-hydroxyphenyl)propanamido]-3′-methylbutyl}-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine (JPP6) (19) as a novel KOR selective ligand.116 The key structures featured in 19 that provided the κ potency and selectivity were the isopropyl group attached in the (S)-configuration for R1, a hydrogen substituent for R2, and a p-hydroxyphenylethyl group for R3.117,118
Figure 9.

Structures of 17, 18, and 19
Further modification of lead compound 19 led to the discovery of JDTic, a KOR antagonist, with high potency and selectivity in the sulfur-35 guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) in vitro functional assay.117–119 JDTic had a Ke value of 0.01 nM (pA2 = 10.46) in the inhibition of U69,593-stimulated [35S]GTPγS binding in cloned human KOR.119 In the same test, nor-BNI had a Ke = 0.04 nM at the KOR.120 Similar to nor-BNI, JDTic showed no agonist activity at levels of 10 μM and possessed selectivity for the KOR over the MOR and DOR of 341- and 7930-fold, respectively. Using the same in vitro assays, nor-BNI had 484- and 113-fold selectivity for the KOR relative to the MOR and DOR, respectively.
A structure-activity relationship (SAR) study revealed that the KOR potency and selectivity exhibited by JDTic result from a combination of (a) the (3R,4R) configuration of the trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine core structure; (b) the isoquinoline amino group and 7-hydroxy group held in a rigid orientation by the 1,2,3,4 tetrahydroisoquinoline structure in its 3R attachment to the amide carboxyl; (c) an S-configuration of the 2-methylpropyl group in the spacer between the piperidine ring and the D-hydroxy Tic acyl group; and (d) the lack of a substituent on the amide nitrogen. More details can be found in Thomas and co-workers.119
JDTic was initially evaluated for its ability (a) to antagonize the κ opioid selective agonist enadoline- and the μ-selective opioid agonist sufentanil-induced antinociception in ICR male mice;121 (b) to antagonize the κ selective agonist U50,488-induced antinociception in squirrel monkey using the shock titration procedure;121 (c) to antagonize U50,488-induced diuresis in SD rats;121 and (d) to affect the morphine (20)-induced physical dependence in adult SD rats.122
Pretreatment of mice with s.c. or orally-administered JDTic antagonized effects of enadoline-induced antinociception in the tail-flick test.121 In contrast to nor-BNI, JDTic has no effect on the μ selective agonist sufentanil-induced antinociception in the tail-flick test regardless of the pretreatment time.121 Unlike nor-BNI, orally administered JDTic also antagonized endoline-induced antinociception. JDTic also potently antagonized the U50,488-induced antinociception in squirrel monkeys and was more potent than nor-BNI in antagonizing diuresis induced by U50,488 in rats.121 JDTic, like nor-BNI and GNTI, shows long duration of antagonist activity in these animal tests. Opiate-derived κ antagonist nor-BNI has been reported to potentiate certain overt withdrawal signs in morphine-dependent rats.123 In contrast, the trans-2,4-dimethyl-4-(3-hydroxyphenyl)piperidine derived KOR antagonist JDTic was without significant adverse effect on morphine-induced physical dependence in rats.122 Importantly, JDTic was able to reduce several symptoms of opiate withdrawal and thus shows potential to be useful for alleviating symptoms of opiate withdrawal in humans.122
In the naltrexone-related compounds section, it was pointed out that central administration of nor-BNI and GNTI produced antidepressant-like behavioral effects in the Porsolt rat FST.29,39 When tested in parallel, there appear to be few differences between the effects of nor-BNI and JDTic, although JDTic may be slightly more potent.45 For example, JDTic was found to decrease immobility and increase swimming time at doses as low as 0.3 mg/kg (s.c.) in the rat FST, which is consistent with a profile demonstrated by typical antidepressants such as imipramine (lowest active dose was 5.6 mg/kg).38 Interestingly, these and other KOR antagonists were shown to have anxiolytic-like effects that accompany their antidepressant-like effects.45 Systemic administration of JDTic and nor-BNI increases open arm exploration in the EPM anxiolytic test and decreases conditioned fear in the fear-potentiated startle paradigm in rats, both anxiolytic-like effects.45 This is particularly important since KOR antagonists do not possess the reward-related78 or sedative effects45 that contribute to the abuse liability of benzodiazepines. The fact that the κ opioid antagonist JDTic and nor-BNI had anxiolytic-like effects that accompanied their antidepressant effects is very interesting since acute administration of serotonin selective uptake inhibitors (SSRIs), which are often used to treat depression and anxiety-related disorders in humans, produce anxiogenic effects.45 The ability of KOR antagonists to produce acute anxiolytic-like effects together with antidepressant effects differentiates them from standard antidepressants. This behavioral profile suggests that if KOR antagonists were ultimately developed to treat depressive disorders, they might be better tolerated by patients, particularly at the beginning of a treatment regimen.
Depressive disorders are reported to be the most common co-morbid condition among individuals with cocaine abuse disorders, and studies with depressed cocaine abusers report that they have poorer treatment prognosis.124 Thus, cocaine abusers with depressive symptoms may be especially vulnerable to relapse due to increased cocaine effects. In some studies with imipramine and desipramine, improvement in the reduction of cocaine use was noted.125,126
A rat model that addresses relapse to cocaine-taking behavior has been developed. When cocaine is withheld in rats trained to self-administer the cocaine intravenously, they gradually reduce and eventually cease their self-administration behavior. When they are subsequently stressed with foot-shock or primed with a dose of cocaine, reinstatement (relapse) of cocaine-seeking (self-administration) behavior returns. Pretreatment of the rats with the KOR antagonist JDTic using oral administration significantly reduced the stress-induced reinstatement but did not reduce cocaine-primed reinstatement, demonstrating that JDTic specifically targets stress-related effects that regulate relapse to cocaine use.38
JDTic was evaluated for its effect on alcohol-seeking behavior, alcohol relapse, and maintenance responding for alcohol in P-rats.127 Pretreatment with JDTic was effective at decreasing alcohol-seeking and alcohol relapse in P-rats using the Pavlovian Spontaneous Recovery (PSR) and alcohol deprivation models, respectively. However, JDTic did not decrease alcohol maintenance responding in P-rats.127 In a separate study, both JDTic and nor-BNI were shown to decrease alcohol self-administration in male Wistar rats.128 Moreover, pretreatment with JDTic dose dependently reversed acute alcohol withdrawal-induced anxiety (hangover anxiety) and decreased cue-induced reinstatement of alcohol seeking.128 Surprisingly, pretreatment with JDTic had no effect on stress-induced reinstatement of alcohol seeking in male Wistar rats.128
Nicotine is reported to produce a dose-dependent increase in DYN in the striatum129 and, similar to other substances of abuse, to elevate DA levels in the NAc.130 In addition, as pointed out in the Introduction, activation of KOR/DYN systems produces aversion, dysphoria, anhedonia, depression, and stress responses in humans and rodent models thought to reflect these effects.11,23,24,39,78,131,132 Indeed other studies suggested that chronic nicotine exposure changes KOR levels to enhance negative effects.133 In agreement with these reports, pretreatment with JDTic or nor-BNI blocked spontaneous nicotine withdrawal-induced anxiety-like behavior in the EPM and somatic signs of withdrawal.134 In addition, both JDTic and nor-BNI blocked the expression of mecamylamine-precipitated nicotine withdrawal.134
Disruption in perception and cognition is characteristic of psychiatric conditions such as schizophrenia. The KOR agonist salvinorin A (21) and the non-competitive N-methyl-D-aspartic acid (NMDA) receptor antagonist ketamine (22) are two compounds known to alter perception and cognition in humans. Using the 5-choice serial reaction time task (5CSRTT), a food-motivated test that quantifies attention in rodents, salvinorin A and ketamine were shown to produce the same pattern of disruptive effects characterized by increases in signs often associated with reduced motivation (omission errors) and deficits in processing (elevated latencies to respond correctly).135 Pretreatment with JDTic blocked all salvinorin A effects and some ketamine effects. Since binding and functional studies revealed that ketamine is a less potent full agonist at KOR, these studies provided evidence that KORs might be involved in some of the cognitive abnormalities observed in psychiatric disorders such as schizophrenia. Thus, modulation of KOR function may represent a unique approach for treating core symptoms of schizophrenia. However, it is also possible to conceptualize the effects of salvinorin A on attention as reflecting a prodepressive- or aversive-like state, especially when considering ketamine’s agonist effects at KORs.135 Work from the Chavkin group has shown that at least some of the aversive effects of KOR agonists are mediated via interactions with corticotropin-releasing factor (CRF).51 Indeed, it has now been shown that JDTic blocks the cognitive-disrupting effects of CRF,53 again suggesting interactions between KOR and stress systems that may open the door for new indications for which there are currently no medications.
JDTic also proved useful for obtaining a crystal of the human KOR suitable for X-ray analysis.136 Elucidation of the high-resolution crystal structure of the human KOR in complex with JDTic will be helpful in the design of new κ antagonists (Figure 11). The representation in Figure 11A shows the large receptor pocket and the tight fit of JDTic into the computer-generated KOR binding pocket. There is a large pocket for the 3,4-dimethyl-4-(3-hydroxyphenyl) ring, a deep pocket for one of the methyls of the isopropyl group, and a flat pocket for the 7-hydroxytetrahydroisoquinoline. The binding diagram in Figure 11B shows 22 contact residues that are within 4.5 Å from the JDTic structure. Salt bridges and hydrogen bonds are shown as red and blue dotted lines, respectively. Residues that vary among MOR, DOR, and KOR are shown in cyan. The conserved Asp 138 is in orange. This crystal structure provides detailed insight into the atomic details of molecular recognition and selectivity of the KOR and provides critical information for structure-based design of new KOR antagonists with improved pharmacological profiles.
Figure 11.

Binding of JDTic in human KOR crystal structure
(A) JDTic in the binding pocket of the crystal structure.
(B) Diagram of JDTic interaction in the binding pocket side chain at 4.5 Å cut-off. Reprinted with permission from reference 134. Copyright 2010 Nature.
In addition to the X-ray structure of JDTic bound to the KOR, the X-ray structures for the MOR, DOR, and ORL-1 receptors bound to inhibitors were reported.137–139 Knowledge of how inhibitors of the KOR, MOR, DOR, and ORL-1 interact with these receptors will help in the design of better antidepressants, anxiolytics, analgesics, and drugs to treat addiction that lack the undesirable properties of presently available opioid antagonists.
A number of JDTic analogues have been developed which are potent and selective KOR antagonists. Table 1 lists the [35S]GTPγS binding data for six of the analogues that have the best KOR efficacy and selectivity.140,141 All the analogues have sub-nanomolar efficacy for the KOR and greater than 100-fold and 570-fold selectivity relative to the MOR and DOR, respectively.140,141 Compounds 23a–e were reported to antagonize U50,488-induced antinociception in a tail-withdrawal test using male C57BL/6 mice22 and, like JDTic and nor-BNI, 23b and 23c had a long duration of action. In contrast, the duration of action for 23a, 23d, and 23e lasted for 1 day or less (Table 1).22 The effect of 23f on the antagonism of κ agonist-induced antinociception has not been reported.
Table 1.
Inhibition of Agonist Stimulated [35S]GTPγS Binding by Compounds in Cloned Human MOR, DOR, and KOR
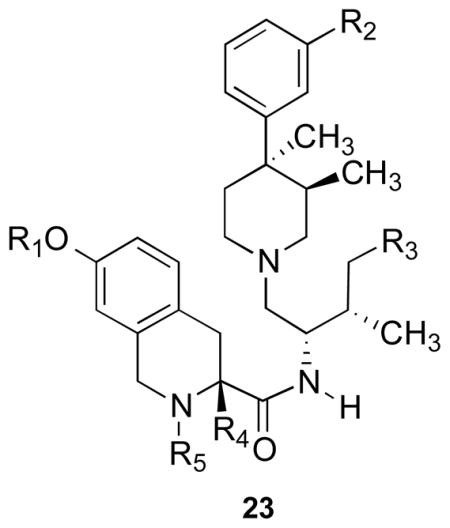
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R4 | R5 | Ke (nM)
|
μ/κ | δ/κ | ||
| DAMGO, μ | DPDPE, δ | U69,593, κ | ||||||||
| 2 (nor-BNI) | 26 | 29 | 0.05 | 520 | 580 | |||||
| 3 (JDTic) | H | OH | H | H | H | 3.41 | 79.3 | 0.01 | 341 | 7930 |
| 23a | CH3 | OH | H | H | H | 51 | 118 | 0.06 | 850 | 1966 |
| 23b | H | OCH3 | H | H | H | 24 | 21.2 | 0.037 | 649 | 573 |
| 23c | H | OH | CH3 | H | H | 3 | 24 | 0.03 | 100 | 800 |
| 23d | H | OH | H | CH3 | H | 3.6 | 854 | 0.03 | 120 | 28500 |
| 23e | H | OH | H | H | CH3 | 210 | 491 | 0.16 | 1313 | 3070 |
| 23f | H | CONH2 | H | H | H | 21 | 480 | 0.12 | 175 | 400 |
Compounds 23c and 23f antagonized U50,488-induced diuresis in rats after i.p. administration.142 In addition, 23c also blocked U50,488-induced diuresis and prevented footshock-induced reinstatement of cocaine-seeking in rats after oral administration.142
N-Substituted 4β-Methyl-5-(3-hydroxyphenyl)morphans
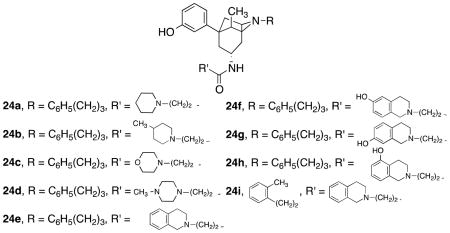
In a preliminary study, N-substituted 4β-methyl-5-(3-hydroxyphenyl)morphans (24a–i) were reported to be opioid receptor pure antagonists with [35S]GTPγS binding properties similar to those of the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine (17) class (Table 2). The morphans 24 can be viewed as conformationally rigid analogues of 17. A preliminary study identified N-[(1R,4S,5S,7R)-5-(3-hydroxyphenyl)-4-methyl-2-(3-phenylpropyl)-2-azabicyclo[3.3.1]non-7-yl]-3-(1-piperidinyl)propanamide [(−)-KAA1] (24a) with a Ke = 0.24 nM at the KOR and 175- and 138-fold selectivity for the κ relative to the MOR and DOR as a potent and selective KOR antagonist.143 In a follow-up study a number of analogues of 24a were synthesized where the R and R′ groups were varied and evaluated in the [35S]GTPγS binding assays.144 Table 2 lists data for nor-BNI, JDTic, 24a, and eight of the analogues that showed good potency and selectivity for the KOR. Even though the data on these compounds are currently limited to only [35S]GTPγS results, the unique structures of these compounds suggest that physiochemical properties and animal behavioral studies need to be conducted to determine if one or more of these KOR antagonists should be developed.
Table 2.
Inhibition of Agonist Stimulated [35S]GTPγS Binding by Compounds in Cloned Human MOR, DOR, and KOR
| |||||
|---|---|---|---|---|---|
| Compd | Ke (nM)
|
μ/κ | δ/κ | ||
| DAMGO, μ | DPDPE, δ | U69,593, κ | |||
| 2 (nor-BNI | 26 | 29 | 0.05 | 520 | 580 |
| 3 (JDTic) | 25 | 76 | 0.02 | 1250 | 3800 |
| 24a (KAA1) | 42 | 33 | 0.24 | 175 | 138 |
| 24b | 176 | 313 | 0.47 | 374 | 666 |
| 24c | 259 | 77 | 2.2 | 118 | 35 |
| 24d | 82 | 186 | 0.87 | 94 | 214 |
| 24e | 48 | 13 | 0.09 | 533 | 144 |
| 24f | 26 | 32 | 0.30 | 87 | 107 |
| 24g | 52 | 62 | 0.09 | 578 | 689 |
| 24h | 4.6 | 25 | 0.07 | 66 | 357 |
| 24i | 28 | 25 | 0.04 | 700 | 625 |
Peptides
During the timeframe 1988–2005, a few dynorphin A peptide KOR-selective antagonists were reported, but none of these early developed peptides has been widely studied in animal behavioral test. Four of these peptides are listed below (see ref. 65 for a review).
The peptide in this group that was studied the most was Ac[Phe1,2,3,Arg4,D-Ala8]dynorphin A-(1–11) amide (arodyn) (28). Pretreatment with 0.3 or 1 nmol, i.c.v., of arodyn (Figure 12) two hours prior to testing antagonized the antinociceptive effects of U50,488. Pretreatment with 0.3 nmol, i.c.v., of arodyn also prevented stress-induced CPP but not cocaine-induced CPP.46
Figure 12.

Structures of opioid peptides 25–28
Zyklophin (29) (Figure 13) is a cyclic dynorphin A analogue that was developed as the first dynorphin A-based antagonist with modification in the C-terminal “address” domain.145 Zyklophin had Ki values of 30.3, 5880, and >10,000 nM at KOR, MOR, and DOR, respectively, using Chinese hamster ovary (CHO) cells stably expressing KOR, MOR, and DOR.145 Radioligands [3H]diprenorphine, [3H]DAMGO, and [3H]DPDPE were used for KOR, MOR, and DOR, respectively. For comparison, dynorphin A-(1–11)NH2 has Ki values of 0.57, 1.85, and 6.18 at KOR, MOR, and DOR, respectively. Zyklophin also antagonized dynorphin A-(1–13)NH2 at KOR in an adenylyl cyclase assay with a KB = 84 nM.
Figure 13.
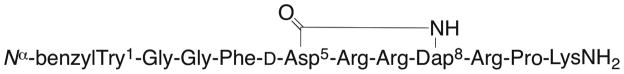
Structure of zyklophin (29)
Pretreatment with zyklophin at doses of 1–3 mg/kg, s.c., or 0.3, 1, or 3 nmol, i.c.v., antagonized the antinociception by the selective KOR agonist U50,488 in C57BL/6J mice tested in the 55 °C warm-water, tail-withdrawal test but had no effect on morphine or SNC-80-mediated antinociception.146 Zyklophin lacked any antinociception effects after either s.c. or i.c.v. administration. These results suggest that zyklophin selectively antagonized KOR in vivo. A pretreatment dose of 3 mg/kg, s.c., of zyklophin also prevented stress-induced reinstatement of cocaine-seeking behavior in a CPP test. Pretreatment with zyklophin at 1 and 3 mg/kg, s.c., had no effect on cocaine-induced reinstatement of CPP.
Saito and co-workers147 isolated the cyclic peptide cyclo(Phe-D-Pro-Phe-Trp) (30) (CJ-15,208) (Figure 14) from the fermentation broth of a fungus, Ctenomyces serratus ATCC15502, and by spectroscopic and chemical means deduced it to be the cyclic tetrapeptide cyclo(Phe-D-Pro-Phe-XXXX), where XXXX was a tryptophan residue with undefined stereochemistry. Seale and co-workers148 established that the Trp had the L-configuration, and thus, the structure of 30 was cyclo(Phe-D-Pro-Phe-Trp) by total synthesis.148 Using a guinea pig brain membrane preparation for binding studies, Saito and co-workers reported that 30 had IC50 values of 47, 260, and 2600 nM at the KOR, MOR, and DOR, respectively. Thus, 30 has some preference for the KOR.
Figure 14.
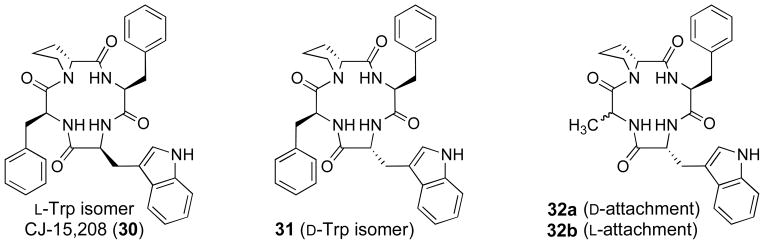
Structures of CJ-15,208 and analogues 31 and 32a–b
Compound 30 was reported to be a weak KOR antagonist by showing that 30 reversed the KOR agonist asimadoline-suppressed twitch response in a rabbit vas deferens assay. These interesting studies led two groups to synthesize 30 and to evaluate the compound in different binding and efficacy assays.149,150 One group,149 using cloned human, KOR, MOR, and DOR expressed in separate cell lines and [3H]diprenorphine as the radioligand, found Ki = 29, 130, and 2000 for inhibition of binding at the KOR, MOR, and DOR, respectively. The second group,150 using membranes from CHO cells stably expressing cloned rat KOR and MOR and mouse DOR and [3H]diprenorphine, [3H]DAMGO, and [3H]DPDPE radioligands for KOR, MOR, and DOR, respectively, reported Ki = 35.4, 619, and 4150 nM for inhibition of KOR, MOR, and DOR binding, respectively. Thus, both groups using different assay conditions confirmed the modest KOR selectivity of 30 reported by Saito and co-workers.147 However, both groups found that 30 had significantly higher potency for MOR over KOR in [35S]GTPγS binding efficacy assay. Dolle and co-workers149 reported IC50 = 440 and 25 nM at KOR and MOR, respectively, using human cell lines stimulated by U50,488 and loperamide for KOR and MOR, respectively, and Ross and co-workers150 reported KB = 62.8 and 10.3 at KOR and MOR, respectively, using rat cell lines and dynorphin and DAMGO for stimulation of KOR and MOR.
Both groups149,150 also synthesized and studied the properties of the D-Trp isomer of 30, cyclo(Phe-D-Pro-Phe-D-Trp) (31) (Figure 14). Dolle and co-workers reported Ki = 3.8, 30, and >1000 nM for inhibition of radioligand binding at KOR, MOR, and DOR, respectively. Ross and co-workers found Ki = 30.6, 259, and 2910 nM at KOR, MOR, and DOR, respectively. Even though Dolle and co-workers found that 31 had higher efficacy at KOR than 30, both groups reported that 31 had modest KOR selectivity similar to that of 30. In contrast, the two groups reported quite different results for the [35S]GTPγS binding assay of 31. Using the [35S]GTPγS binding assay, Dolle and co-workers reported IC50 values of 140 and 21 nM at KOR and MOR, respectively, IC50 values similar to those for 30. Ross and co-workers found KB values of 22.8 for KOR and no antagonist efficacy at 300 nM at MOR in [35S]GTPγS assays. This difference could be due to the fact that Dolle and co-workers used human cell lines, whereas Ross and co-workers used rat cell lines. A recent study has shown that there can be differences between human and rat KORs.151 However, additional studies will be needed to explain the different results. In their study Dolle and co-workers found that cyclo(Ala-D-Pro-Phe-D-Trp) (32a) (Figure 14) was a dual KOR/MOR antagonist with 10-fold greater in vitro functional efficacy (IC50 = 5 nM, KOR; 48 nM, MOR) relative to cyclo(Phe-D-Pro-Phe-D-Trp) (31). Dolle and co-workers also were able to obtain an X-ray crystal structure of 32a, which showed that at least this structure might yield a suitable complex with the human KOR suitable for analysis. In more recent studies Aldrich and co-workers152 reported that cyclo(Ala-D-Pro-Phe-Trp) (32b) was also a dual KOR/MOR antagonist with KB = 2.6 and 7.3 nM at KOR and MOR, respectively, in a [35S]GTPγS assay. However, 32b exhibited antinociceptive potencies (ED50 = 1.49 nM, i.c.v.) in the warm-water, tail-withdrawal test similar to that of 30 which was almost completely blocked by the MOR-selective irreversible antagonist β-funaltrexamine (β-FNA). Thus, even though 32b showed no agonist efficacies in the [35S]GTPγS assay, it was a potent MOR agonist in the warm-water tail withdrawal test.
Ross and co-workers150 evaluated 31 in the warm-water, tail-withdrawal assay using C57B1/6J mice. Pretreatment of 31 with 3-nmol, i.c.v., doses 80 min before agonist administration significantly antagonized the antinociception effects of the KOR-selective agonist U50,488 but not of the MOR and DOR agonist morphine and SNC-80, respectively. The D-Trp isomer of 30 also had about 10% antinociceptive activity at an i.c.v. dose of 10 nmol. Somewhat surprisingly, 30 had an EC50 = 3.71 nmol as an agonist in the antinociceptive test. The results suggested that the compound acted as a combined KOR and MOR receptor agonist. Thus, 30 was a selective antagonist of U50,488-induced antinociception but with weaker potency than that of 31.
Compound 31 was also evaluated in a CPP model of stress-induced reinstatement of cocaine-seeking behavior using conditions similar to those used for zyklophin. Pretreatment (daily for 2 days) with 31 (3 nmol, i.c.v.) prevented stress-induced reinstatement of cocaine place preference. However, pretreatment with 31 (3 mmol, i.c.v.) was ineffective at blocking cocaine-induced reinstatement.
Amidosulfonylbiphenylamines
Using a high throughput screening process, a group from Pfizer PharmaTherapeutics Research and Development identified two compounds with Ki values for inhibition of [3H]diprenorphine of CHO cells expressing KOR and MOR. The first compound 33 (Figure 15) had a Ki = 2.49 nM and 1.60 nM at KOR and MOR, respectively.153 The second compound 34 had Ki = 9 and 21 nM for KOR and MOR, respectively. Compound 33 was a functional agonist in the [35S]GTPγS binding assay, whereas 34 was a functional antagonist. Lead optimizations of 33 and 34 to improve KOR potency and selectivity relative to the MOR as well as improve drug-like properties led to the selection of 2-methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine (PF-04455242) (35) for preclinical development.154 Compound 35 has Ki values of 3 and 65 nM in a radioligand binding assay using CHO cell membranes expressing human KOR and MOR, respectively. Thus, 35 has 21-fold selectivity for κ relative to μ. The compound has negligible affinity for DOR in the human DOR (Ki = >4 μM). Compound 35 was reported to have Ki antagonist values of 1.23 and 10 nM at the KOR and MOR, respectively, using a [35S]GTPγS binding assay. The assay used CHO cell membranes expressing the human KOR and MOR and U50,488 and morphine for stimulation of the KOR and MOR, respectively. When tested at 100 nM, 35 has no agonist activity. The physicochemical properties—P-glucoprotein (P-gp) liability and human liver microsomal clearance—for 35 were improved over those for the lead structures 33 and 34.153 Ex vivo and in vivo radioligand binding studies in rats were used to show that 35 had significantly higher KOR occupancy compared to MOR and to help establish the dose for use in animal behavioral studies.
Figure 15.
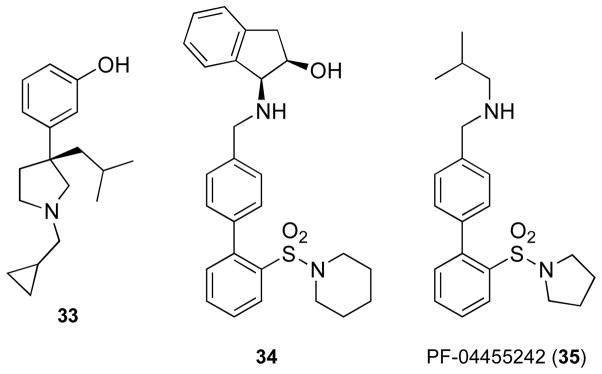
Structures of 33, 34, and PF-04455242 (35)
Using male ICR mice and the procedure reported by D’Amour and Smith,155 35 was shown to antagonize the effects of U50,488 or morphine with AD50 values of 0.67 and 12.03 mg/kg, respectively.153 In a rat tail-flick test 35 had AD50 values of 1.5 and 9.8 mg/kg for antagonist of κ and μ agonist-induced antinociception. Compound 35 at 10 and 32 mg/kg did not have any agonist effect in mice on latency to tail withdrawal. Pretreatment with 35 (1 h, s.c.) reduced immobility in mice with a minimal effective dose of 3.2 mg/kg in the FST and was also active in the social deficit stress assay.154 Pretreatment with 35 attenuated stress-induced reinstatement of cocaine CPP in mice but did not demonstrate rewarding or aversive effect directly.154
Aminobenzyloxyarylamides
In a study directed toward preclinical development of an opioid receptor occupancy tracer, some interesting KOR antagonists were identified.156 Radioligand binding studies using [3H]diprenorphine and membranes from CHO cells expressing human κ and μ or HEK293 cells expressing DORs, respectively, showed that 4-{4-[2-(3,5-dimethylphenyl)pyrrolidin-1-ylmethyl]phenoxy}-3-fluorobenzamide (LY2456302) (36a) (Figure 16) had a Ki value of 0.949 at the KOR and was 24- and 175-fold selective for the KOR relative to the MOR and DOR, respectively.156
Figure 16.

Structures of aminobenzyloxyarylamides 36a–c and 37a–c
In an in vitro inhibition of agonist stimulants, [35S]GTPγS binding study using membranes from CHO cells expressing human KOR and MOR and KOR-selective U69,593 and MOR-selective agonist DAMGO, respectively, for stimulation and using membranes from HEK293 cells expressing the human DOR and selective δ agonist DPDPE, 36a had a Ke = 0.813 with selectivity of 21- and 135-fold, respectively.156 Compounds 36b and 36c (Figure 16) with Ki values of 0.722 and 0.565 nM, respectively, were slightly more potent than 36a. Compound 36b was 36- and 212-fold selective for KOR relative to MOR and DOR, respectively. Compound 36c with a 63- and 373-fold selectivity for KOR over MOR and DOR, respectively, was the most KOR-selective compound in the radioligand binding assay. Compounds 36b and 36c had Kb values at the KOR of 0.632 and 1.57 nM, respectively, which are slightly lower and larger, respectively, than the value for 36a. Compounds 36b and 36c have 132- and 187-fold selectivity for KOR over DOR, respectively, but like 36a have very low (11- and 14-fold selectivity) relative to MOR. Enantiomers 37a–c of 36a–c (Figure 16) showed much lower KOR potency in both the radioligand binding and the [35S]GTPγS assays.
Compound 36c antagonized the antinociceptive activity of 1 mg/kg, s.c., of the KOR-selective agonist U69,593 with an ED50 = 0.24 mg/kg, orally (p.o.). Compound 36c had an ED50 of 30 mg/kg for antagonizing a 10-mg/kg, s.c., dose of morphine. Thus, even though 36c has very low selectivity for KOR relative to MOR in vitro, it is KOR-selective in the rat formalin test.
In a separate study,157 new Kb values were determined for 36a using the [35S]GTPγS functional assay in cloned human MOR, DOR, and KOR. For antagonist assays, DAMGO, DPDPE, and U69,593 were used for stimulation of MOR, DOR, and KOR, respectively. The Kb values found for 36a were 40.1, 2.12, and 264 nM at the MOR, KOR, and DOR, respectively. In vitro binding using high guanosine 5′-diphosphate sodium salt (Na+/GDP), Ki values for 36a were given as 16.4, 122, and 0.597 nM for MOR, DOR, and KOR, respectively. Using a liquid chromatography-tandem mass spectrometry (LC/MS/MS) procedure, 36a was shown to have good KOR occupancy (κ opioid receptor occupancy (KRO)) in rat brain in the striatum. No significant occupancy of either μ or δ receptor (measured in the striatum) at doses up to 30 mg/kg was demonstrated. Thus, the ED50 is greater than 30 mg/kg at μ and δ receptors.
Compound 36a at doses of 1 and 3 mg/kg, p.o., decreased the amount of ethanol consumed in an ethanol-drinking maintenance test using female P rats.157 Using a female P rat progressive ratio responding for ethanol test, pretreatment with 36a at 10 mg/kg, p.o., reduced the number of active lever responses, and the quantity of ethanol consumed and the breakpoint were also reduced. Results from this study were evidence of a reduction in motivation to work for access to ethanol despite their extensive operant history for ethanol.157
Compound 36a was evaluated in the mouse FST.157 Similar to the rat FST, this test measures immobility of mice after pretreatment with test compounds. The less immobility compared to vehicle, the more pronounced the antidepressant-like effect. At 10 mg/kg, p.o., 36a showed a decrease in immobility relative to vehicle but did not show a decrease at 1 and 3 mg/kg, p.o.157
Combined doses of 36a and imipramine were also evaluated in the mouse FST.157 An inactive 3-mg/kg, p.o., dose of 36a combined with an inactive 5-mg/kg, i.p., dose of imipramine was as efficacious as an active 15 mg/kg, i.p., of imipramine alone relative to vehicle, imipramine 5 mg/kg, i.p., alone and 3 mg/kg, p.o. of 36a alone. In addition a 1-mg/kg, p.o., dose of 36a combined with a 5-mg/kg, i.p., dose of imipramine decreased immobility relative to vehicle control, imipramine 5 mg/kg, i.p., alone and 1 mg/kg, p.o., of 36a alone. These results suggest that the decrease in immobility of 36a and imipramine operate through different mechanisms.
Compound 36a was evaluated for its reversal of U69,593-induced and morphine-induced prepulse inhibition (PPI).157 PPI is a neurological phenomenon in which a weaker prestimulus (prepulse) inhibits the reaction of an animal (or human) to a subsequent strong startling stimulus (pulse). Compound 36a reverses the sensory motor gating deficiencies induced by U69,593 and morphine in male Sprague Dawley rats.
8-Azabicyclo[3.2.1]octan-3-yloxybenzamides
Through the process of screening their corporate compound collection, a group at AstraZeneca Pharmaceuticals identified 38 (Figure 17) as a structure for the development of a KOR antagonist. Compound 38 had a Ki = 0.5 nM at the KOR for inhibition of radioligand binding and an IC50 = 77 nM for functional antagonism at the KOR using the [35S]GTPγS assay. Functional antagonism at the MOR and DOR were >30 μM. An SAR study led to 3-[[(3-endo)-8-[(5-methyl-2-thienyl)methyl]-8-azabicyclo[3.2.1]oct-3-yl]oxy]-benzamide (AZ-MTAB) (39) (Figure 17) which had IC50 values of 20, 722, and 8306 nM at KOR, MOR, and DOR, respectively, in the [35S]GTPγS functional assays.
Figure 17.

Structures of 38 and AZ-MTAB
The AstraZeneca compound 39 was compared to the Eli Lilly compound 36a, nor-BNI, and JDTic for their ability to antagonize a 2.5-mg/kg, s.c., dose of U50,488-induced diuresis. All four compounds dose-dependently blocked U50,488-induced diuresis on the initial test (day 0, 5-h urine collection). As previously reported,158 nor-BNI and JDTic (no additional administration) also antagonized a 2.5-mg/kg, s.c., dose of U50,488 on day 7.
The AstraZeneca compound 39 and the Eli Lilly compound 36a were evaluated for anxiolytic-like activity in prenatal-stress rat using the EPM test. This test is based on evidence that the effects of early-life stress can be persistent into adulthood and can lead to mood disorders,159 a test that can be used to model anxiety and depression.160,161 The test begins by stressing female rats for 7 days that comprise the third trimester. The off-spring of these stressed rats were raised to adulthood and then tested for anxiety-like behavior in the EPM. Prenatal stress suppressed percentage time in the open arms and open arm entries. Prenatal stressed rats dosed 2 h prior to testing in the EPM with 39 (30 μmol/kg) and 36a (24 μmol/kg) increased time spent in open arms and open arms entries that were suppressed by prenatal stress.
Clinical Studies
There is some clinical evidence that non-selective KOR antagonists may be useful for treating depression. In a double-blind study the κ antagonist partial μ agonist buprenorphine (40) (Figure 18) induced antidepressant effects in patients with endogenous depression.162 In addition, Kosten and co-workers163 reported that depressive symptoms were significantly decreased with buprenorphine treatment in heroin (41)-addicted patients who were depressed at intake.163 Buprenorphine was also shown to be effective in treatment of refractory depression.164
Figure 18.

Structures of buprenorphine, heroin, and methadone
In initial human studies using opiate-dependent patients, Kosten and co-workers reported that patients maintained on buprenorphine showed a greater reduction in cocaine use than those on methadone165,166 (42). Even though a larger-scale comparison of buprenorphine to methadone did not show significant superiority of buprenorphine to methadone,163 the initial studies were suggestive that reduction in cocaine use seen in the initial studies could be due to the KOR antagonist properties of buprenorphine.
As a way to isolate the κ antagonism properties of buprenorphine without concomitant μ partial agonist properties, combinations of buprenorphine and the μ/κ antagonist naltrexone were studied (see ref. 167 for a review). Rothman and co-workers168 was the first to study this combination. These authors reasoned that the high relapse rate of opiate-dependent patients on withdrawal from methadone was due to the existence of a protracted abstinence syndrome, characterized by a dysphoric mood. They suggested that increases in brain dynorphin levels might be functioning as a homeostatic system to oppose the mood-enhancing and reinforcing effects, an effect opposite to that of morphine as depicted in Figure 2 in the Introduction. These authors hypothesized “that κ overdrive may contribute to the dysphoric mood observed in the protracted abstinence syndrome and that a KOR antagonist might help prevent relapse in abstinent opioid-dependent patients.”169,170 Since there was no clinically available selective KOR antagonist to use, Rothman and co-workers used a combination of buprenorphine with naltrexone to obtain a functional KOR antagonist absent of μ partial agonist activity for their clinical test study. The study showed a substantial reduction in cocaine and opiate use during a three-month trial.168
In a more recent open-label study, Gerra and co-workers171 showed that a combination of buprenorphine and naltrexone significantly enhanced compliance and drug abstinence from heroin and cocaine compared with naltrexone alone in heroin-dependent patients. Similar to the study by Rothman and co-workers,168 these results were postulated to be attributable to less aversive responses using the buprenorphine/naltrexone combination compared with naltrexone alone. Both the Rothman168 and Gerra171 studies support the hypothesis of the therapeutic potential of KOR antagonist in drug addiction.
In a recent review, McCann suggested that a buprenorphine/naltrexone combination might also be useful for treating alcohol, methamphetamine, and nicotine dependence.167 In addition, McCann suggested that a buprenorphine/naltrexone combination might be useful for patients using various combinations of drugs of abuse such as cocaine and alcohol.167 Since buprenorphine has NOP (nociceptin receptor) agonist activity, McCann pointed out that the beneficial effects of the buprenorphine/naltrexone combination might be due in part to this NOP agonist activity.167
At this point no selective KOR antagonist has been tested for treatment of mood disorders or drug abuse. However, Phase I clinical studies have been completed for the Eli Lilly compound 36a, JDTic, and the Pfizer compound 35. Phase I studies with JDTic172 and the Pfizer compound173 (35) were terminated; in the case of JDTic, the reason for termination is still undergoing analysis, and Pfizer has not yet shared the reason for termination of their studies. The Phase I clinical study with 36a was to assess the KOR occupancy after single oral administration of 36a as measured by positron emission tomography (PET) with PET imaging radioligand LY2879788 in healthy subjects with the condition listed as alcohol dependence.174 The last update of the study was May 5, 2011. No new studies for 36a are listed in ClinicalTrials.gov.
Summary and Perspective
The hypothesis that KOR antagonists might have use for the treatment of depression and drug abuse developed from molecular pharmacology and animal behavioral model studies demonstrating that stress or repeated exposure to drugs of abuse can activate the KOR system via the endogenous ligand dynorphin which can be blocked by KOR antagonists.
Numerous studies have shown that activation of the KOR/DYN system plays a major role in stress-induced reinstatement of drug seeking. Moreover, KOR function has a profound influence on behaviors that reflect motivational and emotional states in animal behavioral models. Blockade of KOR receptors with selective KOR antagonists is a new strategy for protecting individuals from relapse to drug addiction. In addition, KOR antagonists may have potential for treatment of psychiatric disorders, especially those that are produced or exacerbated by stress. Studies showed that KOR antagonists attenuate stress-induced reinstatement of cocaine-seeking in rats38 and stress-induced cocaine CPP in mice.40 These studies suggested that activation of the KOR/DYN leads to depression and cocaine relapse, and the decreases of the KOR system may be effective treatments of depression and drug abuse.
It has been difficult to evaluate the hypothesis that KOR antagonists would have therapeutic effects in clinical efficacy trials because of limitations with the currently available compound, as described above. However, during the last few years, there has been increased interest in the KOR/DYN system, and more effort has been directed toward the development of improved selective KOR antagonists. The studies have provided information showing that various scaffolds can lead to potent and selective KOR antagonists. In addition, studies with these compounds have provided additional information concerning their activities in various animal behavioral models of mood disorders and drug abuse. This information is reviewed in this perspective along with the encouraging results that three KOR antagonists—JDTic, the Pfizer compound 35, and the Eli Lilly compound 36a—have reached Phase 1 clinical studies. In addition, studies using a buprenorphine/naltrexone combination to generate a functional KOR antagonist have given promising results in patients addicted to cocaine. It was recently reported at a conference that another propriety formulation that reveals the KOR antagonist effects of buprenorphine has antidepressive effects in humans.175 These studies are broadly consistent with preclinical studies in laboratory animals suggesting that KOR antagonists have untapped potential as therapeutic agents despite the current lack of a first-in-class lead molecule.
Even though much has been learned from studies with the new KOR antagonists, considerable research will be required before the details of the mechanisms giving these effects are fully understood. More information about the part that CRF, P-38 MAPK, serotonin, norepinephrine, and the hypocretin/orexin system will provide new insights into how the KOR regulates mood states and drug addiction. In addition, research to determine how CREB regulates stress-induced reinstatement of drug seeking, depression, and sensitivity to both award and aversion needs to be pursued, particularly to its modulation of DYN and KOR activation.
Even though a few better drug-like KOR antagonists than nor-BNI (2) and GNTI (7) have been developed, new KOR antagonists possessing novel scaffolds are still needed both as potential pharmacotherapies and as pharmacological tools to better understand the KOR/DYN system. Considering that most of the studies have been conducted with prototypical KOR antagonists, all of which have long (or irreversible) durations of action despite apparent chemical dissimilarities, the availability of non-peptidergic short-acting compounds will provide crucial insight into the intracellular consequences of these agents that might lead to more effective high-throughput screening (HTS) procedures. In this regard, a recent study involving a broad range of KOR-blocking agents found a positive correlation between duration of action and JNK activation.22 While not yet broadly implemented, it is conceivable that secondary screens could be utilized to quantify JNK activation in early hits from HTS procedures to identify optimal chemical scaffolds that serve as the basis for short-acting KOR antagonists. One potential complication is that the biased agonist effects of KOR antagonists have been most thoroughly characterized using rodents, and there is evidence of species differences in KOR function.151 A long duration of action may ultimately prove to be a desirable characteristic once a drug has been proven to be both safe and efficacious; indeed, the development of sustained-action formulations is common for psychiatric medications (e.g., Buprenorphine SR™).
The elucidation of the high-resolution crystal structure of the human KOR in complex with JDTic will be helpful in the design of new κ antagonists (Figure 11). This crystal structure provides detailed insight into the atomic details of molecular recognition and selectivity of the KOR and provides critical information for structure-based design of new KOR antagonists with improved pharmacological profiles. Regardless, the process of developing of KOR antagonists into medications for neuropsychiatric illness has strong origins in preclinical discoveries of how the brain encodes states such as stress, anhedonia, and dysphoria.
Figure 10.

Structures of morphine, Salvinorin A, and ketamine
Acknowledgments
This work was in part supported by the National Institute on Mental Health grant MH063266 (to WAC) and the National Institute on Drug Abuse grant DA09045 (to FIC) and DA021002 (to FIC). The authors thank Hernán A. Navarro and Scott P. Runyon for helpful suggestions and to S. Wayne Mascarella for Figure 11.
ABBREVIATIONS
- KOR
κ opioid receptor
- GPCRs
G-protein-coupled receptors
- VTA
ventral tegmental area
- NAc
nucleus accumbens
- PFC
prefrontal cortex
- HPC
hippocampus
- ST
striatum
- AMYG
amygdala
- LC
locus coeruleus
- SN
substantia nigra
- DRN
dorsal raphe nucleus
- HL
hypothalamus
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- JNK
c-Jun amino-terminal kinase
- MOR
μ opioid receptor
- DYN
dynorphine
- DA
dopamine
- cAMP
cyclic adenosine monophosphate
- CREB
cAMP response element-binding protein
- FST
forced-swim test
- CPP
conditioned place preference
- nor-BNI
norbinaltorphimine
- JDTic
(3R)-1,2,3,4-tetrahydro-7-hydroxy-N-[(1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl]-3-isoquinolinecarboxamide
- TENA
1,8-bis(β-naltrexamino)-3,6-dioxaoctane
- GNTI
5′-guanidonaltrindole
- i.c.v
intracerebroventricularly
- ANTI
5′-acetamidinoethylnaltrindole
- ICSS
intracranial self-stimulation
- FSS
forced-swim stress
- s.c
subcutaneous
- EPM
elevated plus maze
- i.p
intraperitoneal
- THC
Δ9-tetracannabinoid
- (−)-UPHIT
(1S,2S)-trans-2-isothiocyanato-4,5-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide)
- DIPPA
2-(3,4-dichlorophenyl)-N-methyl-N-[(1S)-1-(3-isothiocyanatophenyl)-2-(1-pyrrolidinyl)ethyl]acetamide
- DAMGO
[D-Ala2, NMe-Phe4, Gly-ol5]enkephalin
- DPDPE
[D-Pen2,5]Enkephalin, [D-Pen2,D-Pen5]enkephalin
- WKY
Wistar Kyoto
- SD
Sprague Dawley
- JPP6
N-{(2′S)-[3-(4-hydroxyphenyl)propanamido]-3′-methylbutyl}-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine
- [35S]GTPγS
sulfur-35 guanosine-5′-O-(3-thio)triphosphate
- SAR
structure-activity relationship
- SSRI
serotonin selective uptake inhibitor
- PSR
Pavlovian Spontaneous Recovery
- NMDA
N-methyl-D-aspartic acid
- CRF
corticotropin-releasing factor
- 5CSRTT
5-choice serial reaction time task
- KAAI
N-[(1R,4S,5S,7R)-5-(3-hydroxyphenyl)-4-methyl-2-(3-phenylpropyl)-2-azabicyclo[3.3.1]non-7-yl]-3-(1-piperidinyl)propanamide
- DOR
δ opioid receptor
- CHO
Chinese hamster ovary
- P-gp
P-glucoprotein
- p.o
by mouth
- Na+/GDP
guanosine 5′-diphosphate sodium salt
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- KRO
κ opioid receptor occupancy
- PPI
prepulse inhibition
- PET
positron emission tomography
- NOP
nociceptin receptor
Biographies
F. Ivy Carroll, Ph.D. received the B.S. degree in chemistry from Auburn University in 1957 and was awarded the Ph.D. in chemistry by the University of North Carolina at Chapel Hill in 1961. He joined the research staff of the Research Triangle Institute as a Research Chemist and rose steadily to the position of Vice President of the Chemistry and Life Sciences Group, a position he held from 1996–2001. Dr. Carroll also served as Director of the Center for Organic and Medicinal Chemistry from 1975–2007. He is presently Distinguished Fellow for Medicinal Chemistry. Dr. Carroll has varied research interests, but since 1990, a major thrust of his research efforts involved development of pharmacotherapies for substance abuse (cocaine, nicotine, methamphetamine, opioids, and ethanol) and other CNS disorders.
William A. Carlezon, Jr., Ph.D. is a Professor of Psychiatry at Harvard Medical School, Director of the Behavioral Genetics Laboratory at McLean Hospital, and Editor-in-Chief of Neuropsychopharmacology. His research focuses on nature/nurture issues as they relate to the brain. He and his team study the basic processes by which the brain develops and is modified in response to experience, including exposure to stress, drugs, trauma, or toxins. His work is applicable to a variety of psychiatric and neurologic conditions, including stress-related disorders, addiction, and autism.
References
- 1.Pert CB, Snyder SH. Opiate receptor: Demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- 2.Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic (3H) Etorphine to rat-brain homogenate. Proc Natl Acad Sci US A. 1973;70:1947–1949. doi: 10.1073/pnas.70.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terenius L. Characteristics of the “receptor” for narcotic analgesics in synaptic plasma membrane fraction from rat brain. Acta Pharmacol Toxicol. 1973;33:377–384. doi: 10.1111/j.1600-0773.1973.tb01539.x. [DOI] [PubMed] [Google Scholar]
- 4.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 5.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The δ-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci US A. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 7.Minami M, Toya T, Katao Y, Maekawa K, Nakamura S, Onogi T, Kaneko S, Satoh M. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993;329:291–295. doi: 10.1016/0014-5793(93)80240-u. [DOI] [PubMed] [Google Scholar]
- 8.Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, Mattei MG, Charron G, Bloch B, Kieffer B. κ-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci US A. 1995;92:7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng F, Xie GX, Thompson RC, Mansour A, Goldstein A, Watson SJ, Akil H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc Natl Acad Sci US A. 1993;90:9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreibich A, Reyes BA, Curtis AL, Ecke L, Chavkin C, Van Bockstaele EJ, Valentino RJ. Presynaptic inhibition of diverse afferents to the locus ceruleus by kappa-opiate receptors: a novel mechanism for regulating the central norepinephrine system. J Neurosci. 2008;28:6516–6525. doi: 10.1523/JNEUROSCI.0390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- 12.Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, Hamon M International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- 13.Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Ann Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 14.Schwarzer C. 30 years of dynorphins--new insights on their functions in neuropsychiatric diseases. Pharmacol Ther. 2009;123:353–370. doi: 10.1016/j.pharmthera.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 16.Piros ET, Hales TG, Evans CJ. Functional analysis of cloned opioid receptors in transfected cell lines. Neurochem Res. 1996;21:1277–1285. doi: 10.1007/BF02532368. [DOI] [PubMed] [Google Scholar]
- 17.Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ. Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. 2005;280:27662–27669. doi: 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kam AY, Chan AS, Wong YH. Kappa-opioid receptor signals through Src and focal adhesion kinase to stimulate c-Jun N-terminal kinases in transfected COS-7 cells and human monocytic THP-1 cells. J Pharmacol Exp Ther. 2004;310:301–310. doi: 10.1124/jpet.104.065078. [DOI] [PubMed] [Google Scholar]
- 21.Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melief EJ, Miyatake M, Carroll FI, Beguin C, Carlezon WA, Jr, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Duration of Action of a Broad Range of Selective kappa-Opioid Receptor Antagonists Is Positively Correlated with c-Jun N-Terminal Kinase-1 Activation. Mol Pharmacol. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 24.Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- 25.Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spanagel R, Herz A, Shippinberg TA. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci US A. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maisonneuve IM, Archer S, Glick SD. U50,488, a kappa opioid receptor agonist, attenuates cocaine-induced increases in extracellular dopamine in the nucleus accumbens of rats. Neurosci Lett. 1994;181:57–60. doi: 10.1016/0304-3940(94)90559-2. [DOI] [PubMed] [Google Scholar]
- 28.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2009 doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlezon WA, Jr, Beguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther. 2009;123:334–343. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 33.Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci. 2002;22:10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 36.Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 37.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- 39.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C. Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology. 2006;31:1241–1248. doi: 10.1038/sj.npp.1300872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muschamp JW, Van’t Veer A, Carlezon WA., Jr Tracking down the molecular substrates of stress: new roles for p38alpha MAPK and kappa-opioid receptors. Neuron. 2011;71:383–385. doi: 10.1016/j.neuron.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muschamp JW, Van’t Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, Meloni EG, Carlezon WA., Jr Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31:3095–3103. doi: 10.1523/JNEUROSCI.5973-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, Lucki I. Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology. 2010;35:752–763. doi: 10.1038/npp.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-Like Effects of κ-Opioid Receptor Antagonists in Models of Unlearned and Learned Fear in Rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 46.Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101(Suppl 1):23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 49.Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA., Jr Kappa Opioid Receptor Signaling in the Basolateral Amygdala Regulates Conditioned Fear Anxiety in Rats. Biol Psychiatry. 2011;70:425–433. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruchas MR, Land BB, Lemos JC, Chavkin C. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One. 2009;4:e8528. doi: 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van’t Veer A, Yano JM, Carroll FI, Cohen BM, Carlezon WA., Jr Corticotropin-Releasing Factor (CRF)-Induced Disruption of Attention in Rats Is Blocked by the kappa-Opioid Receptor Antagonist JDTic. Neuropsychopharmacology. 2012;37:2809–2816. doi: 10.1038/npp.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koob GF. Stress, corticotropin-releasing factor, and drug addiction. Ann N Y Acad Sci. 1999;897:27–45. doi: 10.1111/j.1749-6632.1999.tb07876.x. [DOI] [PubMed] [Google Scholar]
- 55.Koob GF. Cocaine reward and dopamine receptors: love at first site. Arch Gen Psychiatry. 1999;56:1107–1108. doi: 10.1001/archpsyc.56.12.1107. [DOI] [PubMed] [Google Scholar]
- 56.Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Husbands SM. Kappa-opioid receptor ligands. Expert Opin Ther Patents. 2004;14:1725–1741. [Google Scholar]
- 59.Berrocoso E, Sanchez-Blazquez P, Garzon J, Mico JA. Opiates as antidepressants. Curr Pharm Des. 2009;15:1612–1622. doi: 10.2174/138161209788168100. [DOI] [PubMed] [Google Scholar]
- 60.Aguilar MA, Rodriguez-Arias M, Minarro J. Neurobiological mechanisms of the reinstatement of drug-conditioned place preference. Brain Res Rev. 2009;59:253–277. doi: 10.1016/j.brainresrev.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 61.Yahyavi-Firouz-Abadi N, See RE. Anti-relapse medications: preclinical models for drug addiction treatment. Pharmacol Ther. 2009;124:235–247. doi: 10.1016/j.pharmthera.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCurdy CR, Prisinzano TE. Opioid Receptor Ligands. In: Abraham DJ, Rotella DP, editors. Burger’s Medicinal Chemistry, Drug Discovery and Development. 7. Vol. 8. John Wiley & Sons, Inc; Hoboken, NJ: 2010. pp. 569–735. CNS Disorders. [Google Scholar]
- 66.Erez M, Takemori AE, Portoghese PS. Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. Evidence for bridging between proximal recognition sites. J Med Chem. 1982;25:847–849. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 67.Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor-binaltorphimine, potent and selective κ-opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 68.Portoghese PS, Lipkowski AW, Takemori AE. Bimorphinans as highly selective, potent kappa opioid receptor antagonists. J Med Chem. 1987;30:238–239. doi: 10.1021/jm00385a002. [DOI] [PubMed] [Google Scholar]
- 69.Takemori AE, Ho BY, Naeseth JS, Portoghese PS. Nor-binaltorphimine, a highly selective kappa-opioid antagonist in analgesia and receptor binding assays. J Pharmacol Exp Ther. 1988;246:255–258. [PubMed] [Google Scholar]
- 70.Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch Int Pharmacodyn Ther. 1992;316:30–42. [PubMed] [Google Scholar]
- 71.Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- 72.Broadbear JH, Negus SS, Butelman E, de Costa BR, Woods JH. Differential effects of systemically administered nor-binaltorphimine (nor-BNI) on κ-opioid agonists in the mouse writhing assay. Psychopharmacology. 1994;115:311–319. doi: 10.1007/BF02245071. [DOI] [PubMed] [Google Scholar]
- 73.Jones RM, Portoghese PS. 5′-Guanidinonaltrindole, a highly selective and potent kappa-opioid receptor antagonist. Eur J Pharmacol. 2000;396:49–52. doi: 10.1016/s0014-2999(00)00208-9. [DOI] [PubMed] [Google Scholar]
- 74.Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Mutational evidence for a common kappa antagonist binding pocket in the wild-type kappa and mutant mu[K303E] opioid receptors. J Med Chem. 1998;41:4911–4914. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- 75.Stevens WC, Jr, Jones RM, Subramanian G, Metzger TG, Ferguson DM, Portoghese PS. Potent and selective indolomorphinan antagonists of the kappa-opioid receptor. J Med Chem. 2000;43:2759–2769. doi: 10.1021/jm0000665. [DOI] [PubMed] [Google Scholar]
- 76.Negus SS, Mello NK, Linsenmayer DC, Jones RM, Portoghese PS. Kappa opioid antagonist effects of the novel kappa antagonist 5′-guanidinonaltrindole (GNTI) in an assay of schedule-controlled behavior in rhesus monkeys. Psychopharmacology (Berl) 2002;163:412–419. doi: 10.1007/s00213-002-1038-x. [DOI] [PubMed] [Google Scholar]
- 77.Potter DN, Damez-Werno D, Carlezon WA, Jr, Cohen BM, Chartoff EH. Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry. 2011;70:744–753. doi: 10.1016/j.biopsych.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA., Jr Effects of kappa-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology (Berl) 2004;172:463–470. doi: 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- 79.Leshner AI. Addiction is a brain disease, and it matters. Science. 1997;278:45–47. doi: 10.1126/science.278.5335.45. [DOI] [PubMed] [Google Scholar]
- 80.de Wit H, Stewart J. Reinstatement of cocaine-reinforced responding in the rat. Psychopharmacology (Berl) 1981;75:134–143. doi: 10.1007/BF00432175. [DOI] [PubMed] [Google Scholar]
- 81.de Wit H, Stewart J. Drug reinstatement of heroin-reinforced responding in the rat. Psychopharmacology (Berl) 1983;79:29–31. doi: 10.1007/BF00433012. [DOI] [PubMed] [Google Scholar]
- 82.Gerber GJ, Stretch R. Drug-induced reinstatement of extinguished self-administration behavior in monkeys. Pharmacol Biochem Behav. 1975;3:1055–1061. doi: 10.1016/0091-3057(75)90016-7. [DOI] [PubMed] [Google Scholar]
- 83.Stretch R, Gerber GJ. Drug-induced reinstatement of amphetamine self-administration behaviour in monkeys. Can J Psychol. 1973;27:168–177. doi: 10.1037/h0082466. [DOI] [PubMed] [Google Scholar]
- 84.Bossert JM, Ghitza UE, Lu L, Epstein DH, Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: an update and clinical implications. Eur J Pharmacol. 2005;526:36–50. doi: 10.1016/j.ejphar.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 85.Epstein DH, Preston KL, Stewart J, Shaham Y. Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl) 2006;189:1–16. doi: 10.1007/s00213-006-0529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 87.Szmuszkovicz J. U-50,488 and the kappa receptor: a personalized account covering the period 1973 to 1990. Prog Drug Res. 1999;52:167–195. doi: 10.1007/978-3-0348-8730-4_4. [DOI] [PubMed] [Google Scholar]
- 88.Williams KL, Woods JH. Oral ethanol-reinforced responding in rhesus monkeys: effects of opioid antagonists selective for the mu-, kappa-, or delta-receptor. Alcohol Clin Exp Res. 1998;22:1634–1639. doi: 10.1111/j.1530-0277.1998.tb03960.x. [DOI] [PubMed] [Google Scholar]
- 89.Holter SM, Henniger MS, Lipkowski AW, Spanagel R. Kappa-opioid receptors and relapse-like drinking in long-term ethanol-experienced rats. Psychopharmacology (Berl) 2000;153:93–102. doi: 10.1007/s002130000601. [DOI] [PubMed] [Google Scholar]
- 90.Mitchell JM, Liang MT, Fields HL. A single injection of the kappa opioid antagonist norbinaltorphimine increases ethanol consumption in rats. Psychopharmacology (Berl) 2005;182:384–392. doi: 10.1007/s00213-005-0067-7. [DOI] [PubMed] [Google Scholar]
- 91.Kovacs KM, Szakall I, O’Brien D, Wang R, Vinod KY, Saito M, Simonin F, Kieffer BL, Vadasz C. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- 92.Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2007;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berger AL, Williams AM, McGinnis MM, Walker BM. Affective cue-induced escalation of alcohol self-administration and increased 22-kHz ultrasonic vocalizations during alcohol withdrawal: role of kappa-opioid receptors. Neuropsychopharmacology. 2012 doi: 10.1038/sj.npp.1301628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology (Berl) 2010;210:199–209. doi: 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- 95.Walker BM, Zorrilla EP, Koob GF. Systemic kappa-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol. 2011;16:116–119. doi: 10.1111/j.1369-1600.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valdez GR, Harshberger E. Kappa opioid regulation of anxiety-like behavior during acute ethanol withdrawal. Pharmacol Biochem Behav. 2012;102:44–47. doi: 10.1016/j.pbb.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C. Stress-induced activation of the dynorphin/kappa-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci. 2012;32:1488–1495. doi: 10.1523/JNEUROSCI.2980-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jackson KJ, McLaughlin JP, Carroll FI, Damaj MI. Effects of the kappa opioid receptor antagonist, norbinaltorphimine, on stress and drug-induced reinstatement of nicotine conditioned place preference. Psychopharmacology (Berl) 2012 doi: 10.1007/s00213–00012-02716-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okada-Ogawa A, Kurose M, Meng ID. Attenuation of cannabinoid-induced inhibition of medullary dorsal horn neurons by a kappa-opioid receptor antagonist. Brain Res. 2010;1359:81–89. doi: 10.1016/j.brainres.2010.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Welch SP. Blockade of cannabinoid-induced antinociception by norbinaltorphimine, but not N,N-diallyl-tyrosine-Aib-phenylalanine-leucine, ICI 174,864 or naloxone in mice. J Pharmacol Exp Ther. 1993;265:633–640. [PubMed] [Google Scholar]
- 101.de Costa BR, Band L, Rothman RB, Jacobson AE, Bykov V, Pert A, Rice KC. Synthesis of an affinity ligand (‘UPHIT’) for in vivo acylation of the kappa-opioid receptor. FEBS Lett. 1989;249:178–182. doi: 10.1016/0014-5793(89)80619-2. [DOI] [PubMed] [Google Scholar]
- 102.Chang AC, Takemori AE, Ojala WH, Gleason WB, Portoghese PS. κ Opioid receptor selective affinity labels: Electrophilic benzeneacetamides as κ-selective opioid antagonists. J Med Chem. 1994;37:4490–4498. doi: 10.1021/jm00052a008. [DOI] [PubMed] [Google Scholar]
- 103.Chang AC, Takemori AE, Portoghese PS. 2-(3,4-Dichlorophenyl)-N-methyl-N-[(1S)-1-(3-isothiocyanatophenyl)-2-(1-pyrrolidi nyl)ethyl]acetamide: an opioid receptor affinity label that produces selective and long-lasting kappa antagonism in mice. J Med Chem. 1994;37:1547–1549. doi: 10.1021/jm00037a001. [DOI] [PubMed] [Google Scholar]
- 104.Horan P, de Costa BR, Rice KC, Porreca F. Differential antagonism of U69,593- and bremazocine-induced antinociception by (−)-UPHIT: evidence of kappa opioid receptor multiplicity in mice. J Pharmacol Exp Ther. 1991;257:1154–1161. [PubMed] [Google Scholar]
- 105.Carr GV, Lucki I. Comparison of the kappa-opioid receptor antagonist DIPPA in tests of anxiety-like behavior between Wistar Kyoto and Sprague Dawley rats. Psychopharmacology (Berl) 2010;210:295–302. doi: 10.1007/s00213-010-1832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zimmerman DM, Leander JD. Selective opioid receptor agonists and antagonists: Research tools and potential therapeutic agents. J Med Chem. 1990;33:895–902. doi: 10.1021/jm00165a002. [DOI] [PubMed] [Google Scholar]
- 107.Mitch CH, Leander JD, Mendelsohn LG, Shaw WN, Wong DT, Cantrell BE, Johnson BG, Reel JK, Snoddy JD, Takemori AE, Zimmerman DM. 3,4-Dimethyl-4-(3-hydroxyphenyl)piperidines: Opioid antagonists with potent anorectant activity. J Med Chem. 1993;36:2842–2850. doi: 10.1021/jm00072a002. [DOI] [PubMed] [Google Scholar]
- 108.Mitch CH, Zimmerman DM, Snoddy JD, Reel JK, Cantrell BE. Synthesis and Absolute Configuration of LY255582, a potent opioid antagonist. J Org Chem. 1991;56:1660–1663. [Google Scholar]
- 109.Zimmerman DM, Gidda JS, Cantrell BE, Schoepp DD, Johnson BG, Leander JD. Discovery of a potent, peripherally selective trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist for the treatment of gastrointestinal motility disorders. J Med Chem. 1994;37:2262–2265. doi: 10.1021/jm00041a003. [DOI] [PubMed] [Google Scholar]
- 110.Zimmerman DM, Leander JD, Cantrell BE, Reel JK, Snoddy J, Mendelsohn LG, Johnson BG, Mitch CH. Structure-activity relationships of the trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine antagonists for μ and κ opioid receptors. J Med Chem. 1993;36:2833–2841. doi: 10.1021/jm00072a001. [DOI] [PubMed] [Google Scholar]
- 111.Zimmerman DM, Smits S, Nickander R. Further investigation of novel 3-methyl-4-phenylpiperidine narcotic antagonists. Proceedings of the 40th Annual Scientific Meeting of the Committee on Problems of Drug Dependence; Rockville, MD, USA: National Institute on Drug Abuse; 1978. pp. 237–247. [Google Scholar]
- 112.Zimmerman DM, Smits SE, Hynes MD, Cantrell BE, Leander JD, Mendelsohn LG, Nickander R. Picenadol. Drug Alcohol Depend. 1985;14:381–401. doi: 10.1016/0376-8716(85)90069-9. [DOI] [PubMed] [Google Scholar]
- 113.Zimmerman DM, Nickander R, Horng JS, Wong DT. New structural concepts for narcotic antagonists defined in a 4-phenylpiperidine series. Nature. 1978;275:332–334. doi: 10.1038/275332a0. [DOI] [PubMed] [Google Scholar]
- 114.Zimmerman DM, Smits SE, Hynes MD, Cantrell BE, Reamer M, Nickander R. Structural requirements for affinity and intrinsic activity at the opiate receptor defined in 4-phenylpiperidine and related series. In: Harris LS, editor. Problems of Drug Dependence, 1981: Proceedings of 43rd Annual Scientific Meeting of the Committee on Problems of Drug Dependence, Inc. National Institute on Drug Abuse; Rockville, MD, USA: 1982. pp. 112–118. NIDA Research Monograph 41. [PubMed] [Google Scholar]
- 115.Thomas JB, Mascarella SW, Rothman RB, Partilla JS, Xu H, McCullough KB, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Investigation of the N-substituent conformation governing potency and mu receptor subtype-selectivity in (+)-(3R, 4R)-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonists. J Med Chem. 1998;41:1980–1990. doi: 10.1021/jm980063g. [DOI] [PubMed] [Google Scholar]
- 116.Thomas JB, Fall MJ, Cooper JB, Rothman RB, Mascarella SW, Xu H, Partilla JS, Dersch CM, McCullough KB, Cantrell BE, Zimmerman DM, Carroll FI. Identification of an opioid κ receptor subtype-selective N-substituent for (+)-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine. J Med Chem. 1998;41:5188–5197. doi: 10.1021/jm980511k. [DOI] [PubMed] [Google Scholar]
- 117.Carroll FI. 2002 Medicinal Chemistry Division Award address: monoamine transporters and opioid receptors. Targets for addiction therapy. J Med Chem. 2003;46:1775–1794. doi: 10.1021/jm030092d. [DOI] [PubMed] [Google Scholar]
- 118.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J Med Chem. 2001;44:2687–2690. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 119.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 120.Black SL, Jones AR, Brandt W, Lewis JW, Husbands SM. The role of the side chain in determining relative δ- and κ-affinity in C5′-substituted analogues of naltrindole. J Med Chem. 2003;46:314–317. doi: 10.1021/jm020997b. [DOI] [PubMed] [Google Scholar]
- 121.Carroll FI, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: A novel κ-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 122.Carroll FI, Harris LS, Aceto MD. Effects of JDTic, a selective kappa-opioid receptor antagonist, on the development and expression of physical dependence on morphine using a rat continuous-infusion model. Eur J Pharmacol. 2005;524:89–94. doi: 10.1016/j.ejphar.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 123.Maldonado R, Negus S, Koob GF. Precipitation of morphine withdrawal syndrome in rats by administration of mu-, delta- and kappa-selective opioid antagonists. Neuropharmacology. 1992;31:1231–1241. doi: 10.1016/0028-3908(92)90051-p. [DOI] [PubMed] [Google Scholar]
- 124.Brown RA, Monti PM, Myers MG, Martin RA, Rivinus T, Dubreuil ME, Rohsenow DJ. Depression among cocaine abusers in treatment: relation to cocaine and alcohol use and treatment outcome. Am J Psychiatry. 1998;155:220–225. doi: 10.1176/ajp.155.2.220. [DOI] [PubMed] [Google Scholar]
- 125.Nunes EV, McGrath PJ, Quitkin FM, Ocepek-Welikson K, Stewart JW, Koenig T, Wager S, Klein DF. Imipramine treatment of cocaine abuse: possible boundaries of efficacy. Drug Alcohol Depend. 1995;39:185–195. doi: 10.1016/0376-8716(95)01161-6. [DOI] [PubMed] [Google Scholar]
- 126.Carroll KM, Nich C, Rounsaville BJ. Differential symptom reduction in depressed cocaine abusers treated with psychotherapy and pharmacotherapy. J Nerv Ment Dis. 1995;183:251–259. doi: 10.1097/00005053-199504000-00012. [DOI] [PubMed] [Google Scholar]
- 127.Deehan GA, Jr, McKinzie DL, Carroll FI, McBride WJ, Rodd ZA. The long-lasting effects of JDTic, a kappa opioid receptor antagonist, on the expression of ethanol-seeking behavior and the relapse drinking of female alcohol-preferring (P) rats. Pharmacol Biochem Behav. 2012;101:581–587. doi: 10.1016/j.pbb.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schank JR, Goldstein AL, Rowe KE, King CE, Marusich JA, Wiley JL, Carroll FI, Thorsell A, Heilig M. The kappa opioid receptor antagonist JDTic attenuates alcohol seeking and withdrawal anxiety. Addict Biol. 2012;17:634–647. doi: 10.1111/j.1369-1600.2012.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Isola R, Zhang H, Tejwani GA, Neff NH, Hadjiconstantinou M. Acute nicotine changes dynorphin and prodynorphin mRNA in the striatum. Psychopharmacology (Berl) 2009;201:507–516. doi: 10.1007/s00213-008-1315-4. [DOI] [PubMed] [Google Scholar]
- 130.DiChiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci US A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carlezon WA, Jr, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Blockade of cannabinoid-induced antinociception by norbinaltorphimine, but not N,N-diallyl-tyrosine-Aib-phenylalanine-leucine, ICI 174,864 or naloxone in mice. J Pharmacol Exp Ther. 2006;316:440–447. [PubMed] [Google Scholar]
- 132.Zimmer A, Valjent E, Konig M, Zimmer AM, Robledo P, Hahn H, Valverde O, Maldonado R. Absence of h-9-tetrahydrocannabinol dysphoric effects in dynorphin-deficient mice. J Neurosci. 2001;21:9499–9505. doi: 10.1523/JNEUROSCI.21-23-09499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tejeda HA, Natividad LA, Torres OV, Castaneda ED, O’Dell LE. The behavioral and neurochemical effects produced by kappa-opioid receptor stimulation are diminished in nicotine-dependent adolescent versus adult rats. Soc Neurosci Abstr. 2008;360:14. [Google Scholar]
- 134.Jackson KJ, Carroll FI, Negus SS, Damaj MI. Effect of the selective kappa-opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology (Berl) 2010:285–294. doi: 10.1007/s00213-010-1803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nemeth CL, Paine TA, Rittiner JE, Beguin C, Carroll FI, Roth BL, Cohen BM, Carlezon WA., Jr Role of kappa-opioid receptors in the effects of salvinorin A and ketamine on attention in rats. Psychopharmacology (Berl) 2010;210:263–274. doi: 10.1007/s00213-010-1834-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cai TB, Zou Z, Thomas JB, Brieaddy L, Navarro HA, Carroll FI. Synthesis and in vitro opioid receptor functional antagonism of analogues of the selective kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) J Med Chem. 2008;51:1849–1860. doi: 10.1021/jm701344b. [DOI] [PubMed] [Google Scholar]
- 141.Cueva JP, Cai TB, Mascarella SW, Thomas JB, Navarro HA, Carroll FI. Synthesis and In Vitro Opioid Receptor Functional Antagonism of Methyl-Substituted Analogues of (3R)-7-Hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-hyroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) J Med Chem. 2009;52:7463–7472. doi: 10.1021/jm900756t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beardsley PM, Pollard GT, Howard JL, Carroll FI. Effectiveness of analogs of the kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxami de (JDTic) to reduce U50,488-induced diuresis and stress-induced cocaine reinstatement in rats. Psychopharmacology (Berl) 2010;210:189–198. doi: 10.1007/s00213-010-1846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas JB, Atkinson RN, Namdev N, Rothman RB, Gigstad KM, Fix SE, Mascarella SW, Burgess JP, Vinson NA, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Discovery of an opioid kappa receptor selective pure antagonist from a library of N-substituted 4beta-methyl-5-(3-hydroxyphenyl)morphans. J Med Chem. 2002;45:3524–3530. doi: 10.1021/jm020084h. [DOI] [PubMed] [Google Scholar]
- 144.Carroll FI, Melvin MS, Nuckols MC, Mascarella SW, Navarro HA, Thomas JB. N-substituted 4β-methyl-5-(3-hydroxyphenyl)-7α-amidomorphans are potent, selective kappa opioid receptor antagonists. J Med Chem. 2006;49:1781–1791. doi: 10.1021/jm058264p. [DOI] [PubMed] [Google Scholar]
- 145.Patkar KA, Yan X, Murray TF, Aldrich JV. [Nalpha-benzylTyr1, cyclo(D-Asp5, Dap8)]-dynorphin A-(1–11)NH2 cyclized in the “address” domain is a novel kappa-opioid receptor antagonist. J Med Chem. 2005;48:4500–4503. doi: 10.1021/jm050105i. [DOI] [PubMed] [Google Scholar]
- 146.Aldrich JV, Patkar KA, McLaughlin JP. Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc Natl Acad Sci US A. 2009;106:18396–18401. doi: 10.1073/pnas.0910180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Saito T, Hirai H, Kim YJ, Kojima Y, Matsunaga Y, Nishida H, Sakakibara T, Suga O, Sujaku T, Kojima N. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J Antibiot (Tokyo) 2002;55:847–854. doi: 10.7164/antibiotics.55.847. [DOI] [PubMed] [Google Scholar]
- 148.Seale PW, Stead P, Jaxa-Chamiec A. In: In Peptides 2000; Proceedings of the European Peptide Symposium, 26th, Montpellier, France, 2000. Martinex J, Fehrentz J-A, editors. Editions EDK; Montpellier, France: 2000. pp. 271–272. [Google Scholar]
- 149.Dolle RE, Michaut M, Martinez-Teipel B, Seida PR, Ajello CW, Muller AL, DeHaven RN, Carroll PJ. Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg Med Chem Lett. 2009;19:3647–3650. doi: 10.1016/j.bmcl.2009.04.105. [DOI] [PubMed] [Google Scholar]
- 150.Ross NC, Reilley KJ, Murray TF, Aldrich JV, McLaughlin JP. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting kappa opioid receptor antagonism. Br J Pharmacol. 2011;165:1097–1108. doi: 10.1111/j.1476-5381.2011.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schattauer SS, Miyatake M, Shankar H, Zietz C, Levin JR, Liu-Chen LY, Gurevich VV, Rieder MJ, Chavkin C. Ligand Directed Signaling Differences Between Rodent and Human Kappa Opioid Receptors. J Biol Chem. 2012;287:41595–41607. doi: 10.1074/jbc.M112.381368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Aldrich JV, Kulkarni SS, Senadheera SN, Ross NC, Reilley KJ, Eans SO, Ganno ML, Murray TF, McLaughlin JP. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15,208. ChemMedChem. 2011;6:1739–1745. doi: 10.1002/cmdc.201100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Verhoest PR, Sawant Basak A, Parikh V, Hayward M, Kauffman GW, Paradis V, McHardy SF, McLean S, Grimwood S, Schmidt AW, Vanase-Frawley M, Freeman J, Van Deusen J, Cox L, Wong D, Liras S. Design and Discovery of a Selective Small Molecule kappa Opioid Antagonist (2-Methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine, PF-4455242) J Med Chem. 2011;54:5868–5877. doi: 10.1021/jm2006035. [DOI] [PubMed] [Google Scholar]
- 154.Grimwood S, Lu Y, Schmidt AW, Vanase-Frawley MA, Sawant-Basak A, Miller E, McLean S, Freeman J, Wong S, McLaughlin JP, Verhoest PR. Pharmacological characterization of 2-methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine (PF-04455242), a high-affinity antagonist selective for kappa-opioid receptors. J Pharmacol Exp Ther. 2011;339:555–566. doi: 10.1124/jpet.111.185108. [DOI] [PubMed] [Google Scholar]
- 155.D’Amour FE, Smith DL. 74A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 156.Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, Shi Q, Canada EJ, Kahl SD, Statnick MA, McKinzie DL, Benesh DR, Rash KS, Barth VN. Discovery of aminobenzyloxyarylamides as kappa opioid receptor selective antagonists: application to preclinical development of a kappa opioid receptor antagonist receptor occupancy tracer. J Med Chem. 2011;54:8000–8012. doi: 10.1021/jm200789r. [DOI] [PubMed] [Google Scholar]
- 157.Buezo ND, McKinzie DL, Mitch CH, Pedregal-Tercero C. Eli Lilly and Company; Indianapolis, IN: US Patent 8,173,695 B2. Kappa Selective Opioid Receptor Antagonist. 2012
- 158.Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 159.Pechtel P, Pizzagalli DA. Effects of early life stress on cognitive and affective function: an integrated review of human literature. Psychopharmacology (Berl) 2011;214:55–70. doi: 10.1007/s00213-010-2009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Secoli SR, Teixeira NA. Chronic prenatal stress affects development and behavioral depression in rats. Stress. 1998;2:273–280. doi: 10.3109/10253899809167291. [DOI] [PubMed] [Google Scholar]
- 161.Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- 162.Emrich HM, Vogt P, Herz A. Possible antidepressive effects of opioids: action of buprenorphine. Ann N Y Acad Sci. 1982;398:108–122. doi: 10.1111/j.1749-6632.1982.tb39483.x. [DOI] [PubMed] [Google Scholar]
- 163.Kosten TR, Morgan C, Kosten TA. Depressive symptoms during buprenorphine treatment of opioid abusers. J Subst Abuse Treat. 1990;7:51–54. doi: 10.1016/0740-5472(90)90035-o. [DOI] [PubMed] [Google Scholar]
- 164.Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15:49–57. doi: 10.1097/00004714-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 165.Kosten TR, Kleber HD, Morgan C. Treatment of cocaine abuse with buprenorphine. Biol Psychiatry. 1989;26:637–639. doi: 10.1016/0006-3223(89)90090-5. [DOI] [PubMed] [Google Scholar]
- 166.Schottenfeld RS, Pakes J, Ziedonis D, Kosten TR. Buprenorphine: dose-related effects on cocaine and opioid use in cocaine-abusing opioid-dependent humans. Biol Psychiatry. 1993;34:66–74. doi: 10.1016/0006-3223(93)90258-f. [DOI] [PubMed] [Google Scholar]
- 167.McCann DJ. Potential of buprenorphine/naltrexone in treating polydrug addiction and co-occurring psychiatric disorders. Clin Pharmacol Ther. 2008;83:627–630. doi: 10.1038/sj.clpt.6100503. [DOI] [PubMed] [Google Scholar]
- 168.Rothman RB, Gorelick DA, Heishman SJ, Eichmiller PR, Hill BH, Norbeck J, Liberto JG. An open-label study of a functional opioid kappa antagonist in the treatment of opioid dependence. J Subst Abuse Treat. 2000;18:277–281. doi: 10.1016/s0740-5472(99)00074-4. [DOI] [PubMed] [Google Scholar]
- 169.Rothman RB. A review of the role of anti-opioid peptides in morphine tolerance and dependence. Synapse. 1992;12:129–138. doi: 10.1002/syn.890120206. [DOI] [PubMed] [Google Scholar]
- 170.Rothman RB, Long JB, Bykov V, Xu H, Jacobson AE, Rice KC, Holaday JW. Upregulation of the opioid receptor complex by the chronic administration of morphine: a biochemical marker related to the development of tolerance and dependence. Peptides. 1991;12:151–160. doi: 10.1016/0196-9781(91)90182-o. [DOI] [PubMed] [Google Scholar]
- 171.Gerra G, Fantoma A, Zaimovic A. Naltrexone and buprenorphine combination in the treatment of opioid dependence. J Psychopharmacol. 2006;20:806–814. doi: 10.1177/0269881106060835. [DOI] [PubMed] [Google Scholar]
- 172.ClinicalTrials.gov. First in Humans Study of JDTic. http://www.clinicaltrials.gov/ct2/show/NCT01431586?term=JDTic&rank=1, last updated May 9: 2011.
- 173.ClinicalTrials.gov. A Study Of Kappa Opioid Receptor Occupancy Of PF-04455242, Using PET (Positron Emission Tomography) http://www.clinicaltrials.gov/ct2/show/NCT00939887?term=PF-04455242&rank=3, last updated February 4: 2010.
- 174.ClinicalTrials.gov. A Study of Brain Receptor Occupancy in Healthy Subjects. http://www.clinicaltrials.gov/ct2/show/NCT01232439?term=ly2456302&rank=1, last updated May 5, 2011.
- 175.Ehrich EW. 1.3 New Clinical Research in Opioid Modulation Indicates Novel Utility in Treating Resistant Depression. Neuropsychopharmacology. 2012;38:S1. [Google Scholar]