

Abstract
Liu, X., L. He, B. Dinger, L. Stensaas, and S. Fidone. Sustained exposure to cytokines and hypoxia enhances excitability of oxygen-sensitive type I cells in rat carotid body: Correlation with the expression of HIF-1α protein and adrenomedullin. High Alt Med Biol 14:53–60, 2013.—Recent studies in our laboratory demonstrated that chronic hypoxia (CH) induces a localized inflammatory response in rat carotid body that is characterized by macrophage invasion and increased expression of inflammatory cytokines. Moreover, CH-induced increased hypoxic sensitivity is blocked by concurrent treatment with the common anti-inflammatory drugs, ibuprofen and dexamethasone. The present study examines the hypothesis that selected cytokines enhance the excitability of oxygen-sensitive type I cells in the carotid body, and that downstream effects of cytokines involve upregulation of the transcription factor, hypoxia inducible factor-1α (HIF-1α). Cultured type I cells were exposed for 24 h to hypoxia and/or a cocktail of cytokines consisting of interleukin-1β, interleukin-6, and tumor necrosis factor-α. Subsequent evaluation of hypoxia-evoked intracellular Ca2+-responses showed that previous exposure to cytokines plus hypoxia resulted in a 110% (p<0.001) increase in cell excitability, whereas exposure to cytokines or hypoxia alone elicited smaller increases of 22% (not significant) and 35% (p<0.01), respectively. These changes were correlated with increased immunostaining for HIF-1α in similarly treated type I cells, where exposure to cytokines plus hypoxia promoted the nuclear translocation of the transcription factor. Moreover, treatment with cytokines and/or hypoxia elevated the expression of the HIF-1-regulated gene, adrenomedullin. These in vitro results are supported by studies which show that elevated type I cell sensitivity following in vivo CH is blocked by concurrent treatment with ibuprofen. The data suggest that CH-induced adaptation in arterial chemoreceptors may in part be mediated by cytokine/hypoxia-induced upregulation of HIF-1α, and consequent enhanced expression of specific hypoxia-sensitive genes in type I cells.
Key Words: inflammation, ibuprofen, cell calcium, tyrosine hydroxylase, oxygen-sensitive genes
Introduction
Oxygen-sensing in carotid body chemoreceptors is initiated in specialized type I glomus cells where hypoxia-evoked depolarization results in Ca2+ entry, the release of multiple neurotransmitters, and increased nerve impulse traffic in the carotid sinus nerve (CSN) (Fidone and Gonzalez, 1986; Fidone et al., 1997). An adaptive feature of carotid body function is the development of enhanced sensitivity to acute hypoxic challenges following chronic hypoxia (CH). The 3–10 day time-course of adaptation parallels the development of ventilatory acclimatization to hypoxia (VAH), a sustained increase in ventilation that is dependent on intact carotid body function (Powell et al., 1998). Multiple studies have demonstrated CH-induced phenotypic changes in type I cells, including altered expression of K+- and Na+-channel proteins, and increased levels of specific neurotransmitters and neuromodulators (Bisgard, 2000; Wang and Bisgard, 2002; Caceres et al., 2007). The cellular and molecular mechanisms that regulate these functional adjustments are only partially understood.
Numerous studies within the last decade have established hypoxia-inducible factor-1α (HIF-1α) as a critical mediator of hypoxia-induced phenotypic adjustments in diverse cell types in multiple tissues (Semenza, 2000a; Semenza, 2001). The effect of CH on HIF-1α protein levels in carotid body has not been reported. However, studies in other tissues have consistently demonstrated increased levels of HIF-1α following hypoxia. In addition, mice partially deficient in HIF expression (i.e., HIF-1α+/-) do not develop an increased hypoxic ventilatory response (HVR) during CH, suggesting that the transcription factor is critical for increasing chemoreceptor sensitivity (Kline et al., 2002b). Genes regulated by HIF-1α include endothelin-1 (ET-1), vascular endothelial growth factor (VEGF), neuronal- and inducible nitric oxide synthase (nNOS and iNOS), and adrenomedullin (ADM), all of which are expressed by type I cells (Garayoa et al., 2000; Powell and Fu, 2008).
Complex cellular mechanisms regulate levels of HIF-1α. Early studies showed that the transcription factor is produced constitutively, and that lowering tissue Po2 leads to inhibition of O2-dependent hydroxylases, which in normoxia hydroxylate specific proline residues, resulting in ubquitination and proteosomal degradation of HIF-1 (Dery et al., 2005). More recently, transcriptional and translational mechanisms have been discovered. In particular, the inflammatory cytokines, interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα), have been shown to promote translation of HIF-1α mRNA (Hellwig-Burgel et al., 1999; Hellwig-Burgel et al., 2005). Moreover, interleukin-6 (IL-6) and IL-1β facilitate the nuclear translocation of HIF-1α, suggesting that specific cytokines can enhance HIF-mediated gene expression (Haddad, 2002; Ramadori et al., 2010).
Multiple studies have demonstrated that CH, as well as chronic intermittent hypoxia (CIH), induces inflammation in rat carotid body, resulting in macrophage invasion and increased expression of proinflammatory cytokines (Liu et al., 2009; Del Rio et al., 2011; Liu et al., 2011b; Del Rio et al., 2012; Lam et al., 2012). Moreover, concurrent treatment with the nonsteroidal anti-inflammatory drug, ibuprofen, or the steroid, dexamethasone, blocked CH-induced inflammation and chemoreceptor adaptation (Liu et al., 2009). Other studies in our laboratory suggest that adaptation involves cytokine-mediated changes in the functional properties of the afferent terminals of primary sensory chemoreceptor neurons (Liu et al., 2011a; Liu et al., 2011b). But it is also possible that increased sensitivity to hypoxia occurs in type I cells, which have been shown to express specific cytokine receptors (Wang et al., 2002; Wang et al., 2006). Thus, the current experiments were designed to test the hypothesis that inflammatory cytokines play critical roles in mediating phenotypic adjustments in type I cells during sustained hypoxia. One set of experiments examined the effect of concurrent ibuprofen treatment on increased type I cell excitability elicited by in vivo CH. In separate experiments, we examined excitability following exposure of cultured type I cells to a cocktail of inflammatory cytokines. As was noted earlier, numerous previous studies have demonstrated that hypoxia depolarizes type I cells, leading to the activation of voltage-sensitive Ca2+-channels, elevated levels of intracellular Ca2+, and the release of multiple neurotransmitters (Gonzalez et al., 1995; Fidone et al., 1997). Moreover, Carroll and his colleagues (Wasicko et al., 2006) have demonstrated a robust correlation between the degree membrane depolarization and the intensity of the Ca2+-response in rat type I cells. We have therefore used conventional Ca2+-imaging techniques to evaluate changes in cell excitability. In parallel experiments, we employed standardized immunocytochemical methods to quantify the effect of cytokines on the level of HIF-1α, as well as the expression of the HIF-1-sensitive gene, ADM in cultured type cells (Frede et al., 2005; Ishimitsu et al., 2006).
Methods
Animals and exposure to chronic hypoxia
Animal protocols were approved by the University of Utah Institutional Animal Care and Use Committee. Rats exposed in a hypobaric chamber were housed in standard rodent cages with food and water. Pressures were reduced from ambient BP at the University of Utah (i.e., BP ∼630 Torr; 1500 m) until a selected pressure equivalent to ∼5500 m (380 Torr) was reached, and maintained for a specified period. Control, normal animals were maintained outside the chamber in ambient conditions.
Dissociation and culture of carotid body type I cells
Twenty-six young adult rats (∼120–140 g) were anesthetized with ketamine (10 mg/kg, i.m.) plus xylazine (0.9 mg/kg, i.m.). The carotid artery bifurcation was surgically excised and placed into ice-cold modified Tyrode's solution containing, in mM: NaCl, 112; KCl, 4.7; CaCl2, 2.2; MgCl2, 1.1; Na glutamate, 42; glucose 5.6, and HEPES buffer, 5 (pH 7.43 @ 37°C) and equilibrated with 100% O2. Carotid bodies were dissected free of surrounding connective tissue and transferred to Ham's F-12 medium (Ca2+- and Mg2+-free) containing 0.2% collagenase and 0.2% trypsin. Each carotid body was cut into 6–12 pieces and incubated for 40 min in a CO2 incubator (5% CO2, 95% air) at 36.5°C. Tissue fragments were rinsed (2×10 min, room temperature) in F-12 medium (Ca2+- and Mg2+-free), transferred to poly-l-lysine coated glass coverslips, and triturated in a small volume of medium, plus 10% fetal calf serum and 5 μg/mL insulin. For normoxia, coverslips containing O2-sensitive type I cells were maintained in the incubator with 5% CO2, 95% air, at 36.5°C. Separate groups of cells were exposed to hypoxia in a Ruskin Invivo 400 Hypoxia Workstation, set to maintain O2 at 5.1% (PO2 ∼32 Torr at the University of Utah), with 5% CO2, and the balance N2 (36.5°C).
Intracellular [Ca2+] measurements
As was described previously (He et al., 2000), freshly dissociated type I cells attached to coverslips were incubated in F-12 medium containing 0.5 μM fura-2 AM for 10–15 min in a CO2 incubator at 36.5°C. Coverslips were placed in a flow chamber where they were superfused at 0.75–1.0 ml/min with modified Tyrode solution equilibrated with air. The temperature was maintained at 35°–36.5°C. The chamber was mounted on the stage of a Zeiss inverted microscope incorporated into a Zeiss/Attofluor workstation equipped with an excitation wavelength selector (filter changer) and an intensified charge-coupled device camera system. Fura-2 fluorescent emission was measured at 520 nm in response to alternating excitation wavelengths of 334 and 380 nm. Measurements of basal [Ca2+]I were made in solution equilibrated with air (Po2 ∼120 Torr); hypoxic solutions were adjusted to Po2 ∼36–40 Torr. Data were collected and analyzed using Attofluor Ratiovision software (version 6.0).
Immunocytochemistry on cultured type I cells
Cells were fixed in either ice-cold 4% paraformaldehyde in 0.1 M PBS for 5 min (for fixation of adrenomedullin); or in acetone/methanol (1:1; for fixation of nuclear HIF-1α and tyrosine hydroxylase) at −20°C, rinsed 3X3 min in PBS and incubated in 5% normal goat serum plus Triton X-100 for 20 min at room temperature (RT). Primary antibodies diluted in 0.1 M PBS plus 2% serum and 0.1% Triton X-100 were added and incubation continued for 2–3 hours at RT or overnight at 4°C. Cells were rinsed 3X3 min in 0.1 M PBS and incubated in fluorescent secondary antibodies for 30 min at RT, and then rinsed 3X3 min in PBS. Primary antibodies included mouse anti-human HIF-1α (Novus Biologicals; diluted 1:400), rabbit anti-rat tyrosine hydroxylase (Chemicon; diluted 1:500), and rabbit anti-rat adrenomedullin (Phoenix Pharmaceuticals; diluted 1:200). Secondary antibodies and dilutions were rhodamine-conjugated goat-anti-rabbit IgG (1:200), fluorescein-conjugated donkey-anti-mouse IgG (1:200), and fluorescein-conjugated donkey-anti-rabbit IgG (1:200). Images were obtained on an Olympus FV1000 laser scanning confocal microscope, and immunofluorescence intensities were quantified with ImageTool.
Quantitative reverse transcriptase-polymerase chain reaction
The method has been described in detail previously (Liu et al., 2009; Liu et al., 2011a). Briefly, in accord with the kit instructions (RNAqueous-Micro, Ambion, Austin, TX), total RNA was extracted from tissue samples pooled from groups of 5 rats for each experimental condition. Following removal of contaminating DNA (DNase treatment), first strand complementary DNA was synthesized from 1 μg total RNA (quantified with a NanoDrop ND-1000 spectrophotometer) using RETROscript (Ambion). Aliquots of cDNA corresponding to 2 ng of total RNA were introduced into a SybrGreen reaction mix (25 μL; Qiagen) containing ‘upstream’ and ‘downstream’ primers for the ADM gene. Quantitative reverse transcriptase-polymerase chain reaction (qPCR) was conducted in an MJ Research PTC-200 equipped with a Chromo4 detector. Reactions were initiated at 95°C for 15 min, followed by 40 cycles consisting of 15 sec at 94°C, 15 sec at 58°C, and 15 sec at 72°C, with the final cycle extended to 5 min at 72°C, followed by melting curve determination; samples were stabilized at 4°C. Sample comparisons were based on data normalized to 18SrRNA. Amplifications without the RT step were performed to exclude possible contamination with genomic DNA.
Statistical analysis
Data were analyzed using Student's t-test, non-parametric ANOVA with Dunn's multiple comparisons post-tests, or standard ANOVA with Bonferroni multiple comparison post-tests, as appropriate. P values<0.05 were considered to indicate significant differences between groups.
Results
Two to three hours after incubation in air-equilibrated media, the mean basal [Ca2+]I in type I cells harvested from normal animals was 44.06 nM (±1.24 nM; SEM). The resting [Ca2+]I in cells from normoxic rats treated with ibuprofen was similarly 42.01 nM (p>0.05). CH slightly elevated the basal [Ca2+]I to 50.03 nM; this value was not significantly different from normal (p>0.05), but it was judged to be higher than in cells from normoxic rats treated with ibuprofen (p<0.01). The resting [Ca2+]I in cells from CH animals treated with ibuprofen was 43.51 nM, consistent with normal levels (p>0.05). Figure 1 shows hypoxia-evoked (bath PO2 ∼36–40 Torr for 60 sec) [Ca2+]I –responses in type I cells. The upper panel consists of representative traces of [Ca2+]I for four experimental conditions; the lower panel summarizes data averaged from samples ranging in size from 194 to 276 cells for each condition. Because the standard deviations of the four data sets were considered not equal (Bartlett test; p<0.0001), the data were evaluated using the Kruskal-Wallis non-parametric ANOVA. CH (7 or 8 days @ 380 Torr) elicited a highly significant 74% increase (p<0.001 vs. normal cells) in the response, consistent with elevated excitability. Treatment of normal animals with ibuprofen (4 mg/kg/day) did not alter the [Ca2+]I-response of type I cells (p>0.05, normal vs. normal+ibuprofen). However, concurrent treatment of CH animals with the drug completely prevented enhancement of the response in type I cells (p<0.001 vs. CH).
FIG. 1.

Intracellular [Ca2+]I-responses in type I cells harvested from normal rats (N), normal rats treated with ibuprofen (4 mg/kg/day for 8–10 days; N+I), rats exposed to hypoxia (380 Torr) for 8–10 days (CH), and CH rats treated with ibuprofen(CH+I). (A) Representative traces of [Ca2+]I from type I cells exposed to hypoxia (Po2 ∼36–40 Torr indicated by solid line). (B) Summary data (mean±SEM from 194–276 cells in each group; ***p<0.001 versus normal.
Figure 2 shows the effects of a 24 h exposure to 5.1% O2 (hypoxia) and/or a cocktail of inflammatory cytokines (50 ng/mL IL-1β; 50 ng/mL IL-6, and 25 ng/mL TNFα) on type I cells. At similar concentrations these agents have been shown to inhibit K+-currents and increase [Ca2+]I in type I cells (Shu et al., 2007; Fan et al., 2009), and enhance gene expression/protein synthesis and/or excitability in cultured sensory neurons (Binshtok et al., 2008; Melemedjian et al., 2010). The present experiments evaluated the increase in [Ca2+]I evoked by an acute hypoxic challenge (60 sec @ bath PO2 ∼40 Torr) in cells following a 24 h culture in the presence or absence of the cocktail. These assessments were made 30–60 min following removal of the cells from exposure to cytokines and/or hypoxia. Again, representative traces of evoked changes in [Ca2+]I for four experimental conditions are shown in the upper panel; the lower panel presents data averaged from 38 to 56 cells for each condition. Basal [Ca2+]I was not significantly altered by sustained hypoxia or exposure to cytokines. In 24 h normoxic cells, the acute hypoxic challenge increased [Ca2+]I to ∼112.3±7.1 nM above the resting concentration (p<0.001). Exposure of normoxic cells to the cytokine cocktail for 24 h resulted in an insignificant 22% increase in the response to acute hypoxia. The response in cells incubated in 5% O2 was 35% greater (p<0.01) than the normoxic cells. Concurrent exposure of cells to hypoxia plus cytokines resulted in a robust 110% increase in the hypoxic response (p<0.001 vs. normal and CH) indicating that the stimuli are acting synergistically to enhance cell sensitivity.
FIG. 2.

Intracellular [Ca2+]I in cultured type I cells following a 24 h exposure to media equilibrated with air (20% O2); air-equilibrated media plus a cocktail of inflammatory cytokines (cyt; see text for details); media equilibrated with 5% O2; or 5% O2 media plus cytokines. Top panel shows typical responses evoked by an acute hypoxic challenge (PO2 ∼36–40 Torr) following incubation in each specified condition. Bottom panel shows summary data from 38–56 cells in each group; ** and *** indicate p<0.01 and p<0.001, respectively, versus normal; +++ indicates p<0.001 versus 5% O2.
Fluorescence images presented in Figure 3A show the effect of the cytokine cocktail alone, or in combination with hypoxia (CH, 5.1% O2, 24 h) on immunostaining intensity of HIF-1α (green) and tyrosine hydroxylase (TH; red) in cultured type I cells. TH, a cytosolic enzyme, is an established marker of type I cell cytoplasm. Note that the centers of the cells are virtually devoid of red TH staining, suggesting that cytoplasm is distributed around, but not above the nucleus, consistent with the flattened morphology commonly observed in cultured cells (Bonney et al., 1974; Sattler et al., 1978). In normoxia, exposure of type I cells to cytokines for 24 h evokes a noticeable increase in the staining intensity of HIF-1α and TH in cell cytoplasm. Moreover, hypoxia, or hypoxia in the presence of cytokines, resulted in further increases in cytoplasmic TH and HIF-1α. Quantification of gray-scale intensities (n=20–29 cells in each group), summarized in Figure 3C show that exposure to hypoxia or cytokines elicited significant (p<0.001) increases in cytoplasmic staining intensity for TH and HIF-1α (p<0.001); whereas the combination of hypoxia plus cytokines caused a further (nonsignificant; p>0.05 vs. CH) increase in HIF-1α and TH cytoplasmic staining intensity. In addition to prominent cytoplasmic immunostaining for HIF-1α in these preparations, enlarged images in panel B show that cytokine treatment enhances nuclear translocation of HIF. Quantification of nuclear staining (Fig. 3C, bottom) indicates that hypoxia plus cytokines evoke a robust increase in the level of nuclear HIF-1α which is significantly (p<0.001) greater than the effect of hypoxia or cytokines alone.
FIG. 3.

Effect of inflammatory cytokines and/or hypoxia on expression of HIF-1α and tyrosine hydroxylase (TH) in cultured type I cells. (A) immunofluorescence images of cells stained for TH and HIF-1; N: normoxia (24 h exposure in media equilibrated with 20% O2); CH: 24 h exposure @ 5.1% O2; Cy: 24 h exposure in media containing IL-1β (50 ng/mL), IL-6 (50 ng/mL), and TNFα (25 ng/mL). Enlargements in (B) show that low levels of nuclear HIF-1α staining in cells exposed to CH are substantially enhanced following treatment with CH+Cy. Gray scale quantification of staining intensities in (C) shows effects of CH and/or Cy on levels of cytoplasmic HIF and TH, and on the presence of HIF in cell nuclei. *** and +++ indicate p<0.001 versus normoxia and CH, respectively.
An increased presence of HIF-1α predicts elevated expression of HIF-regulated genes. Data in Figure 4 show the effect of hypoxia (5.1% O2) and/or the cocktail of inflammatory cytokines on immunostaining for adrenomedullin (ADM) in cultured type I cells. The low level of ADM staining in normal, untreated cells was significantly elevated following a 24 h treatment with the cytokine cocktail. Likewise, 24 h at 5.1% O2 evoked a robust response, which was further enhanced by the combined presence of cytokines plus hypoxia. Because the effect of hypoxia on ADM levels in carotid body have not been reported previously, we also measured mRNA levels for the peptide in carotid bodies harvested from rats exposed to hypobaric hypoxia (380 Torr) for 3 and 7 days. Data in Figure 5 show that in vivo CH evokes more than a 2-fold increase in ADM expression at both time points, consistent with the effect of hypoxia on protein immunostaining intensity in cultured type I cells.
FIG. 4.
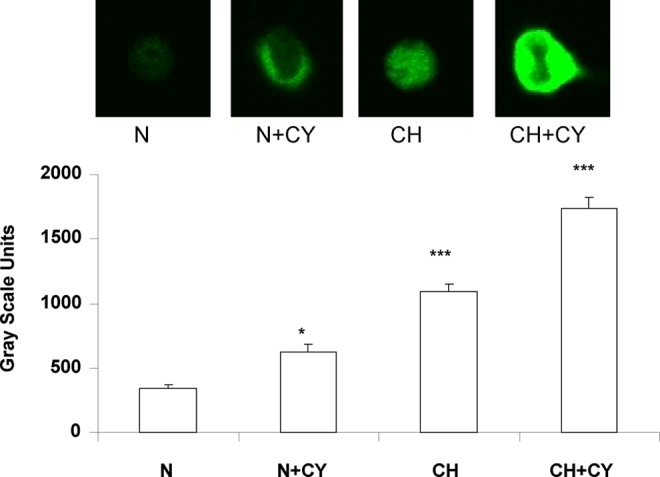
Effect of inflammatory cytokines and/or hypoxia on adrenomedullin (ADM) expression in cultured type I cells. Upper panel shows immunofluorescence staining for ADM in cells incubated for 24 h in nomoxia (N: media equilibrated with air), a cocktail of inflammatory cytokines (CY), or hypoxia (CH: media equilibrated with 5.1% O2. Lower panel shows gray scale quantification of cytoplamic staining intensities for various experimental conditions. N=24–59 cells in each group. * and *** indicate p<0.05 and 0.001 versus normoxia, respectively.
FIG. 5.

Effect of chronic hypoxia on adrenomedullin mRNA expression in rat carotid body. Rats were exposed to hypobaric hypoxia @ 380 Torr for 3 or 7 days; normoxic controls were maintained at ∼640 Torr (ambient barometric pressure at the University of Utah) for 7 days. ***p<0.001 versus control group.
Discussion
Studies within the last decade have demonstrated that type I cells express receptors for IL-1β and IL-6 (Wang et al., 2002; Wang et al., 2006). Moreover, acute application of the former cytokine has been shown to inhibit K+-current and elevate [Ca2+]I in type I cells (Shu et al., 2007). The present study extends these important findings by showing for the first time that sustained exposure to inflammatory cytokines increases levels of HIF-1α and the HIF-1-regulated peptide, ADM, suggesting that CH-induced inflammation in carotid body contributes to phenotypic adjustments in type I cells that are associated with chemoreceptor adaptation to hypoxia (Kline et al., 2002a; Martinez et al., 2003). Moreover, our data show that cytokine treatment elevates Ca2+-responses evoked by subsequent acute hypoxic challenges. These findings concur with our recent demonstration that CH-evoked enhancement of chemoreceptor excitability is blocked by the common anti-inflammatory drugs, ibuprofen and dexamethasone. Importantly, these drugs also blocked CH-induced gene expression for IL-1β, IL-6, and TNFα in carotid body (Liu et al., 2009). A role for inflammation in elevating type I cell excitability is further indicated by the findings presented in Figure 1, which show that concurrent CH plus ibuprofen treatment in vivo prevent CH-induced increases in [Ca2+]I-responses evoked by a subsequent acute hypoxic challenge in vitro.
Elevated immunostaining intensity for HIF-1α in type I cells following exposure to low-O2 plus cytokines is consistent with previous findings in other cell types, where hypoxia and cytokines increase HIF-1 levels via diverse mechanisms, involving post-translational proline hydroxylases, versus translation of HIF-1 mRNA, respectively (Dery et al., 2005; Hellwig-Burgel et al., 2005). Our data also indicate that nuclear levels of HIF-1α are likewise substantially increased following exposure to hypoxia and cytokines, consistent with transcription factor activation of specific hypoxia-sensitive genes.
The increased presence of ADM following exposure to CH in vivo, as well as sustained hypoxia and cytokines in culture, provides further evidence that chemoreceptor adaptation involves highly specified HIF-1-directed phenotypic adjustments in type I cells, including the expression of known hypoxia-sensitive genes. Immunocytochemical experiments have previously localized ADM in these cells, and exposure of whole carotid bodies to the peptide elicits a dose-dependent release of dopamine (Martinez et al., 2003). CH is known to increase expression of TH, the rate-limiting enzyme for dopamine synthesis in carotid body (Gonzalez et al., 1979; Fidone and Gonzalez, 1982; Fidone and Gonzalez, 1982). Following CH, acute hypoxia evokes an abnormally high amount of dopamine release (Fidone and Gonzalez, 1982; Gonzalez-Guerrero et al., 1993), consistent with an increased presence of ADM. Available evidence suggests that acute hypoxia evokes the release of multiple neurotransmitter agents from type I cells (Nurse, 2010). Thus, the physiological influence of elevated ADM expression may occur as one of multiple adaptations within a highly complex neurotransmitter system. In addition to its putative effects on synaptic function, other studies have shown that ADM is a potent vasodilator (Heaton et al., 1995; Kobayashi et al., 2003), and in this regard, increased levels of ADM may participate in carotid body vascular expansion, a phenomenon which has been extensively documented following CH (Hellstrom and Pequignot, 1982; Pequignot et al., 1990; Chen et al., 2007).
Interestingly, our data also indicate that cytokine exposure enhances TH expression in type I cells. It is well established that hypoxia elevates TH mRNA levels primarily via the transcription factor, activator protein-1 (AP-1) (Semenza, 2000b; Yuan et al., 2004). Evidence indicates that the signaling pathway for AP-1 involves cell depolarization, Ca2+-entry, and the immediate early gene, c-fos (Yuan et al., 2004). Our finding that a cocktail of inflammatory cytokines elevate TH levels are consistent with the recent demonstration that IL-1β, acting via IL-1 receptors on type I cells, evokes depolarization and increased [Ca2+]I (Shu et al., 2007; Fan et al., 2009). Our data also show that the effect of cytokines plus hypoxia are additive for expression of HIF-1α and TH, suggesting that these stimuli may act via separate mechanisms that elevate [Ca2+]I and/or downstream mediators of gene induction.
Finally, it is important to consider that our experiments utilized a cocktail of cytokines that was selected based upon their demonstrated increased expression in carotid body following CH (Liu et al., 2009). The concentrations used are consistent with measured levels in other inflamed tissues (Faustino et al., 2011). However, our studies in carotid body show that the time-course of CH-induced expression is distinct for each substance (Liu et al., 2009), suggesting that the experimental conditions used in the present study may not exactly mimic the in vivo milieu. Moreover, multiple other factors are absent in the culture environment, including a variety of neurotransmitter and neuromodulators, and possibly other pro-inflammatory and anti-inflammatory cytokines. The importance of these putative differences may be indicated by the finding that the cytokine cocktail increased TH immunostaining in isolated type I cells, whereas we previously reported that TH expression is not altered when CH-induced cytokine expression is blocked by concurrent treatment with ibuprofen (Liu et al., 2009). Differences between culture and in vivo environments notwithstanding, the present results are consistent with the hypothesis that CH-induced inflammation contributes to elevated type I cell sensitivity, which may in part be mediated by HIF-1α.
Acknowledgment
This work was supported by National Heart, Lung and Blood Institute Grant HL-086508.
Author Disclosure Statement
No competing financial interests exist.
References
- Binshtok AM. Wang H. Zimmermann K. Amaya F. Vardeh D. Shi L. Brenner GJ. Ji RR. Bean BP. Woolf CJ. Samad TA. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgard GE. Carotid body mechanisms in acclimatization to hypoxia. Resp Physiol. 2000;121:237–3246. doi: 10.1016/s0034-5687(00)00131-6. [DOI] [PubMed] [Google Scholar]
- Bonney RJ. Becker JE. Walker PR. Potter VR. Primary monolayer cultures of adult rat liver parenchymal cells suitable for study of the regulation of enzyme synthesis. In Vitro. 1974;9:399–413. doi: 10.1007/BF02615992. [DOI] [PubMed] [Google Scholar]
- Caceres AI. Obeso A. Gonzalez C. Rocher A. Molecular identification and functional role of voltage-gated sodium channels in rat carotid body chemoreceptor cells. Regulation of expression by chronic hypoxia in vivo. J Neurochem. 2007;102:231–245. doi: 10.1111/j.1471-4159.2007.04465.x. [DOI] [PubMed] [Google Scholar]
- Chen J. He L. Liu X. Dinger B. Stensaas L. Fidone S. Effect of the endothelin receptor antagonist bosentan on chronic hypoxia-induced morphological and physiological changes in rat carotid body. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1257–L1262. doi: 10.1152/ajplung.00419.2006. [DOI] [PubMed] [Google Scholar]
- Del Rio R. Moya EA. Iturriaga R. Differential expression of pro-inflammatory cytokines, endothelin-1 and nitric oxide synthases in the rat carotid body exposed to intermittent hypoxia. Brain Res. 2011;1395:74–85. doi: 10.1016/j.brainres.2011.04.028. [DOI] [PubMed] [Google Scholar]
- Del Rio R. Moya EA. Parga MJ. Madrid C. Iturriaga R. Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. Eur Respir J. 2012;39:1492–1500. doi: 10.1183/09031936.00141511. [DOI] [PubMed] [Google Scholar]
- Dery MA. Michaud MD. Richard DE. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int J Biochem Cell Biol. 2005;37:535–540. doi: 10.1016/j.biocel.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Fan J. Zhang B. Shu HF. Zhang XY. Wang X. Kuang F. Liu L. Peng ZW. Wu R. Zhou Z. Wang BR. Interleukin-6 increases intracellular Ca2+ concentration and induces catecholamine secretion in rat carotid body glomus cells. J Neurosci Res. 2009;87:2757–2762. doi: 10.1002/jnr.22107. [DOI] [PubMed] [Google Scholar]
- Faustino JV. Wang X. Johnson CE. Klibanov A. Derugin N. Wendland MF. Vexler ZS. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31:12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidone S. Gonzalez C. Catecholamine synthesis in rabbit carotid body in vitro. J Physiol. 1982;333:69–79. doi: 10.1113/jphysiol.1982.sp014439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidone SJ. Gonzalez C. Initiation and control of chemoreceptor activity in the carotid body. In: Fishman AP, editor. Handbook of Physiology—The Respiratory System II. Part I. II. Am Physiol Soc Bethesda; 1986. pp. 247–312. [Google Scholar]
- Fidone SJ. Gonzalez C. Almaraz L. Dinger B. Cellular mechanisms of peripheral chemoreceptor function. In: Crystal RG, editor; West JB, editor. The Lung: Scientific Founda1tions. Lippincott-Raven Publishers; Philadelphia: 1997. pp. 1725–1746. [Google Scholar]
- Frede S. Freitag P. Otto T. Heilmaier C. Fandrey J. The proinflammatory cytokine interleukin 1beta and hypoxia cooperatively induce the expression of adrenomedullin in ovarian carcinoma cells through hypoxia inducible factor 1 activation. Cancer Res. 2005;65:4690–4697. doi: 10.1158/0008-5472.CAN-04-3877. [DOI] [PubMed] [Google Scholar]
- Garayoa M. Martinez A. Lee S. Pio R. An WG. Neckers L. Trepel J. Montuenga LM. Ryan H. Johnson R. Gassmann M. Cuttitta F. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: A possible promotion mechanism of carcinogenesis. Mol Endocrinol. 2000;14:848–862. doi: 10.1210/mend.14.6.0473. [DOI] [PubMed] [Google Scholar]
- Gonzalez C. Dinger BG. Fidone SJ. Mechanisms of carotid body chemoreception. In: Dempsey JA, editor; Pack AI, editor. Regulation of Breathing. Marcel Dekker, Inc.; New York: 1995. pp. 391–471. [Google Scholar]
- Gonzalez C. Kwok Y. Gibb J. Fidone S. Effects of hypoxia on tyrosine hydroxylase activity in rat carotid body. J Neurochem. 1979;33:713–719. doi: 10.1111/j.1471-4159.1979.tb05216.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Guerrero PR. Rigual R. Gonzalez C. Effects of chronic hypoxia on opioid peptide and catecholamine levels and on the release of dopamine in the rabbit carotid body. J Neurochem. 1993;60:1769–1776. doi: 10.1111/j.1471-4159.1993.tb13402.x. [DOI] [PubMed] [Google Scholar]
- Haddad JJ. Recombinant human interleukin (IL)-1 beta-mediated regulation of hypoxia-inducible factor-1 alpha (HIF-1 alpha) stabilization, nuclear translocation and activation requires an antioxidant/reactive oxygen species (ROS)-sensitive mechanism. Eur Cytokine Netw. 2002;13:250–260. [PubMed] [Google Scholar]
- He L. Dinger B. Fidone S. Cellular mechanisms involved in carotid body inhibition produced by atrial natriuretic peptide. Am J Physiol Cell Physiol. 2000;278:C845–C852. doi: 10.1152/ajpcell.2000.278.4.C845. [DOI] [PubMed] [Google Scholar]
- Heaton J. Lin B. Chang JK. Steinberg S. Hyman A. Lippton H. Pulmonary vasodilation to adrenomedullin: A novel peptide in humans. Am J Physiol. 1995;268:H2211–H2215. doi: 10.1152/ajpheart.1995.268.6.H2211. [DOI] [PubMed] [Google Scholar]
- Hellstrom S. Pequignot J-M. Morphometric studies on intact and sympathectomised carotid bodies of long-term hypoxic rats. A light and electron microscopial study. In: Pallot DJ, editor; Croom Helm., editor. The Peripheral Arterial Chemoreceptors. London; 1982. pp. 293–301. [Google Scholar]
- Hellwig-Burgel T. Rutkowski K. Metzen E. Fandrey J. Jelkmann W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999;94:1561–1567. [PubMed] [Google Scholar]
- Hellwig-Burgel T. Stiehl DP. Wagner AE. Metzen E. Jelkmann W. Review: Hypoxia-inducible factor-1 (HIF-1): A novel transcription factor in immune reactions. J Interferon Cytokine Res. 2005;25:297–310. doi: 10.1089/jir.2005.25.297. [DOI] [PubMed] [Google Scholar]
- Ishimitsu T. Ono H. Minami J. Matsuoka H. Pathophysiologic and therapeutic implications of adrenomedullin in cardiovascular disorders. Pharmacol Ther. 2006;111:909–927. doi: 10.1016/j.pharmthera.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Kline DD. Peng Y-J. Manalo DJ. Semenza GL. Prabhakar NR. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1a. PNAS. 2002a;99:821–826. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD. Peng YJ. Manalo DJ. Semenza GL. Prabhakar NR. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci USA. 2002b;99:821–826. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H. Yanagita T. Yokoo H. Wada A. Pathophysiological function of adrenomedullin and proadrenomedullin N-terminal peptides in adrenal chromaffin cells. Hypertens Res. 2003;2:S71–S78. doi: 10.1291/hypres.26.s71. [DOI] [PubMed] [Google Scholar]
- Lam SY. Liu Y. Ng KM. Lau CF. Liong EC. Tipoe GL. Fung ML. Chronic intermittent hypoxia induces local inflammation of the rat carotid body via functional upregulation of proinflammatory cytokine pathways. Histochem Cell Biol. 2012;137:303–317. doi: 10.1007/s00418-011-0900-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. He L. Dinger B. Fidone SJ. Chronic hypoxia-induced acid-sensitive ion channel expression in chemoafferent neurons contributes to chemoreceptor hypersensitivity. Am J Physiol Lung Cell Mol Physiol. 2011a;301:L985–L992. doi: 10.1152/ajplung.00132.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. He L. Dinger B. Gonzalez C. Stensaas L. Fidone S. A chronic pain: Inflammation-dependent chemoreceptor adaptation in rat carotid body. Respir Physiol Neurobiol. 2011b;178:362–369. doi: 10.1016/j.resp.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. He L. Stensaas L. Dinger B. Fidone S. Adaptation to chronic hypoxia involves immune cell invasion and increased expression of inflammatory cytokines in rat carotid body. Am J Physiol Lung Cell Mol Physiol. 2009;296:L158–L166. doi: 10.1152/ajplung.90383.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A. Saldise L. Ramirez MJ. Belzunegui S. Zudaire E. Luquin MR. Cuttitta F. Adrenomedullin expression and function in the rat carotid body. J Endocrinol. 2003;176:95–102. doi: 10.1677/joe.0.1760095. [DOI] [PubMed] [Google Scholar]
- Melemedjian OK. Asiedu MN. Tillu DV. Peebles KA. Yan J. Ertz N. Dussor GO. Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30:15113–15123. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse CA. Neurotransmitter and neuromodulatory mechanisms at peripheral arterial chemoreceptors. Exp Physiol. 2010;95:657–667. doi: 10.1113/expphysiol.2009.049312. [DOI] [PubMed] [Google Scholar]
- Pequignot J-M. Hellstrom S. Hertzgerg T. McDonald DM. Long-term hypoxia and hypercapnia in the carotid body: A review. In: Eyzaguirre C, editor; Fidone SJ, editor; Fitzgerald RS, editor; Lahiri S, editor; Arterial Chemoreception. Springer-Verlag; New York: 1990. pp. 100–114. [Google Scholar]
- Powell FL. Fu Z. HIF-1 and ventilatory acclimatization to chronic hypoxia. Respir Physiol Neurobiol. 2008;164:282–287. doi: 10.1016/j.resp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL. Milsom WK. Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Ramadori P. Ahmad G. Ramadori G. Cellular and molecular mechanisms regulating the hepatic erythropoietin expression during acute-phase response: A role for IL-6. Lab Invest. 2010;90:1306–1324. doi: 10.1038/labinvest.2010.85. [DOI] [PubMed] [Google Scholar]
- Sattler CA. Michalopoulos G. Sattler GL. Pitot HC. Ultrastructure of adult rat hepatocytes cultured on floating collagen membranes. Cancer Res. 1978;38:1539–1549. [PubMed] [Google Scholar]
- Semenza GL. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000a;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Oxygen-regulated transcription factors and their role in pulmonary disease. Respir Res. 2000b;1:159–162. doi: 10.1186/rr27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: Control of oxygen homeostasis in health and disease. Pediatr Res. 2001;49:614–617. doi: 10.1203/00006450-200105000-00002. [DOI] [PubMed] [Google Scholar]
- Shu HF. Wang BR. Wang SR. Yao W. Huang HP. Zhou Z. Wang X. Fan J. Wang T. Ju G. IL-1beta inhibits IK and increases [Ca2+]i in the carotid body glomus cells and increases carotid sinus nerve firings in the rat. Eur J Neurosci. 2007;25:3638–3647. doi: 10.1111/j.1460-9568.2007.05586.x. [DOI] [PubMed] [Google Scholar]
- Wang X. Wang BR. Duan XL. Zhang P. Ding YQ. Jia Y. Jiao XY. Ju G. Strong expression of interleukin-1 receptor type I in the rat carotid body. J Histochem Cytochem. 2002;50:1677–1684. doi: 10.1177/002215540205001213. [DOI] [PubMed] [Google Scholar]
- Wang X. Zhang XJ. Xu Z. Li X. Li GL. Ju G. Wang BR. Morphological evidence for existence of IL-6 receptor alpha in the glomus cells of rat carotid body. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:292–296. doi: 10.1002/ar.a.20310. [DOI] [PubMed] [Google Scholar]
- Wang Z-Y. Bisgard GE. Chronic hypoxia-induced morphological and neurochemical changes in the carotid body. Microsc Res Tech. 2002;59:168–177. doi: 10.1002/jemt.10191. [DOI] [PubMed] [Google Scholar]
- Wasicko MJ. Breitwieser GE. Kim I. Carroll JL. Postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol. 2006;154:356–371. doi: 10.1016/j.resp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Yuan G. Adhikary G. McCormick AA. Holcroft JJ. Kumar GK. Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol. 2004;557:773–783. doi: 10.1113/jphysiol.2003.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]