

Sir,
Sialidosis is a rare, autosomal recessive lysosomal storage disorder resulting from deficiency of neuraminidase-1 (also known as lysosomal sialidase) - the enzyme responsible for catalyzing the hydrolysis of the terminal sialic acid residues of sialylated glyco-conjugates. Deficiency of the enzyme leads to progressive intracellular accumulation of sialylated glycopeptides and oligosaccharides. Clinically, sialidosis is classified into two types: the milder ‘normo-somatic’ type I form (also known as the cherry red spot - myoclonus syndrome) and the more severe, earlier onset, dysmorphic type II form1. Type II is further subdivided into the congenital, infantile and juvenile forms, depending upon the age of onset and severity of the disease. The genetic basis of this disorder was elucidated with the mapping of the disease locus, in a patient with combined deficiency of neuraminidase and 21-hydroxylase, followed by characterization of the human neuraminidase-1 gene (NEU1) by Bonten and co-workers in 19962,3. So far, about 40 different mutations in the NEU1 gene have been reported4.
Three novel mutations in the NEU1 gene in two Indian patients of sialidosis, one with the type I form and the other with the infantile type II form, are reported here. Till date, only three families of Indian origin with sialidosis have been reported and to the best of our knowledge there are no NEU1 mutation reports from India5–7.
Patient 1 was from a Christian family from Kerala and patient 2 was from a Hindu family from Tamil Nadu. Both had been evaluated by senior clinicians (patient 1 by a neurologist in Kochi and patient 2 by a medical geneticist at the Medical Genetics Unit of Christian Medical College, Vellore) and were clinically suspected to have sialidosis. Patient 1, diagnosed to have type I sialidosis, was a 14 year old girl, of non-consanguineous parentage, referred for evaluation of progressively worsening gait disturbance, dysarthria and abnormal involuntary movements in the form of hand tremors and myoclonic jerks since last two years. She had fine tremors at rest, dysmetria, nystagmus, gait ataxia and bilateral fundal cherry red spots. Patient 2, diagnosed to have infantile onset type II sialidosis, was a one-and-half year old girl child referred for evaluation of developmental delay with coarse facies. She was the offspring of third degree consanguineous parents. She had macrocephaly, coarse facies, mild corneal haziness, bilateral fundal cherry red spots, a protuberant tongue, gum hypertrophy, generalized hypertrichosis, large Mongolian spots on the back, umbilical hernia, hepatomegaly, bilateral genu valgum, thickening of mitral valve leaflets with mitral valve prolapse/mitral regurgitation and large foamy cells in the bone marrow suggestive of a storage disorder. The urine samples of the two patients were sent to the Centre for DNA Fingerprinting and Diagnostics, Hyderabad, for thin layer chromatography (TLC) for oligosaccharides. The urine TLC pattern in both patients was found to be consistent with that of sialidosis1,5. As the urine TLC was suggestive of sialidosis, EDTA blood samples (5 ml) were obtained from both patients and from the parents of patient 2, with the written informed consent of the respective parents, for molecular genetic study of the NEU1 gene. Patient 1 also underwent a skin biopsy and low neuraminidase-1 levels were documented in the cultured skin fibroblasts for further confirmation of the diagnosis.
Genomic DNA isolation was done from the patients’ and parents’ blood samples using the manual salting out technique. PCR amplification of the 6 exons of the NEU1 gene, including the exon - intron boundaries, was done with sets of primers designed using the PREMIER Primer 3 software (PREMIER Biosoft International, California, USA). Sequence analysis of the amplicons was done with the ABI 3130 sequencer (Applied Biosystems Inc., California, USA) using the Sanger sequencing technique. Results of sequencing were analyzed by alignment of patients’ DNA sequences to wild – type sequences of the human NEU1 gene obtained from the EMBL Nucleotide Sequence database (www.ebi.ac.uk/embl). Using the Clustal 2.1 multiple sequence alignment software (www.clustal.org), the human Neu1 protein sequence was compared against the Neu1 protein sequences of other species (orangutan, cow, rat, mouse and frog) to check for conserved amino-acids at the mutation sites.
Patient 1, with type I sialidosis, was found to be a compound heterozygote with two different mutations: a C880T transition in exon 5 and a 1-base deletion at nucleotide 1191 (c.1191delG) in exon 6 of the NEU1 gene. The C880T missense mutation results in the substitution of arginine to cysteine (R294C) at the 294th amino acid position in the resultant protein. The G base deletion causes frameshift at the 397th amino acid position which extends the length of the resultant protein by 71 amino acids (Fig. A). While an arginine to serine substitution (R294S) at the 294th position of the human neuraminidase-1 protein has been previously reported, an arginine to cysteine substitution (R294C) at the same site has not been reported before, and the 1191delG is a novel, hitherto unreported mutation8. Homology alignment showed that the amino acid arginine was conserved at the 294th position in the Neu1 protein in other mammalian species (orangutan, cow, rat and mouse) also (Fig. B). In fact, the 294th position amino acid lies within the fourth Asp-box motif, one of the five highly conserved motifs within the neuraminidase-1 protein which are essential for neuraminidase function9. Mutations within or near these motifs have been found to significantly impair the activity of the protein8,10. The R294C mutation also belongs to a group of amino acid substitutions (R294S, L270F, V217M, G218A, G227R, F260Y, A298V) which are located on the surface of the neuraminidase-1 molecule. It has been hypothesized that the surface of the neuraminidase-1 molecule, where these amino acids are located, functions as a binding site between sialidase and PPCA (protective protein/ cathepsin A) and/or beta- galactosidase to form a catalytically active high molecular weight complex and mutations in this site can result in disruption of this multi-enzyme complex11,12. However, this R294C mutation like the R294S mutation, most likely does not render the enzyme completely inactive and the mutant protein probably retains some catalytic activity, due to which it produces the less severe type I sialidosis phenotype8. The c.1191delG mutation would result in a mutant protein with disruption of the normal sequence distal to the 397th amino acid position and an added 71 amino acids at the carboxy-terminal end. The exact effect of this mutation at the functional level remains to be elucidated.
Fig.
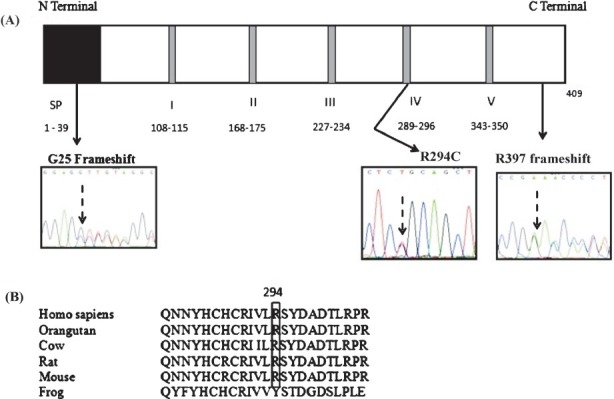
(A). Schematic representation of the human Neu1 protein with the relative positions of the three identified mutations marked by black arrows. The signal peptide region is indicated by a black box marked SP and the five conserved Asp box motifs are denoted by gray bands; the amino acids included in each are mentioned. The chromatograms show the sequences at the mutated sites. (B). Clustal 2.1 multiple sequence alignment of the human Neu1 protein sequence against the Neu1 protein sequences of other species shows that R (arginine) is conserved at the 294th position in other mammalian species also
Patient 2, with infantile onset type II sialidosis, was found to be homozygous for a 1-base deletion at nucleotide 73 (c.73delG) of the NEU1 gene. This deletion causes a frameshift from the 25th amino acid position and introduces a premature TAG termination codon at the 58th amino acid position, thereby resulting in a truncated protein with a truncation by 357 amino acids at the C-terminal end (Fig. A). This mutation would result in a severely truncated protein which is expected to be completely or almost completely inactive thereby producing a severe phenotype. Sequence analysis of exon 1 of the NEU1 gene in both the parents of this proband confirmed that both were heterozygous carriers of this mutation.
Further molecular genetic studies on Indian patients with sialidosis will help to establish whether there is any ethnic preponderance of NEU1 gene mutations in the Indian population and to elucidate further genotype-phenotype correlations in sialidosis.
Acknowledgment
The authors thank Dr Bobby Varkey M, Consultant Neurologist at the Lourdes Hospital, Kochi, for referring the patient with type I sialidosis (patient 1). Authors acknowledge the Indian Council of Medical Research, New Delhi, for financial support.
References
- 1.Thomas GH. Disorders of glycoprotein degradation: α-Mannosidosis, β-Mannosidosis, Fucosidosis and Sialidosis. In: Scriver CR, Beaudet AL, Sly WS, Vaile D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw Hill; 2001. p. 5820. [Google Scholar]
- 2.Harada F, Nishimura Y, Suzuki K, Matsumoto H, Oohira T, Matsuda I, et al. The patient with combined deficiency of neuraminidase and 21-hydroxylase. Hum Genet. 1987;75:91–2. doi: 10.1007/BF00273850. [DOI] [PubMed] [Google Scholar]
- 3.Bonten E, van der Spoel A, Fornerod M, Grosveld G, d’Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;10:3156–69. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- 4.Cardiff (UK): The Institute of Medical Genetics, Cardiff University, UK; 2011. [accessed on April 15, 2011]. The Human Gene Mutation Database. Available from: http://www.hgmd.org . [Google Scholar]
- 5.Young ID, Young EP, Mossman J, Fielder ER, Moore JR. Neuraminidase deficiency: case report and review of the phenotype. J Med Genet. 1987;24:283–90. doi: 10.1136/jmg.24.5.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhigjee AI, Seebaran AR, Petersen EM, Bill PL. Sialidosis type I: first report in the Indian population. A clinical, biochemical and electrophysiological study. Clin Neurol Neurosurg. 1991;93:115–8. doi: 10.1016/0303-8467(91)90050-y. [DOI] [PubMed] [Google Scholar]
- 7.Ganguly S, Gabani RU, Chakraborty S, Ganguly SB. Sialidosis type I (cherry red spot - myoclonus syndrome) J Indian Med Assoc. 2004;102:174–5. [PubMed] [Google Scholar]
- 8.Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9:2715–25. doi: 10.1093/hmg/9.18.2715. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Zaitsev S, Taylor G, d’Azzo A, Bonten E. Protective protein/cathepsin A rescues N-glycosylation defects in neuraminidase-1. Biochim Biophys Acta. 2009;1790:275–82. doi: 10.1016/j.bbagen.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukong KE, Elsliger M, Chang Y, Richard C, Thomas G, Carey W, et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum Mol Genet. 2000;9:1075–85. doi: 10.1093/hmg/9.7.1075. [DOI] [PubMed] [Google Scholar]
- 11.Lukong KE, Elsliger MA, Chang Y, Lefrancois S, Morales CR, Pshezhetsky AV. Mutations in sialidosis impair sialidase binding to the lysosomal multienzyme complex. J Biol Chem. 2001;276:17286–90. doi: 10.1074/jbc.M100460200. [DOI] [PubMed] [Google Scholar]
- 12.Criado GR, Pshezhetsky AV, Becerra AR, de Terreros IG. Clinical variability of type II sialidosis by C808T mutation. Am J Med Genet. 2003;116:368–71. doi: 10.1002/ajmg.a.10710. [DOI] [PubMed] [Google Scholar]