

Abstract
Background & objectives:
Moxifloxacin (MFX) is reported to have promising antimycobacterial activity, and has a potential to shorten tuberculosis (TB) treatment. We undertook this study to examine the influence of rifampicin (RMP) and isoniazid (INH) on the steady state pharmacokinetics of MFX individually in healthy individuals.
Methods:
A baseline pharmacokinetic study of MFX (400 mg once daily) was conducted in 36 healthy adults and repeated after one week of daily MFX with either RMP (450/600 mg) (n = 18) or INH (300 mg) (n = 18). Plasma MFX concentrations were determined by a validated HPLC method.
Results:
Plasma peak concentration and exposure of MFX was significantly lower and plasma clearance significantly higher when combined with RMP (P<0.001). The Cmax to MIC and AUC0-12 to MIC ratios of MFX were significantly lower during concomitant RMP (P<0.001). INH had no significant effect on the pharmacokinetics of MFX.
Interpretation & conclusions:
Concomitant RMP administration caused a significant decrease in Cmax and AUC0-12 of MFX, the mean decreases being 26 and 29 per cent, respectively. It is uncertain whether this decrease would affect the treatment efficacy of MFX. Prospective studies in TB patients are needed to correlate MFX pharmacokinetics with treatment outcomes.
Keywords: Isoniazid, moxifloxacin, pharmacokinetics, rifampicin, tuberculosis
Tuberculosis (TB) continues to remain a major public health problem, globally and in India. Though highly effective regimens have been available for the treatment of TB for the past many years, the long duration of such regimens has posed problems for TB treatment and control. The delivery of TB chemotherapy in the field would be much easier if the duration of treatment could be shortened without sacrificing efficacy. This requires the development of agents with potent bactericidal and/or sterilizing activity against Mycobacterium tuberculosis, which allow shortening the treatment duration, or agents with improved pharmacokinetics which allow lengthening of the dosing interval.
Earlier the fluoroquinolone group of drugs has been demonstrated to have significant therapeutic potential in the management of TB1,2. Among the newer generation of fluoroquinolones, moxifloxacin (MFX), a 8-methoxy fluoroquinolone, is a drug with promising antimycobacterial activity. Using a murine model, Nuermberger et al3,4 have demonstrated that MFX has a potential to shorten TB treatment by 2 months when substituted for isoniazid (INH). Moreover, the activity of MFX against ‘persisters’ was greater than that of INH and other fluoroquinolones in an in vitro model which has been confirmed in human studies where MFX has shown greater extended early bactericidal activity (EBA) than INH5,6. This suggests that MFX may be an ideal drug to be used to shorten the duration of TB treatment. MFX also has an activity similar to rifampicin (RMP) in human subjects with pulmonary TB, suggesting that it should undergo further assessment as part of a short course regimen for the treatment of drug-susceptible TB7.
Rifampicin induces a number of drug-metabolizing enzymes, having the greatest effects on the expression of cytochrome P450 (CYP) 3A4 in the liver and in the small intestine8. In addition, RMP induces some of the drug transporter proteins, and also phase II glucuronidation pathway9. MFX undergoes phase II metabolism by means of sulphate and glucuronide conjugation10. Pharmacokinetic studies done in pulmonary TB patients11 and healthy subjects12 have shown that RMP reduces plasma concentrations of MFX. However, the study was done in patients who were receiving RMP and INH, which did not allow discrimination between an effect of RMP or INH on the metabolism of MFX. The objective of this pharmacokinetic study was, therefore, to examine the pharmacokinetic interaction between MFX and that of RMP and INH individually, and also to study the influence of concomitant MFX on the pharmacokinetics of RMP and INH in healthy subjects.
Material & Methods
Subjects: The study was performed in 36 healthy adults (students of Madras Medical College, Chennai) (age >20 yr) with body weight >45 kg, and not suffering from any illness at the time of the study. All study subjects were contacted by investigator personally and underwent a complete clinical examination by a medical officer; haematology and clinical biochemistry testing, chest X-ray and electrocardiogram were also performed. A total of 38 volunteers were initially contacted and two of them were excluded as they were not willing to stay long in hospital. Only those subjects willing to participate and gave informed written consent were included. Smokers and chronic alcoholics were excluded. Sample size was calculated according to IJTLD 2005 which is 14 to 16 subjects for pharmacokinetic/bioavailability studies. The study protocol was approved by the Institutional Ethics Committee of Madras Medical College, Chennai.
Study design: This was a two-period, open-label, sequential-design pharmacokinetic study. Eligible subjects were administered MFX 400 mg once daily for five consecutive days under supervision. Baseline pharmacokinetic study of MFX was conducted on day 6 (Occasion 1). On day 7, the study subjects were randomly divided in to two groups comprising 18 subjects each, one group received MFX with RMP and the other group received MFX with INH daily under supervision for a period of 6 days. The doses of the drugs were RMP 450 mg for those with body weight <60 kg or 600 mg for those weighing ≥60 kg, and INH 300 mg. The pharmacokinetic study was repeated on day 13 (Occasion 2). By adopting this study design, each subject served as his/her own control. Thus the pharmacokinetics of MFX was determined on two occasions, first when MFX was given alone and second when MFX was given with either RMP (n = 18) or INH (n = 18); the effect of each drug on MFX pharmacokinetics was studied in 18 subjects. All drugs were administered through the oral route under complete supervision throughout the study period. All study subjects were instructed not to consume alcohol, grape juice or drugs that were known to act on the cytochrome P-450 enzyme system.
Conduct of study: The study was carried out at the Pharmacology Ward in Madras Medical College, Chennai, India during April - December 2010. During both occasions, the study participants were requested to get admitted to the ward a day prior to the study day. On the day of the study, a sample of blood (2 ml) was collected in a heparinised vacutainer (pre-dosing) after an overnight fast. The participants were administered either MFX (Occasion I), or MFX with RMP or INH (Occasion 2) under supervision with 200 ml water. Serial blood samples were collected at 1, 2, 4, 6, 8 and 12 h after drug administration. Care was taken to ensure that the study subjects did not take any substance that would interfere with the absorption of the study drugs. The blood samples were centrifuged immediately and plasma stored at -20°C until MFX levels were estimated. MFX levls were estimated in all the plasma samples collected during both occasions, while RMP and INH concentrations were estimated in samples collected during the second occasion of the study.
Drug estimations: Plasma MFX concentrations were determined by HPLC (Shimadzu, Kyoto, Japan) according to the method of Hemanth Kumar et al13. In brief, plasma samples were deproteinized using perchloric acid and analysis of the supernatant was performed using a reversed-phase C18 column (150 mm) and fluorescence detection at an excitation wavelength of 290 nm and an emission wavelength of 460 nm. The assay was specific for MFX and linear from 0.125 to 10.0 μg/ml. The average accuracy of the method was 99.4 per cent. Plasma RMP and INH were estimated by HPLC according to validated methods14,15. Plasma RMP estimation involved deproteinization with acetonitrile and analysis using a reversed-phase C18 column and UV detection at 254 nm. The assay was linear from 0.25-15.0 μg/ml with an average accuracy of 98 per cent. Plasma INH estimation involved deproteinization of plasma with para hydroxy benzaldehyde and trifluoroacetic acid and analysis using a reversed-phase C8 column and UV detection at 267nm. The assay was linear from 0.25 - 10.0 μg/ml with an average accuracy of 102.1 per cent.
Pharmacokinetic analysis: Certain pharmacokinetic variables such as maximum or peak concentration (Cmax), the time to attain Cmax (Tmax), exposure or area under the time concentration curve from 0 to 12 h (AUC0-12), apparent oral clearance (Cl) and half life (t1/2) were calculated by a non-compartmental model using WinNonlin software (Version 5.1) (Pharsight Corporation, Mountain View, CA, USA).
Statistical evaluation: Analysis of data was performed using SPSS (version 14) package (SPSS Inc., Chicago, IL, USA). The significance of differences in the Cmax, AUC0-12 and Cmax to MIC, AUC0-12 to MIC ratios of MFX, when administered alone and in combination with RMP or INH was calculated using paired t-test.
Results
The mean age and body weight of the study subjects who received MFX with RMP were 23.4 ± 2.9 yr and 62.5 ± 9.8 kg, and MFX with INH were 23.4 ± 2.3 yr and 60.0 ± 7.6 kg, respectively. There were 32 males and 4 females. The study subjects in the RMP and INH groups did not significantly differ in age, body weight and baseline pharmacokinetics of MFX. Eight and ten subjects, respectively received 450 and 600 mg RMP doses. The dose per kg body weight did not significantly differ between these two groups of subjects (8.3 vs. 8.7 mg/kg body weight). Hence no subgroup analysis to find the impact of dose variation on kinetic pattern was performed.
The pharmacokinetic variables calculated based on plasma concentrations of MFX alone and in combination with RMP are presented in Table I. RMP co-administration caused significant reduction in Cmax and AUC0-12 of MFX (P<0.001). This was accompanied by a significant increase in the plasma clearance of MFX (P<0.001). The mean decrease in Cmax and AUC0- 12 of MFX during concomitant RMP administration was 26 and 29 per cent, respectively. INH did not cause any significant change in the pharmacokinetics of MFX (Table II). The arithmetic mean ratios of AUC0-12 of MFX when given alone to that combined with RMP and INH was 0.70 and 0.87, respectively.
Table I.
Steady state pharmacokinetics of moxifloxacin (MFX) alone and in combination with rifampicin (RMP)
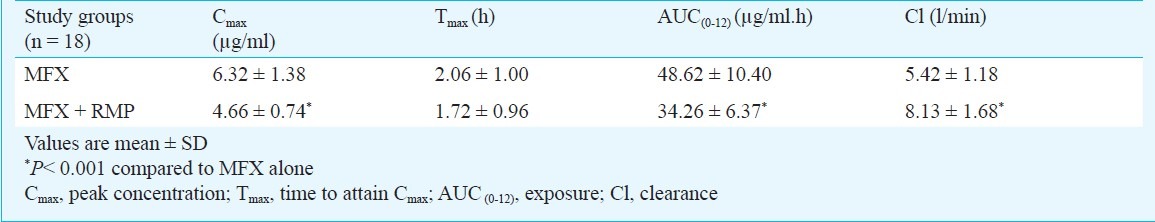
Table II.
Steady state pharmacokinetics of moxifloxcin (MFX) alone and in combination with isoniazid (INH)

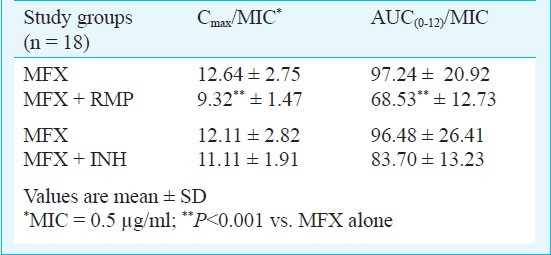
Since there was a huge variation in the number of males and females (16 males vs 2 females in each group), gender based subgroup analysis was not done. However, the mean plasma MFX values were almost similar in males and females. The mean Cmax to MIC and AUC0-12 to MIC ratios of MFX with and without RMP and INH are given in Table III. While concomitant RMP caused a significant decrease in both the ratios (P<0.001), INH did not cause a significant change.
Table III.
Pharmacodynamic values of MFX against M. tuberculosis
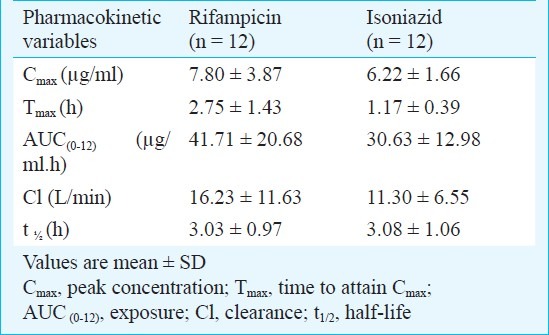
Among the 36 subjects who took part in this study, RMP and INH estimations were undertaken in 12 subjects each. The mean doses of RMP and INH were 8.4 and 4.8 mg/kg body weight, respectively. Table IV gives the steady state pharmacokinetics of RMP and INH.
Table IV.
Steady state pharmacokinetics of rifampicin (RMP) and isoniazid (INH) during moxifloxacin (MFX) coadministration
Discussion
Plasma drug concentrations are among the most important determinants of clinical response accounting for both efficacy and toxicity. Drug interactions could play a significant role when combinations of drugs are used for treatment of illnesses, particularly when any of the drugs are inducers or inhibitors of the hepatic microsomal enzyme pathway, which could thereby decrease or increase blood concentrations. Induction of CYP3A4 by RMP increases the rate of metabolism and decreases plasma MFX concentration. This could result in clinically significant interactions that might lead to altered therapeutic responses, therapeutic failures or toxic reactions16.
In this pharmacokinetic drug interaction study, significant drug interaction was found to exist between MFX and RMP. Reduced plasma peak concentration and exposure and increased clearance of MFX in the presence of RMP were observed. The interaction could probably be due to an increase in phase II metabolism of MFX caused by RMP, which is known to be a strong inducer of CYP450 isoenzymes, and also induces phase II metabolism, specifically, glucuronidation and sulphation9. Increased formation of MFX metabolite, namely, the sulphate metabolite was observed in healthy volunteers, when MFX was co-administered with RMP12. Concomitant RMP administration caused a 29 per cent mean decrease in plasma exposure of MFX, which is almost similar to that reported by Nijland et al (31%) and Weiner et al (27%)11,12. However, the pharmacokinetics of MFX did not significantly change when co-administered with INH, suggesting that RMP was mainly responsible for reducing plasma MFX concentrations.
The TBTC study 27/28 on MFX pharmacokinetics during TB treatment was undertaken by Weiner et al17. This study has compared (i) the pharmacokinetics of MFX alone versus MFX administered with RMP in healthy volunteers, and (ii) the pharmacokinetics of MFX among patients with TB being treated with multidrug therapy (INH or ethambutol, RMP and pyrazinamide) to those of healthy volunteers receiving MFX plus RMP. Although this study has been completed, the findings have not been published.
It has been reported that the pharmacokinetics of MFX exhibits inter-individual variability11,18. In the present study, this variability was taken care of by adopting a two-period sequential study design, in which the same individual was investigated on two occasions, and served as his/her own control.
Pharmacokinetic data and MIC values can be used to assist with the selection of an appropriate dose regimen for clinical trials. The two most relevant pharmacodynamic parameters for the concentration-dependent bactericidal activity of fluoroquinolones are peak concentration to MIC and exposure to MIC ratios. The drug is most effective when these ratios are maximized (Cmax to MIC ratio >10 & AUC0-12 to MIC ratio >100)19. Using a MIC value of 0.5 μg/ml20, we calculated the Cmax to MIC and AUC0-12 to MIC ratios of MFX and observed that these ratios were significantly compromised when MFX was co-administered with RMP. This suggested that concomitant RMP administration could lead to reduced therapeutic efficacy of MFX. In this study, the individuals received daily MFX; intermittent dosing could possibly amplify the extent of this interaction.
In combination therapy, it is important to ensure that adequate plasma concentrations of individual drugs are maintained within the therapeutic range to obtain maximal efficacy. In the absence of a control group, the present study design did not allow a precise evaluation of the effect of MFX on the pharmacokinetics of RMP and INH. However, the pharmacokinetic data of RMP and INH obtained in this study were compared with that of an earlier study done in our Centre21, and found that it compared well with that reported earlier. These findings suggested that MFX did not alter the bioavailability of RMP and INH.
Although certain pharmacokinetic studies including ours, have clearly shown significant reductions in MFX plasma concentrations during RMP co-administration, it is uncertain whether this decrease would affect the treatment efficacy of MFX. It would be useful to undertake prospective studies in TB patients who are undergoing treatment with MFX-containing regimens. The results of the TBTC study 27/28 in TB patients would provide useful information on this aspect17. This would enable to better understand the clinical relevance of the significant pharmacokinetic interaction between MFX and RMP. Tuberculosis patients usually receive a combination of RMP, INH, pyrazinamide (PZA) and ethambutol (EMB), and it would be worthwhile to investigate the additive effect of these drugs on the pharmacokinetics of MFX.
Acknowledgment
The authors thank Drs V. Kumaraswami and A.C. Yegneshwaran for their support. The authors also thank the healthy volunteers who took part in the study. The technical assistance rendered by Ms. V. Sudha and Shri P. Kumar is acknowledged. WinNonlin software was a kind gift provided to Dr A.K. Hemanth Kumar during his training program at the University of Alabama at Birmingham, USA under the NIAID/TRC/ICER program, which was renewed later by Pharsight Corporation, NC, USA.
References
- 1.Gillespie SH, Kennedy N. Fluoroquinolones: a new treatment for TB? Int J Tuberc Lung Dis. 1998;2:265–71. [PubMed] [Google Scholar]
- 2.Tuberculosis Research Centre. Shortening short course chemotherapy: A randomized clinical trial for treatment of smear positive pulmonary tuberculosis patients with regimens using ofloxacin in the intensive phase. Indian J Tuberc. 2002;49:27–38. [Google Scholar]
- 3.Nuermberger EL, Yoshimatsu T, Tyagi S, O’Brien RJ, Vernon AN, Chaisson RE, et al. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am J Respir Crit Care Med. 2004;169:421–6. doi: 10.1164/rccm.200310-1380OC. [DOI] [PubMed] [Google Scholar]
- 4.Nuermberger EL, Yoshimatsu T, Tyagi S, Williams K, Rosenthal I, Vernon AN, et al. Moxifloxacin-containing regimens of reduced duration produce a stable cure in murine tuberculosis. Am J Respir Crit Care Med. 2004;170:1131–4. doi: 10.1164/rccm.200407-885OC. [DOI] [PubMed] [Google Scholar]
- 5.Johnson JL, Hadad DJ, Boom WH, Daley CL, Peloquin CA, Eisenach KD, et al. Early and extended bactericidal activity of levofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2006;10:605–12. [PubMed] [Google Scholar]
- 6.Hu Y, Coates AR, Mitchison DA. Sterilising activities of fluoroquinolones against rifampin-tolerant populations of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47:653–7. doi: 10.1128/AAC.47.2.653-657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gosling RD, Uiso LO, Sam NE, Bongard E, Kanduma EG, Nyindo M, et al. The bactericidal activity of moxifloxacin in patients with pulmonary tuberculosis. Am J Respir Crit Care Med. 2003;168:1342–5. doi: 10.1164/rccm.200305-682OC. [DOI] [PubMed] [Google Scholar]
- 8.Niemi M, Backman JT, Fromm JF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42:819–50. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 9.Gallicano KD, Sahai J, Shukla VK, Seguin I, Pakuts A, Kwok D, et al. Induction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patients. Br J Clin Pharmacol. 1999;48:168–79. doi: 10.1046/j.1365-2125.1999.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stass H, Kubitza D. Pharmacokinetics and elimination of moxifloxacin after oral and intravenous administration in man. J Antimicrob Chemother. 1999;43(Suppl B):83–90. doi: 10.1093/jac/43.suppl_2.83. [DOI] [PubMed] [Google Scholar]
- 11.Nijland HMJ, Ruslami R, Suroto AJ, Burger DM, Alisjahbana B, van Crevel R, et al. Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin Infect Dis. 2007;45:1001–7. doi: 10.1086/521894. [DOI] [PubMed] [Google Scholar]
- 12.Weiner M, Burman W, Luo C, Peloquin CA, Engle M, Goldberg S, et al. Effects of rifampin and multidrug resistance gene polymorphism on concentrations of moxifloxacin. Antimicrob Agents Chemother. 2007;51:2861–6. doi: 10.1128/AAC.01621-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemanth Kumar AK, Ramachandran G. Simple and rapid liquid chromatography method for determination of moxifloxacin in plasma. J Chromatogr B. 2009;877:1205–8. doi: 10.1016/j.jchromb.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 14.Hemanth Kumar AK, Chandra I, Geetha R, Silambuchelvi K, Lalitha V, Prema G. A validated high performance liquid chromatography method for the determination of rifampicin and desacetyl rifampicin in plasma and urine. Indian J Pharmacol. 2004;36:231–3. [Google Scholar]
- 15.Moussa LA, Khassovani CE, Soulaymani R, Jana M, Cassanas G, Alric R, et al. Therapeutic INH monitoring using a simple high performance liquid chromatographic method with ultraviolet detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;766:181–7. doi: 10.1016/s0378-4347(01)00434-0. [DOI] [PubMed] [Google Scholar]
- 16.Finch CK, Chrisman CR, Baciewicz AM, Self TH. Rifampin and rifabutin drug interactions: an update. Arch Intern Med. 2002;162:985–92. doi: 10.1001/archinte.162.9.985. [DOI] [PubMed] [Google Scholar]
- 17.TBTC Study 27/28 PK: Moxifloxacin pharmacokinetics during TB treatment. [accessed on February 9, 2012]. Available from: http://clinicaltrials.gov/ct2/show/NCT00164463 .
- 18.Stass H, Kubzitza D, Schuhly U. Pharmacokinetics, safety and tolerability of moxifloxacin, novel 8-methoxy fluoroquinolone, after repeated oral administration. Clin Pharmacokinet. 2001;40:1–19. doi: 10.2165/00003088-200140001-00001. [DOI] [PubMed] [Google Scholar]
- 19.Berning SE. The role of fluoroquinolones in tuberculosis today. Drugs. 2001;61:9–18. doi: 10.2165/00003495-200161010-00002. [DOI] [PubMed] [Google Scholar]
- 20.Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, et al. Moxifloxacin, ofloxacin, sparfloxacin and ciprofloxacin against Mycobacterium tuberculosis: Evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother. 2007;51:576–82. doi: 10.1128/AAC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurumurthy P, Ramachandran G, Hemanth Kumar AK, Rajasekaran S, Padmapriyadarsini C, Swaminathan S, et al. Decreased bioavailability of rifampin and other antituberculosis drugs in patients with advanced human immunodeficiency virus disease. Antimicrob Agents Chemother. 2004;48:4473–5. doi: 10.1128/AAC.48.11.4473-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]